



Some principles and practices of genetic biobanking studies
- A. K. MacLeod 1 ,
- D. C. M. Liewald 1 ,
- M. M. McGilchrist 2 ,
- A. D. Morris 2 ,
- S. M. Kerr 1 and
- D. J. Porteous 1 , 3
- 1Medical Genetics Section, Centre for Molecular Medicine, University of Edinburgh, Western General Hospital, Edinburgh, 2Health Informatics Centre, University of Dundee, Ninewells Hospital, Dundee, UK, 3on behalf of Generation Scotland; for details of the Generation Scotland Scientific Committee, please see the Acknowledgements section.
- A. K. MacLeod, Molecular Medicine Centre, Western General Hospital, Edinburgh, EH4 2XU, UK, Fax: 44 1316511059. E-mail: andrew.macleod{at}ed.ac.uk
Abstract
Genetic biobanking studies are becoming increasingly common as researchers recognise the need for large samples to identify the genetic basis of susceptibility to complex disease. In the present review, the authors give a brief overview of some of the issues that should be considered when implementing such a large-scale project, from study design to sample management, data coding and storage to the statistical analysis and engagement with the public. Specific solutions to these issues are presented, as implemented in the Generation Scotland projects, but the general principles outlined are relevant to any biobanking study.
- Biobanking
- data acquisition
- data coding
- laboratory information management system
- statistical genetics
SERIES “THE GENETIC AND CARDIOVASCULAR ASPECTS OF OBSTRUCTIVE SLEEP APNOEA/HYPOPNOEA SYNDROME”
Edited by R.L. Riha and W.T. McNicholas
Number 1 in this Series
Several common, complex diseases have genetic components to their susceptibility 1. These diseases may have several genetic risk factors that interact with each other, and the environment, as individual risk factors have small marginal effects which require large samples to detect. This has led, in part, to the development of large-scale biobanking projects attempting to locate and identify genetic effects underlying disease susceptibility 2. In the present review, the authors outline some principles to be considered when designing and implementing such a project, illustrated using examples from Generation Scotland (GS). GS is a multi-institution, cross-disciplinary collaboration between the Scottish University Medical Schools, the National e-Science Centre (Edinburgh and Glasgow, UK), the Scottish School of Primary Care (Dundee, UK), UK Medical Research Council Units in Scotland and the Information Services Division (ISD) of National Health Service (NHS) National Services Scotland (Edinburgh). GS currently comprises four complementary projects: the Scottish Family Health Study (GS:SFHS) 3, Genetic Health in the 21st Century (GS:21CGH), the Donor DNA Databank (GS:3D) and Biomarkers to Battle Chronic Diseases. Together these projects will recruit a cohort of more than 50,000 individuals and family members (approximately 1% of the Scottish population) for genetic analysis, and will establish epidemiological, statistical and informatics infrastructure of benefit to future studies. To accomplish this, GS implements protocols to collect detailed phenotypes relevant to common diseases, including cardiovascular disease, diabetes, obesity and mental health disorders. Consent is also obtained for health developments over the life course (traced through electronic records) to be used for research purposes. Quantitative trait data are also collected 3. Implementations in the current review are presented in the context of the GS projects, but general principles can be applied to any biobanking study.
STUDY DESIGN
Study design will be dictated by the phenotype/disease of interest, and the populations and resources available to researchers. Taking obstructive sleep apnoea/hypopnoea syndrome (OSAHS) as an example of a common disease with complex aetiology, the classical approaches of family, twin and linkage studies have found approximately 40% of trait variation attributable to genetic factors 4, with relative risks of 1.9–2.0 for first degree relatives 5. However, these studies are hampered by the lack of a precise characterisation of OSAHS, and variability in the measures used to quantify breathing interruptions. Genome-wide scans for OSAHS show evidence of linkage to several regions of the genome 6, implying the influence of multiple genetic factors. Some studies have also focused on intermediate phenotypes, such as obesity 7, which are themselves complex traits. Association studies have provided further information on the OSAHS pathogenesis 8, but further studies are necessary to confirm these results. A positive association for a categorical (e.g. disease status) trait represents a significant difference in allele frequencies between case and control groups. Ideally, this would reflect a direct effect of a single nucleotide polymorphism (SNP) on disease susceptibility through a change in protein sequence or regulatory elements, but it is more likely to reflect an indirect association: a SNP in strong linkage disequilibrium (LD) with a causative SNP. LD is a measure of the correlation between alleles at distinct loci, and forms the basis of the HapMap project 9 to catalogue common variation in the human genome by quantifying LD between several thousand SNPs and constructing maps of common haplotypes. Levels of LD in the test population(s) need to be considered when determining which markers to genotype in a specific investigation.
