



Simvastatin and sildenafil combine to attenuate pulmonary hypertension
- L. Zhao 1 ,
- A. Sebkhi 1 ,
- O. Ali 1 ,
- B. Wojciak-Stothard 2 ,
- L. Mamanova 1 ,
- Q. Yang 1 ,
- J. Wharton 1 and
- M. R. Wilkins 1
- 1Experimental Medicine and Toxicology, Imperial College London, Hammersmith Hospital, and 2BHF Laboratories, Dept of Medicine, University College London, London, UK.
- L. Zhao, Experimental Medicine and Toxicology, Imperial College London, Hammersmith Hospital, Ducane Road, London W12 ONN, UK. E-mail: l.zhao{at}imperial.ac.uk
Abstract
Statins have been proposed to be a potential treatment for pulmonary arterial hypertension. If introduced into clinical practice, the statin would have to be used in conjunction with established therapy. We investigated the effects of combining simvastatin with a phosphodiesterase type-5 inhibitor, sildenafil, in the rat model of hypoxia-induced pulmonary hypertension.
Rats were allocated to either: 1) a prevention protocol, to receive simvastatin 20 mg·kg−1·day−1 by intraperitoneal injection or sildenafil 75 mg·kg−1·day−1 orally or the combination (or vehicle) for 2 weeks beginning at the start of exposure to hypoxia (10% inspired oxygen); or 2) a treatment protocol, where the same agents were administered in the last 2 weeks of a 4-week period of hypoxia.
In both protocols, the combination of sildenafil and simvastatin lowered pulmonary artery pressure and produced a significantly greater reduction in right ventricular hypertrophy and pulmonary vascular muscularisation than either drug alone. Moreover, the combination augmented significantly endothelial nitric oxide synthase expression and cGMP levels in the lung and right ventricle above that produced by either drug independently and resulted in greater inhibition of RhoA activity.
These data suggest that simvastatin can be usefully combined with sildenafil in the treatment of pulmonary arterial hypertension to achieve greater therapeutic benefit.
- Animal models
- hypoxia
- pulmonary hypertension
- RhoA
- sildenafil
- simvastatin
There is considerable interest in the pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (statins) on the cardiovascular system beyond the benefits accrued from lowering cholesterol alone. This interest extends to pulmonary arterial hypertension (PAH) 1, 2. While the treatment options for PAH, particularly idiopathic PAH, have improved in recent years, with licensing approval granted to two orally active drugs, phosphodiesterase type-5 (PDE5) inhibitors and endothelin receptor antagonists, in addition to prostanoids, the outlook for PAH in its severest forms is still poor 3, 4. New treatments are required.
In principle, statins may have a beneficial effect on the course of PAH on a number of levels. These include improving or restoring endothelial function, inhibiting the proliferation and migration of vascular smooth muscle cells, and decreasing vascular inflammation and oxidative stress 5–7. Moreover, the effects of statins are not confined to the vasculature. Recent animal and human studies suggest that these drugs may also have direct beneficial effects on the myocardium 6. Statins have been reported to reduce myocardial mass and fibrosis, increase capillary network density and attenuate electrical instability of the hypertrophied heart 8.
The pleiotropic effects of statins depend, in the main, on inhibiting the synthesis of the isoprenoid intermediates farnesylpyrophosphate and geranylgeranylpyrophosphate, which are essential for the post-translational isoprenylation, membrane localisation and activation of Ras and Rho small GTP-binding protein families respectively. These GTPases regulate many cellular functions and couple membrane growth factor receptors to intracellular pathways that affect cell proliferation 6, 7, 9. RhoA and its downstream mediator Rho-kinase have become an attractive target for the treatment of PAH 2, 7. Rho-kinase inhibitors, such as fasudil, inhibit the development of pulmonary hypertension in experimental models 7. Interestingly, cAMP- and cGMP-dependent protein kinases have a key role in regulating Rho activation and expression in vascular smooth muscle cells 10, 11. Inhibition of the RhoA/Rho-kinase signalling pathway may contribute, therefore, to the beneficial effect of established therapies, such as the PDE5 inhibitor sildenafil, in the treatment of pulmonary hypertension 12, 13.
Statins attenuate the development of pulmonary hypertension in experimental animal models 14–18 and have been reported to reverse established pulmonary hypertension and vascular remodelling, induced either by pneumonectomy and monocrotaline treatment 19 or chronic hypoxia 20. This appears to be achieved through increased apoptosis as well as reduced proliferation of smooth muscle cells in obstructive vascular lesions 17, 19. An effect on the mobilisation and homing of bone marrow-derived progentor cells has also been proposed 21. As a result, statins have become an attractive potential therapy for PAH. A recent uncontrolled observational study indicated that simvastatin is well tolerated in patients with different types of pulmonary hypertension, though it was not clear if improvement in their condition could be attributed to the drug treatment 22.