Population-based association studies risk spurious associations, which can give false positive results, arising from unseen population structure or admixture: confounding can occur if the sample populations consist of two or more subpopulations that differ in both allele frequencies and disease prevalence. A well-designed study will minimise such effects, by matching case/control groups for ethnicity or by correcting for observed structure. Methods to combat spurious associations include genomic control 10, which uses unlinked markers to quantify inflation of the test statistic and adjusts it accordingly, or structured association methods 11, which assign individuals probabilistically to a subpopulation, and measure association within these groups. Using family-based controls obviates the needs for such methods by constructing a test that is unaffected by population structure. The transmission disequilibrium test (TDT) 12 returns a positive result when any association between marker and disease status is caused by genuine linkage and not population structure. The TDT was initially developed for parent/affected offspring trios by comparing the frequency of alleles transmitted to affected offspring from heterozygous parents to their Mendelian expectations, with any significant deviation indicating linkage and association between trait and marker. This avoids problems of stratification, as the control group consists of untransmitted alleles within the family, and several extensions to the basic TDT have been implemented. The underlying theory forms the basis of a more general class of family-based association tests 13, which also construct test statistics based on the covariance between genetic and phenotypic residuals, but allow for a more generalised model, incorporating multiple traits and arbitrary family structures in a single test. The family-based data collected in SFHS will lend itself well to these sorts of family-based studies, as well as utilising the other advantages of family studies 14.
ENGAGEMENT
Engagement with the public and patients is essential in any biobanking study, to encourage participation from communities of interest, and engender trust between those communities and scientists. This was an integral consideration of GS from its early stages, beginning before sample collection with a dedicated team of researchers investigating the ethical, legal and social aspects of the study. Consultation initially involved reviews and interviews with focus groups 15 and is ongoing through dialogues with citizen groups, interviews with family members, and exit questionnaires completed by participants after their clinic visit. These processes are outlined in more detail on the consultation pages of the GS website 16. Trust between researchers and communities can also be promoted through the use of primary care specialists as intermediaries between the project and the community 17. SFHS families are initially recruited through general practice lists, allowing interaction between volunteers and trusted professionals.
Engagement between biobanking projects is also desirable. The Public Population Project in Genomics (P3G) 18 is an international consortium set up to promote scientific collaboration between biobanks and to facilitate harmonisation between disparate projects, allowing data to be combined. GS is a charter member of P3G, along with several other biobanking projects, and seeks to work towards commonality between the projects in terms of data collected.
SAMPLE COLLECTION
Biobanks may collect samples from multiple locations and process them in different laboratories. This raises issues of data integration, for which GS has developed bespoke methods to automate collection of phenotypic data to complement other IT systems. This allows the development of a linkage and integration mechanism which gives researchers access to sensitive data held in a dispersed model, aliased to ensure anonymity. Key to this system are the Volunteer Management Service (VMS) and the Appointments Service (APS), developed by the University of Dundee (Dundee, UK). The former manages volunteer identity and contact management, while the latter manages clinic appointment identity. The University of Edinburgh (Edinburgh, UK) GS team has developed a complementary set of flexible, phenotype collection modalities that allow reliable data collection throughout the interaction of a research participant with the data acquisition process (fig. 1⇓). This system integrates with the VMS/APS, maintaining anonymity while providing researchers with transient identifier data, to ensure validity. Anonymity is preserved by using unique sample IDs for each participant: GS labels contain sample IDs in human-readable and barcode format, and are used on all tubes and paperwork linked to the participant, including cryotags suitable for freezing. When received in the laboratory, sample barcodes can be scanned directly into sample inventory software, reducing user input error.

Schematic of information technology behind data acquisition (phenotype) for Generation Scotland: Genetic Health in the 21st Century and the Donor DNA Databank. CHI: community health index; VMS: volunteer management service; LIMS: laboratory information management system. Database servers are represented in yellow, the web server in blue, and desktop or laptop PCs in red.