If statin therapy has a role in the treatment of PAH, it will be used in combination with established treatment, not as a sole therapy. We examined, therefore, the effect of combining simvastatin with a common first choice therapy for PAH, sildenafil, on hypoxia-induced pulmonary hypertension in the rat. Specifically, we investigated the effects of this combination on pulmonary artery pressure (Ppa), right ventricular hypertrophy (RVH) and pulmonary vascular remodelling. In addition, we explored the likely mechanism of any interaction through the endothelial nitric oxide synthase (eNOS)–cGMP and RhoA signalling pathways.
METHODS
Animals and experimental design
Male Sprague–Dawley rats (280–400 g) from Charles River (Margate, UK) were used for all experiments. All studies were conducted in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986 (London, UK).
Animals were divided into four groups (six to eight in each group) for each protocol: 1) normoxia control; 2) hypoxia control (given intraperitoneal vehicle saline 0.2 mL·rat−1·day−1); 3) hypoxia plus simvastatin 20 mg·kg−1·day−1 i.p. 14; 4) hypoxia plus sildenafil 75 mg·kg−1·day−1 (in drinking water); and 5) hypoxia plus simvastatin and sildenafil in combination. Animals were weighed every other day and treatment doses were calculated by using the most recent average body weights for the group. Injection was carried out at approximately the same time each day. The sildenafil dose was monitored by weighing drinking bottles daily, and adjusting concentration to maintain dose if necessary. All animals had free access to food and water and were exposed to a normal 12 h/12 h light/dark cycle.
Rats were exposed to chronic hypoxia in a 10% oxygen normobaric chamber as previously described 23. Two distinct protocols were employed. In one, the prevention study protocol, animals were pre-treated for 2 days and treatment was continued during 2 weeks of exposure to hypoxia. In the other, the treatment study protocol, animals were exposed to hypoxia for 4 weeks, with treatment beginning after 2 weeks of hypoxia exposure.
Haemodynamic measurements
At the end of the experiment, animals were weighed and anaesthetised (Hypnorm 0.25 mL·kg−1; midazolam 25 mg·kg−1 i.p.). Ppa was measured via a pre-curved catheter inserted through the right jugular vein, passed by the right ventricle and into the pulmonary artery. Systemic blood pressure (SBP) was measured by cannulating the carotid artery. These fluid-filled catheters were connected to the pressure transducer of a PowerLab Data Acquisition system (ADInstruments Ltd, Chalgrove, UK) and pressure measurements were recorded from stable tracings.
Following sacrifice, plasma was collected, hearts were harvested and ventricular chambers dissected carefully and weighed. RVH was assessed from the ratio of the right ventricle to the left ventricle, plus the septum mass. The right lungs were snap frozen in liquid nitrogen and stored at -80°C for biochemical measurements as detailed in the following sections. The left lungs were fixed by inflation with 10% formalin in phosphate-buffered saline, embedded in paraffin and sectioned for histology.
Histological analysis
Histological assessment of vascular remodelling was performed as previously described 24. Transverse lung sections were stained using van Gieson's elastic method. The proportion of vessels <100 μm in diameter, with double elastic laminae occupying >50% of the circumference, was quantified in two separate sections from each animal and expressed as a percentage of total vessels counted (per cent muscularised vessels). Counting was performed blind to the treatment of the rat from which the sections were derived.
eNOS Western blotting
Lungs were homogenised in extract buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% non-idet P-40, 1 mM PMSF, 1 mM dithiothreitol, protease inhibitors). Equal amounts of protein lysates (15 μg) were resolved using SDS-PAGE and transferred to nitrocellulose membranes (Immobilon™-P; Millipore, Watford, UK). Membranes were incubated with a purified mouse monoclonal antibody against human eNOS amino acid 1025–1203 (Becton Dickinson UK Ltd, BD Diagnostics, Oxford, UK) diluted (1:1,000) in 6% non-fat milk in PBS-Tween (PBS, 0.1% Tween 20) overnight at 4°C. After washing (6×10 min), they were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies, diluted (1:20,000) in 6% non-fat milk in PBS-Tween for 1 h at room temperature. They were then washed again and proteins visualised by chemiluminescence (SuperSignal; Pierce, Cramlington, UK). Optical densities of individual bands were measured. eNOS expression was normalised to β-actin for the lung and to α-actin for the right ventricle. The data are presented as per cent mean of normoxic control values.