LABORATORY SAMPLE MANAGEMENT
Laboratories play a pivotal role in successful implementation of genetic studies. During the process of study design based on statistical guidelines, the necessary biological samples to be collected should also be considered, along with methods of extraction and storage. Venous blood is a convenient source of DNA, but useful quantities can be obtained from the less invasive method of saliva/mouthwash 19. Serum, plasma and urine allow measurement of biochemical and proteomic phenotypes, which can enrich a genetic study. All samples have high intrinsic value, which may cause delays and increased costs if misplaced or badly stored. In GS this issue has been addressed by the implementation of a laboratory information management system (LIMS).
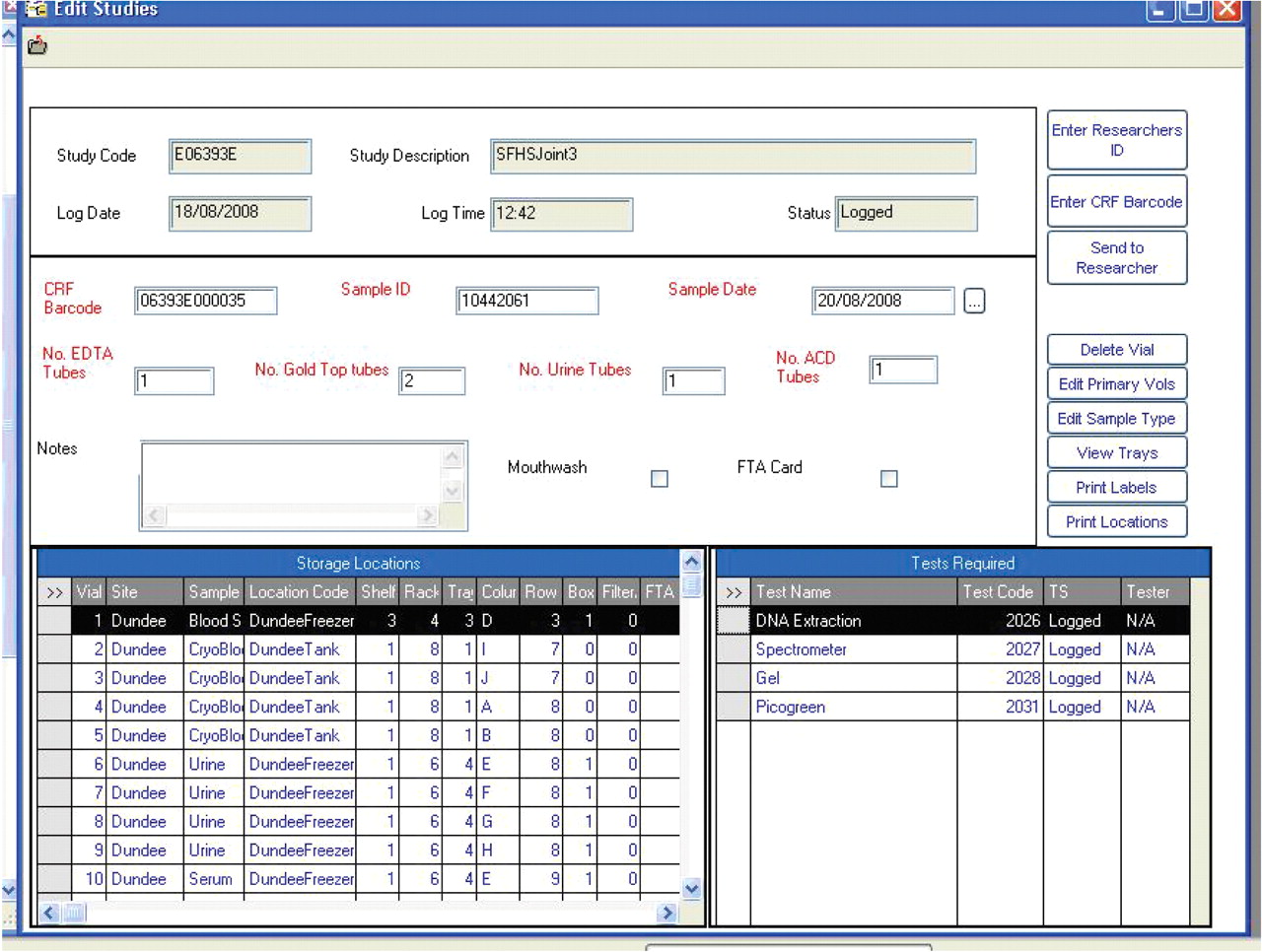
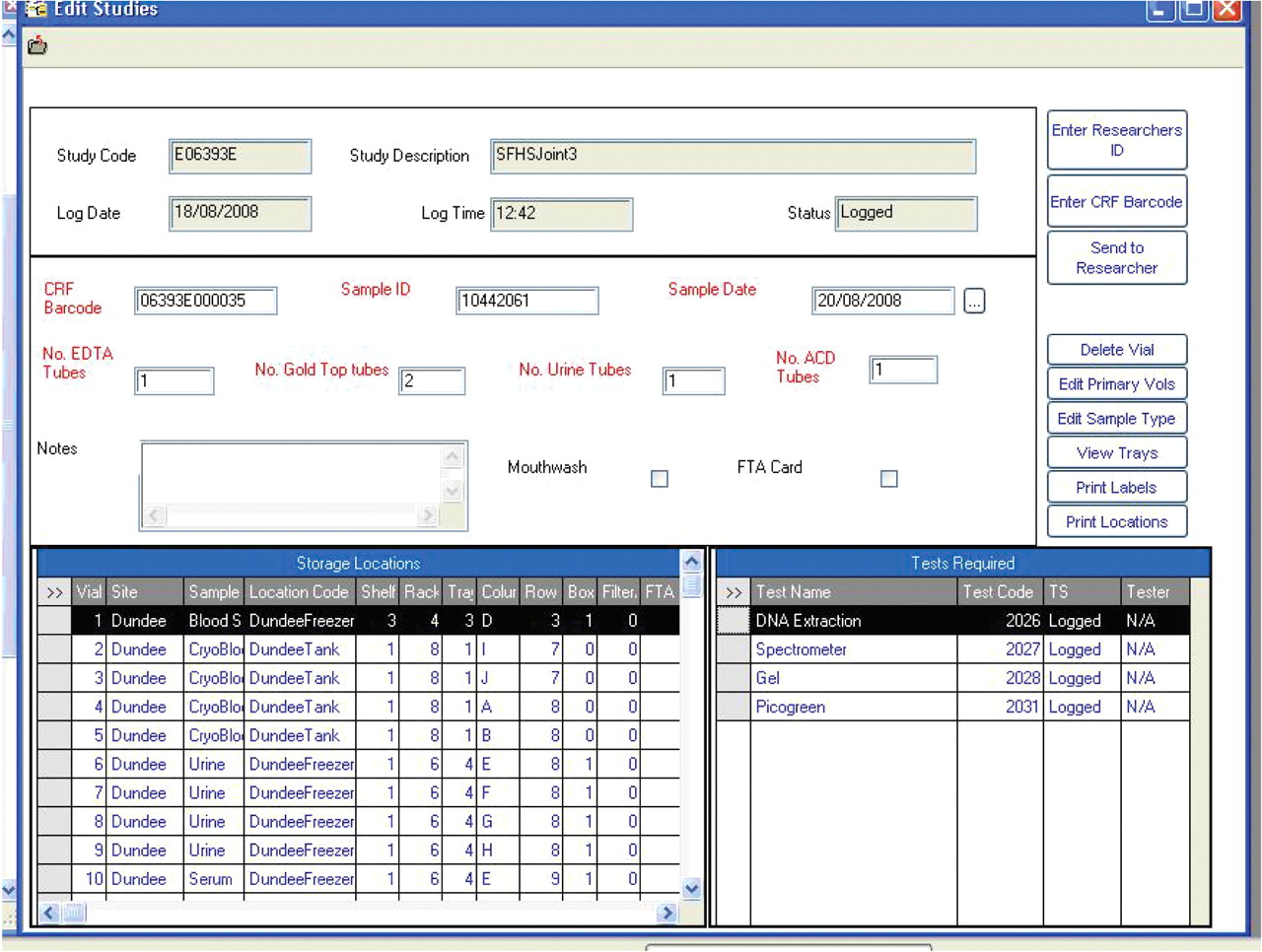
The GS LIMS supplier, Starlims (Hollywood, FL, USA), was chosen after a competitive tender process, and the resulting LIMS provides a method of systematic sample storage, management and tracking. Samples collected across Scotland are booked in on client PCs using remote access to a server in Edinburgh, and storage assigned in freezers and tanks with data validated at point of entry. All laboratories involved in the project use the same sample entry form (fig. 2⇓), as standardising this process facilitates management of samples in multiple locations according to the same protocol. There is validation around each field to minimise data entry errors, and samples are only booked in to the LIMS once they are present in the laboratory. Furthermore, samples can be edited, sent to other researchers or deleted, all with an audit trail necessary for study management and research governance.