Measurement of RhoA expression and activity
An affinity-precipitation assay for endogenous GTP-loaded (active) Rho is commonly regarded as a reliable assay for the measurement of activated protein levels, which reflect tissue RhoA activity. GTP-loaded RhoA levels were measured with recombinant glutathione S-transferase–rhotekin Rho-binding domain (GST-RBD) bound to glutathione beads (GE Healthcare, Uppsala, Sweden), prepared as described previously 25. Fragments of lung and right ventricle were homogenised, suspended in the ice-cold lysis buffer and spun down to remove cell debris. The detailed protocol of preparation of lysis/wash buffer used in Rho pull-down assays was provided by Upstate Biotechnology, Watford, UK (Rho Activation Assay Kit). 100 μg of GST-RBD was added to the supernatant and incubated with rotation for 60 min at 4°C. The beads were collected by centrifugation, washed three times in the lysis/wash buffer, and the affinity-precipitated RhoA was resolved by SDS-PAGE and detected by Western blotting. A mouse monoclonal anti-RhoA antibody (Santa Cruz Biotechnology, Wembley, UK) was used at a dilution of 1:1,000 and a goat polyclonal anti-mouse immunoglobulin G HRP-labelled antibody (Dako, Ely, UK) at 1:2,000. GTP-RhoA expression levels were normalised to total RhoA protein expression levels and presented as per cent mean of normoxia control values.
cGMP measurements
Lung and right ventricular tissue samples were homogenised in cold 6% (weight/volume) trichloroacetic acid at 2–8°C containing IBMX (2.5 mM). After centrifugation, supernatants were washed four times with water-saturated diethyl ether. The aqueous extracts were then lyophilised. Dried extracts were redissolved in the assay buffer and cGMP levels were measured by cGMP Enzymeimmunoassay Biotrak (EIA) System (GE Healthcare, Amersham, UK) according to the manufacturer's instructions.
Statistics
Data are expressed as the mean±sem of individual values. Data were analysed using one-way ANOVA, followed by Turkey's multiple comparison test or with unpaired t-test between two groups, to establish individual group differences. Statistical significance was set at p<0.05.
RESULTS
Prevention and treatment of hypoxia-induced pulmonary hypertension
Exposure to chronic hypoxia for 2 weeks resulted in a two-fold increase in mean Ppa. This was attenuated in animals receiving sildenafil and the combination with simvastatin, but not with simvastatin alone (fig. 1a⇓; table 1⇓).
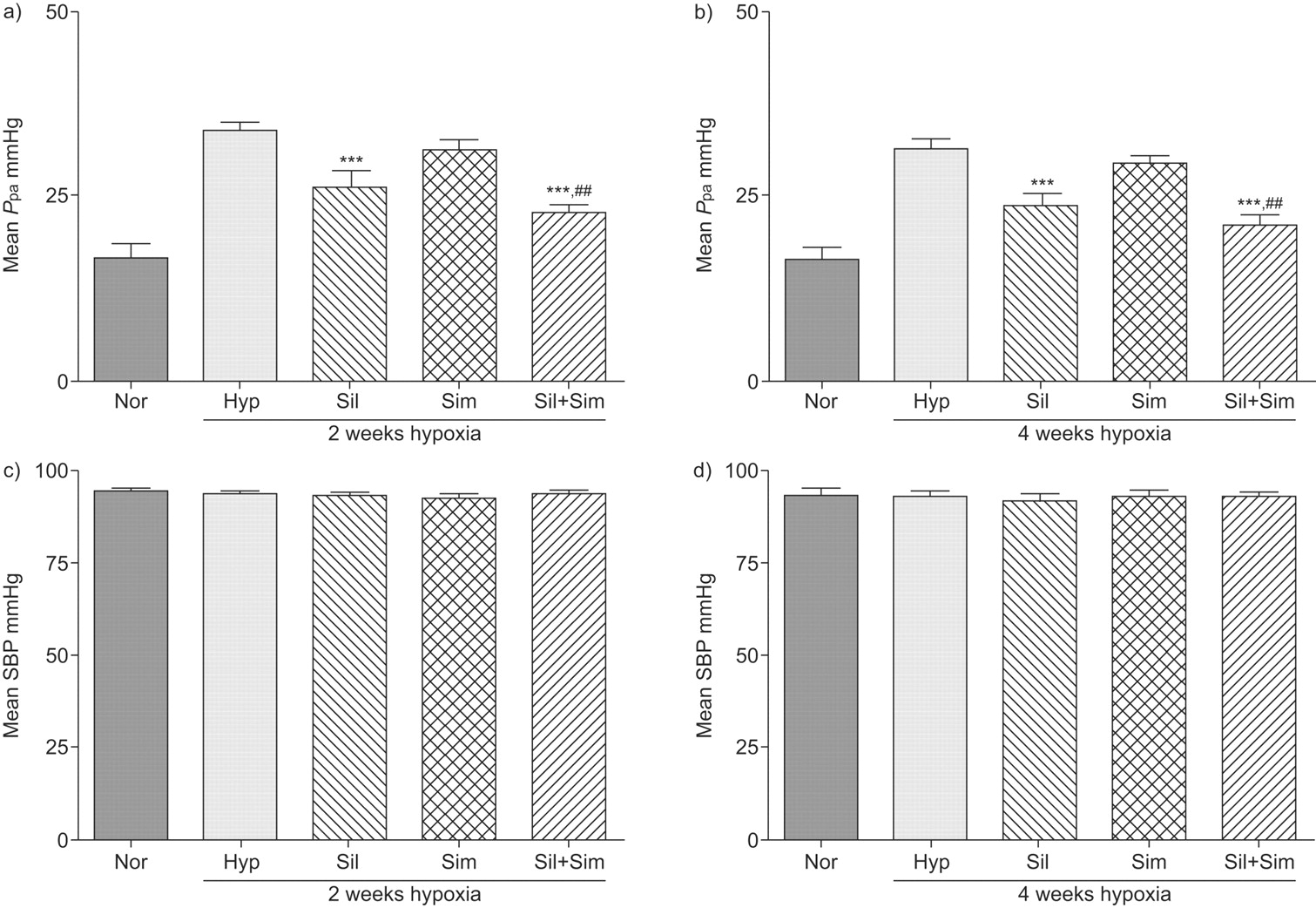
Mean pulmonary artery pressure (Ppa) and mean systolic blood pressure (SBP) in normoxic control rats (Nor) and animals exposed to 2 and 4 weeks of hypoxia (10% oxygen) in the absence (Hyp) and presence of simvastatin (25 mg·kg−1·day−1) (Sim), sildenafil (75 mg·kg−1·day−1) (Sil) or sildenafil plus simvastatin combination (Sil+Sim). Exposure to chronic hypoxia for 2 and 4 weeks resulted in a doubling of Ppa (33.5±1.6 and 31.2±1.3 compared to normoxia control 16.4±1.8 mmHg; p<0.001). In both prevention (a and c) and treatment studies (b and d), sildenafil treatment reduced mean Ppa (prevention 26.1±2.3 and treatment group 23.7±1.5 mmHg) significantly. Simvastatin alone had little effect (prevention 31.0±1.6 and treatment group 29.5±1.0 mmHg), but was effective when combined with sildenafil (prevention 22.6±1.2 and treatment group 20.9±1.7 mmHg), resulting in a mean Ppa comparable to normoxic controls (prevention p = 0.069 and treatment group p = 0.149, compared with normoxia control group). n≥6 for each group. ***: p<0.001 compared with hypoxia group. ##: p<0.01 compared with simvastatin group.
Per cent reduction with sildenafil, simvastatin and combination treatment on hypoxia-induced changes on pulmonary artery pressure (Ppa), right ventricular hypertrophy (RVH), vascular remodelling and haematocrit (Hct)
A similar increase in mean Ppa was observed after 4 weeks of exposure to hypoxia, consistent with our previous experience that the maximum effect is seen at around 2 weeks 23. Treatment with sildenafil alone and in combination with simvastatin produced a marked reduction but simvastatin alone had little effect (fig. 1b⇑; table 1⇑). No significant effect was seen on SBP in either study protocol (fig. 1c⇑ and d).
Prevention and treatment of hypoxia-induced right ventricular hypertrophy
Hypoxia-vehicle treated animals showed significant RVH after both 2- and 4-week hypoxia (fig. 2⇓). Administration of either sildenafil or simvastatin alone was accompanied by a significant reduction in RVH in both protocols and the effect was significantly enhanced following combined treatment with both drugs (fig. 2⇓; table 1⇑).

Right ventricular hypertrophy (RVH) in normoxic control rats (Nor) and animals exposed to 2 and 4 weeks of hypoxia (10% oxygen) in the absence (Hyp) and presence of simvastatin (25 mg·kg−1·day−1) (Sim), sildenafil (75 mg·kg−1·day−1) (Sil) or sildenafil plus simvastatin combination (Sil+Sim). RVH was assessed as the ratio of the right ventricle to the left ventricle plus the septum mass (RV/LV+sep). Hypoxia induced significant RVH after both 2 and 4 weeks of hypoxia (0.56±0.02 and 0.54±0.02 compared with normoxia 0.28±0.01; p<0.001). In both the a) prevention and b) treatment study groups, administration of sildenafil (prevention 0.49±0.02 and treatment 0.48±0.01) and simvastatin (prevention 0.41±0.01 and treatment 0.40±0.01) attenuated RVH significantly, the effect of simvastatin being significantly greater than that seen with sildenafil (p<0.01). The effect was significantly enhanced following combined treatment with both drugs (prevention 0.33±0.01 and treatment 0.33±0.01). n≥6 for each group. *: p<0.05; ***: p<0.001 compared with hypoxia group. ##: p<0.01; ###: p<0.001 compared with simvastatin group.
Prevention and treatment of hypoxia-induced pulmonary vascular remodelling
The proportion of muscularised pulmonary arterioles increased markedly in rats exposed to hypoxia for 2 or 4 weeks (to 75–80%) compared with normoxia controls (15%; p<0.001) (fig. 3a–b⇓). Sildenafil and simvastatin attenuated the development of muscularisation in both study protocols and administered together had a greater effect than either agent given alone (fig. 3a–b⇓).

Percentage muscularised vessels in normoxic control rats (Nor) and animals exposed to 2 and 4 weeks hypoxia (10% oxygen) in the absence (Hyp) and presence of simvastatin (25 mg·kg−1·day−1) (Sim), sildenafil (75 mg·kg−1·day−1) (Sil) or sildenafil plus simvastatin combination (Sil+Sim). The proportion of muscularised pulmonary arteries increased markedly in rats exposed to hypoxia for 2 or 4 weeks (75–80%) compared with normoxia controls (15±0.7%). In the a) prevention and b) treatment study groups, sildenafil (prevention 61.9±3.3 and treatment 63.3±2.0%) and simvastatin (prevention 61.9±3.3 and treatment 59.9±2.7%) both prevented and reversed vascular remodelling and when administered together had a greater effect than either agent alone (prevention 49.5±2.9 and treatment group 45.0±3.0%). **: p<0.01; ***: p<0.001 compared with hypoxia group. ##: p<0.01 compared with simvastatin group or sildenafil group. Scale bars = 50 μm.
Effects on haematocrit
Hypoxia exposure for 2 and 4 weeks induced significant increases in haematocrit (56±3% and 69±2%; p<0.001) compared with normoxia control (40±1%). Treatment with simvastatin, but not sildenafil, was associated with a reduction in hypoxia-induced polycythaemia (table 1⇑).
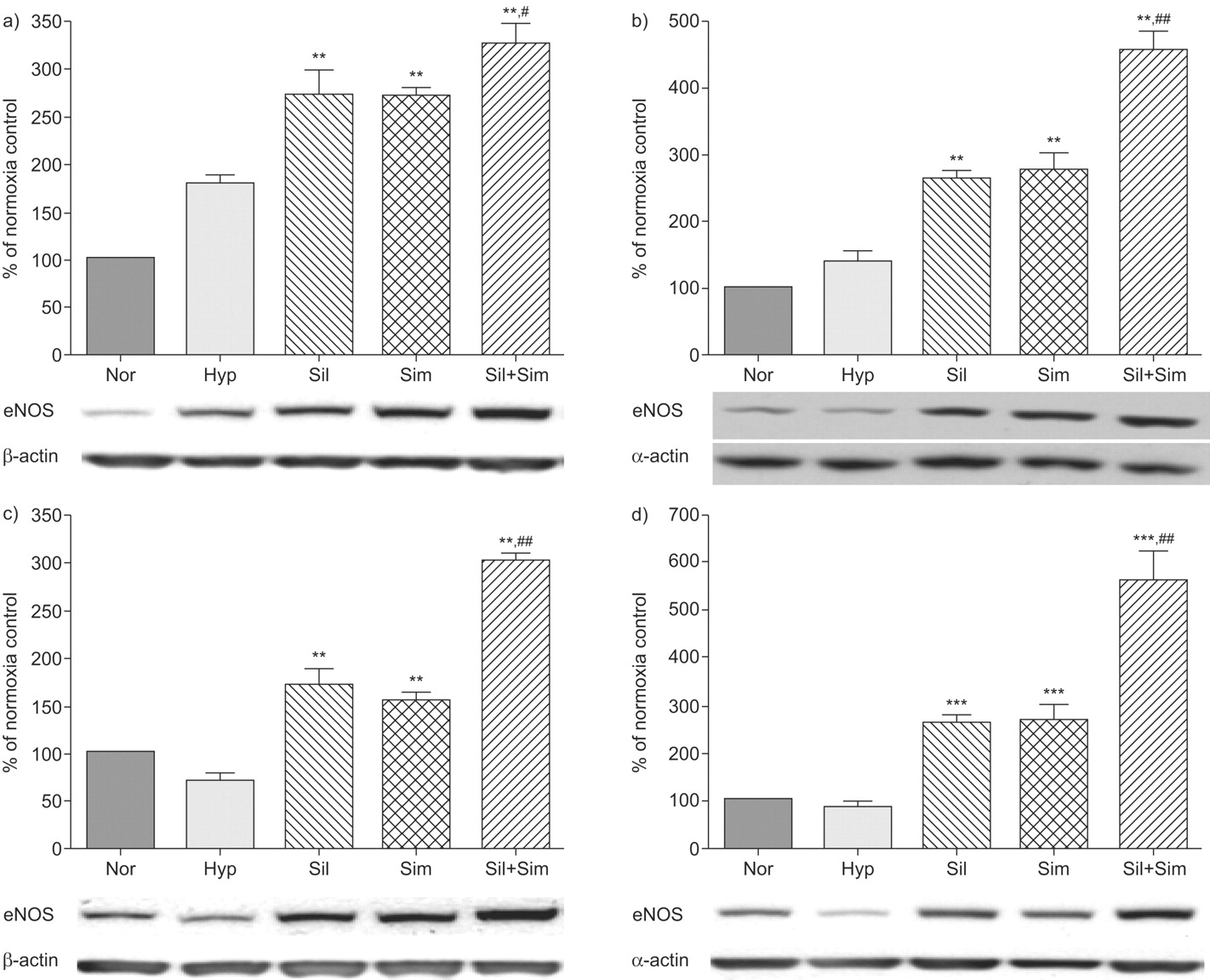
Effects on eNOS expression levels
Levels of eNOS protein expression in both the lung (179±10%; p<0.05) and right ventricle (136±18%; p<0.05) were increased after 2 weeks of hypoxia exposure (fig. 4a–b⇓). Sildenafil and simvastatin both induced a further rise in eNOS expression and when administered together had a greater effect than either agent alone (fig. 4a–b⇓). Following 4 weeks of hypoxia exposure, eNOS levels were decreased in both the lung (72±7%; p<0.05) and right ventricle (85±10%; p<0.05) compared to normoxia controls. Treatment with sildenafil and simvastatin again induced a significant increase, which was enhanced further when the treatments were combined (fig. 4c–d⇓).

Endothelial nitric oxide synthase (eNOS) expression levels in the lung (a and c) and right ventricle (b and d). The data were generated from optical density measurements of individual bands from Western blots and normalised to β-actin for the lung and to α-actin for the right ventricle. Changes in eNOS expression are presented as per cent mean of normoxia control values. a and b) Prevention study group; c and d) treatment study group. Representative examples of Western blots of eNOS, β-actin and α-actin are shown below the graphs. n = 4 for each group. Nor: normoxic controls; Hyp: hypoxia (10% oxygen) group; Sil: sildenafil group; Sim: simvastatin group; Sil+Sim: sildenafil plus simvastatin combination. **: p<0.01; ***: p<0.001 compared with hypoxia group. #: p<0.05; ##: p<0.01 compared with simvastatin or sildenafil group.
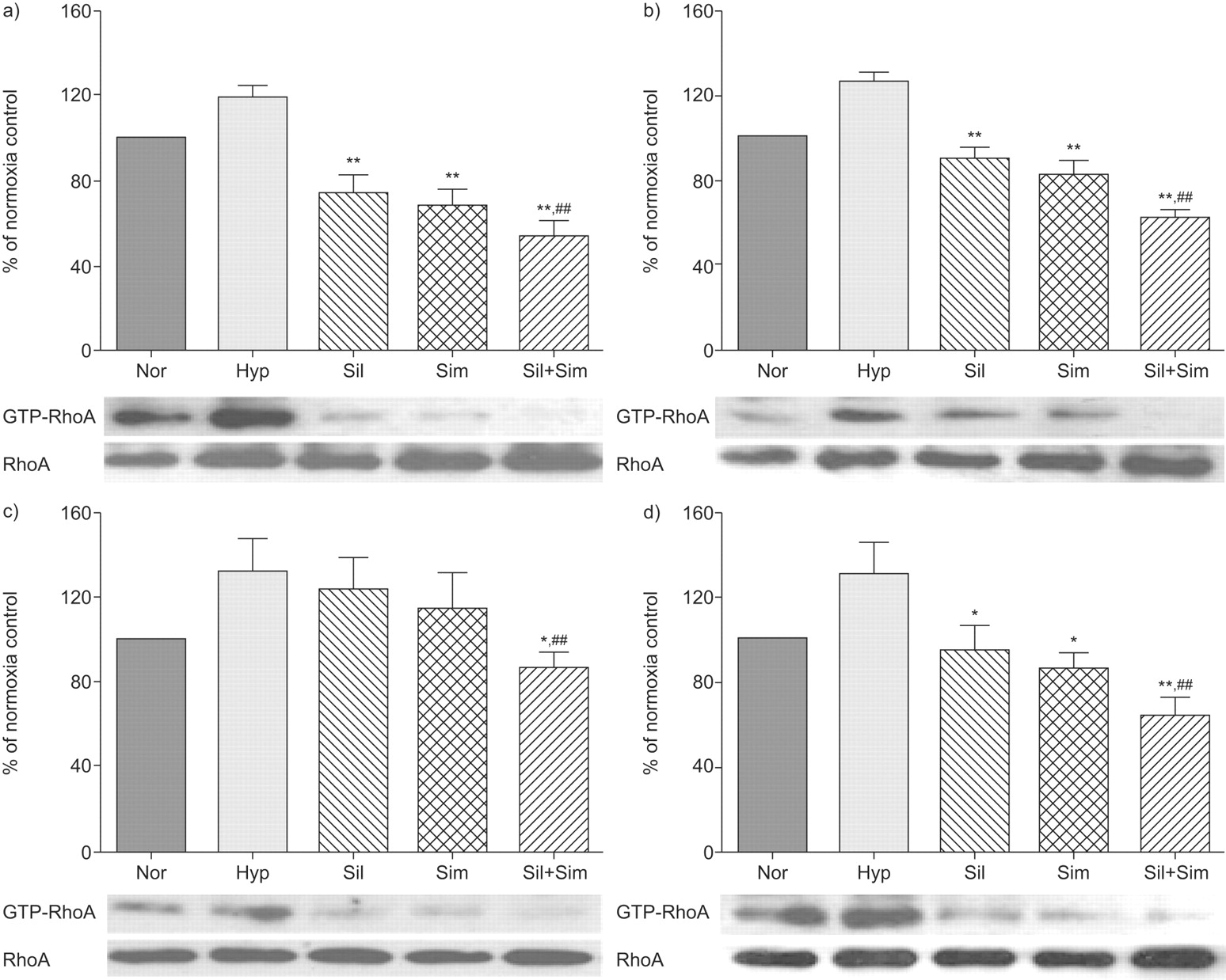
Effects on RhoA expression and activity
Hypoxia exposure for 2 and 4 weeks induced an increase in GTP-RhoA expression levels in the lung and right ventricle compared with normoxic control tissues, without significant changes in RhoA protein expression (fig. 5⇓). In the lung tissues from the prevention protocol, sildenafil or simvastatin alone significantly reduced GTP-RhoA expression levels compared with hypoxic controls, while the combination treatment had the strongest effect (fig. 5a⇓). Similarly in the right ventricle, single treatment with sildenafil or simvastatin was sufficient to reduce GTP-RhoA expression levels to normoxia control levels, while the combination treatment (80±4%) significantly reduced GTP-RhoA expression levels below that measured in animals in a normal atmosphere (fig. 5b⇓). In the treatment protocol, single treatment with sildenafil or simvastatin reduced GTP-RhoA expression levels in the right ventricle, and the combination treatment had the greatest effect (fig. 5c–d⇓).

Effects on GTP-RhoA and RhoA expression levels in the lung (a and c) and right ventricle (b and d). GTP-RhoA levels (a measure of activated RhoA protein) was measured in affinity precipitation assays and resolved by Western blotting. The data were normalised to total RhoA protein and expressed as per cent mean of normoxia controls. a and b) Prevention study group; c and d) treatment study group. Representative examples of Western blots showing changes in GTP-RhoA and RhoA expression levels are shown below the graphs. n = 3 for each group. Nor: normoxic controls; Hyp: hypoxia (10% oxygen) group; Sil: sildenafil group; Sim: simvastatin group; Sil+Sim: sildenafil plus simvastatin combination. *: p<0.05; **: p<0.01 compared with hypoxia group. ##: p<0.01 compared with simvastatin or sildenafil group.
Effects on cGMP levels
The levels of cGMP were not significantly affected by chronic hypoxia following 4 weeks of exposure (fig. 6⇓). Sildenafil and simvastatin both induced a modest increase in cGMP levels in the lung and right ventricle and a marked two- to three-fold rise was observed when the two drugs were administered together (fig. 6⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects on cGMP levels in the lung (a and c) and right ventricle (b and d) of normoxic control rats (Nor) and animals exposed to 2 and 4 weeks of hypoxia (10% oxygen) in the absence (Hyp) and presence of simvastatin (25 mg·kg−1·day−1) (Sim), sildenafil (75 mg·kg−1·day−1) (Sil) or sildenafil plus simvastatin combination (Sil+Sim). a and b) Prevention study group; c and d) treatment study group. *: p<0.05 compared with normoxia control and hypoxia group. ###: p<0.001 compared with all the other groups.
DISCUSSION
The current approach to the treatment of PAH is to combine drugs from different classes in the expectation of achieving greater therapeutic benefit. This study provides compelling evidence to suggest that the combination of sildenafil and simvastatin has therapeutic potential. In a rat model used in the pre-clinical assessment of all drug therapies approved for PAH to date, the combination of simvastatin and sildenafil, whether given at the start of exposure to hypoxia or after 2 weeks of hypoxia-induced pulmonary hypertension, was associated with a greater reduction in RVH and pulmonary vascular remodelling than seen with either drug alone. The combination also showed specificity for the pulmonary circulation. Moreover, these responses were accompanied by a more pronounced increase in lung and right ventricular eNOS expression and cGMP levels and greater reduction in GTP-RhoA levels (a marker of activated RhoA protein) in these tissues: biochemical effects consistent with the known actions of PDE5 and HMG CoA reductase inhibition.
Simvastatin has already been investigated in a number of different experimental models of pulmonary hypertension. Girgis and co-workers 14, 20 have shown previously that simvastatin (20 mg·kg−1·day−1) both prevented and regressed hypoxia-induced pulmonary hypertension, RVH and vascular remodelling in rats. Nishimura and co-workers 17, 19 reported that simvastatin 2 mg·kg−1·day−1 orally both prevented and reversed pulmonary hypertension and neointima formation and improved survival in a challenging rat model in which pulmonary hypertension is induced by monocrotaline and pneumonectomy. Recently, McMurtry et al. 26 found simvastatin (2 mg·kg−1·day−1 by gavage) less impressive in the monocrotaline model, retarding the progression of pulmonary hypertension and RVH at 4 weeks but with no effect on survival. Taraseviciene-Stewart et al. 27 have reported that simvastatin (10 mg·kg−1·day−1 by gavage) led to a significant improvement of the aggressively proliferative form of severe pulmonary hypertension induced by chronic vascular endothelial growth factor receptor blockade (SU5416) in combination with chronic hypoxia. The dose of simvastatin used in our study was at the higher end of the range used in previous studies. Despite this, used alone, we observed modest reductions in Ppa and hypoxia-induced polycythaemia compared with these reports 14, 20. There was a marked effect on RVH and vascular remodelling, although it was not fully reversed. It is possible that the lack of effect on Ppa is masked by reduced pulmonary vascular resistance and an improvement in cardiac output, but neither was measured in this study.
The more pronounced effect of simvastatin on RVH and vascular structure compared with Ppa may reflect a direct antitrophic action and its lack of vasoactive properties, although the relative contribution of vasoconstriction and vascular remodelling to hypoxia-induced pulmonary hypertension is the subject of debate 28. Sildenafil, however, is a more effective vasorelaxant agent, with some antiproliferative properties. Sildenafil has been previously shown to be effective in the rat hypoxia model of pulmonary hypertension but does not regress Ppa, RVH and pulmonary vascular remodelling to normal 23. The addition of simvastatin to sildenafil treatment maintained the reduction in Ppa and demonstrated additional antitrophic effects. Significantly, the dose of sildenafil used in this study (75 mg·kg−1·day−1 orally) is at or near the top of the dose–response curve for this drug in the rat hypoxia model, producing no greater reduction in Ppa than 25 mg·kg−1·day−1. Since increasing the dose of sildenafil has no further effect, the additional benefit gained from combination with simvastatin most likely reflects a true pharmacodynamic effect.
The cardiovascular effects of sildenafil are mediated mainly by cGMP. Both sildenafil and simvastatin increased eNOS expression and the combination of the two drugs had a more marked effect on eNOS expression and a greater than additive effect on tissue cGMP levels. In addition, combination of the two drugs markedly reduced GTP-RhoA expression. Levels of activated RhoA protein were significantly increased in both the lung and right ventricle after 2 and 4 weeks of hypoxia, in agreement with published data 12, 29. Both simvastatin and sildenafil attenuated GTP-RhoA expression in the lung and right ventricle during exposure to hypoxia, an effect augmented by combining the two drugs. RhoA is considered to be the main upstream activator of Rho kinase. RhoA may contribute to pulmonary hypertension by mediating the effects of vasoconstrictors such as angiotensin-II, endothelin-1 and acetylcholine and/or downregulating eNOS expression and nitric oxide production in endothelial cells 30, 31. RhoA and Rho kinase are important for the differentiation and survival of vascular smooth muscle cells and prolonged inhibition induces apoptosis in these cells in vitro 32. Rho proteins can also regulate extracellular matrix production in various cell types 33. RhoA has become a target for drug development in pulmonary hypertension 34. Intravenous administration of the Rho kinase inhibitors fasudil or Y-27632 attenuated pulmonary hypertension and pulmonary vascular remodelling and enhanced eNOS expression in monocrotaline and chronic hypoxia-induced PH in rodents 35–37. Interestingly fasudil was less effective in reducing chronic hypoxia-induced pulmonary hypertension in eNOS-deficient mice compared with wild-type animals, suggesting Rho kinase signalling is eNOS dependent 37. Furthermore, the cardioprotective effects of statins also appear to involve eNOS 38, 39. Our data would support the thesis that eNOS–RhoA–cGMP is a convergent signalling pathway mediating the combined, augmented effects of sildenafil and simvastatin on Ppa, RVH and vascular remodelling in this rat model of pulmonary hypertension.
There are a number of limitations to this study. First, it is not clear whether we have achieved the maximum pharmacological effects of simvastatin in our hypoxia model. Specifically, the effects of simvastatin on Ppa and polycythaemia were less marked than those previously reported, despite using the same dose of simvastatin 14, 20. However, we did note some external surface granularity of the liver in the simvastatin treated animals, suggesting we were close to a dose that might cause liver toxicity. Furthermore, we did not assess pulmonary vascular resistance and cardiac output. Thirdly, a pharmacokinetic interaction between simvastatin and sildenafil needs to be investigated in future studies.
In summary, the addition of simvastatin to sildenafil has a significant protective effect and significantly reverses hypoxia-induced pulmonary hypertension and remodelling. The effects, particularly the trophic effects on RVH and vascular remodelling, are more pronounced when the drugs are combined than when given alone. This response may be explained, at least in part, by an additive effect on eNOS expression and cGMP levels in the lung and right ventricle, and a more marked inhibition of RhoA activity.
Support statement
This work was supported by the British Heart Foundation.
Statement of interest
Statements of interest for J. Wharton and M.R. Wilkins can be found at www.erj.ersjournals.com/misc/statements.dtl
- Received September 19, 2008.
- Accepted March 26, 2009.
- © ERS Journals Ltd