{kind=link}
{kind=link}
Laboratory information management system sample input form showing aliquot storage locations.
The LIMS also allows samples to be shipped and tracked securely from one laboratory to another. For example, 9-mL blood samples are sent to Edinburgh in batches of 500 for DNA extraction, and microtitre plates containing DNA working stocks for genotyping are returned to the originating laboratories. Plate management through the LIMS is vital to keep track of DNA stock solutions and to help with subsequent data management, particularly genotyping. Plates of dilute working stocks of DNA destined for experimental analyses are compiled through the LIMS, eliminating transcription errors of sample IDs and allowing the content of a plate to be shown by scanning its barcode label. Through the access policy, only accredited laboratories with LIMS access are permitted to receive GS DNA plates, allowing detailed tracking of the resource and maintenance of high standards of laboratory practice whether the experimental procedures are commissioned internally or externally. The various rules, validation and reporting functions within the LIMS play a vital role in optimising the quality management of the biological resources within GS. Implementation of a custom LIMS may not be readily available to every laboratory, but other sample inventory software is commercially available in off-the-shelf packages at modest cost. As relevant open-source software becomes available 20, underlying principles of good sample management can be applied using less streamlined systems.
DATA CODING AND MANAGEMENT
Consideration of data coding, acquisition and storage methods before participant recruitment begins will result in a study with more reliable data, and allows easy implementation of subsequent studies once in place. Validation of phenotypic data is essential for the integrity of the study and the efficacy of downstream analyses. GS data are validated in the research clinic at the time of entry via forms on a laptop or PC, to improve accuracy and completeness. Two sets of limits are applied by the system: “Hard Limits”, which preclude entry of physically impossible values, and “Soft Limits”, requiring validation by the user. These limits are decided by the clinical team and set in the data dictionary, which returns an error message if measured values fall outwith the specified limits. Table 1⇓ shows sets of limits for blood pressure measurements, which can be adjusted to take account of physiological states at time of measurement, e.g. sleep, post stress, post operation. This enables acquisition of data that are as error free as possible at the point of collection, to minimise downstream problems.
Examples of hard and soft limits for blood pressure(BP) measurements
GS phenotype collection is a subsystem of an overall infrastructure dealing solely with data from volunteers, referenced by a barcode allocated to each clinic visit. The VMS also has access to the NHS Scotland Community Health Index, via an encrypted version of the volunteer’s unique identifier that allows phenotypes to be linked to healthcare data. The system provides a reliable dataset that minimises harmonisation issues, and has a number of real-time tools that allow study managers to monitor both performance of the internal validation of questionnaires and population sampling in real time. This allows fine tuning of the questionnaire and the study volunteer profile, and monitoring of the accuracy and efficiency of the data acquisition staff in the research clinic.
Before statistical analyses, raw data must be encoded. Problems encoding responses may lead to analytical difficulties, so the validity of questions should be considered: free text will need to be interpreted, which may introduce error, so responses should be pre-encoded wherever possible. Considerations relating to the ultimate use of data are necessary when designing a coding scheme, as published standards may be insufficient to adequately describe data within a particular study, as these systems are often created within a set of parameters that may not apply to such studies. For example, NHS coding systems in the UK generally reflect operational rather than ethnic or political boundaries (table 2⇓), and ethnic classifications make no distinction between Welsh (Celtic) and English (Anglo-Saxon) groupings, but such accuracy may be important in population genetic studies. The NHS/ISD classification of “Other British” therefore may need to be sub-classified to separate Welsh and English. These groups could be recombined for comparison with published NHS data.
Coding standard variations in the measurement of ethnicity
If a study requires integration with other datasets, using the same or a subset of the existing coding system will minimise harmonisation problems. Otherwise, mapping tables will need to be created, which do not always give exact correspondence, possibly leading to data loss. If there is a need for linkage between data from two or more projects, those data should be held in similar form. Transformation errors between numeric and textual codes for main identifiers may prevent accurate linkage, and while probabilistic record linkage will help, prior planning will greatly enhance success. If there is no plan to compare the study with others then measures need not conform to standards and can be purely internal, but once comparison becomes important, these standards and interoperability need to be considered.
DATA COLLECTION, STORAGE AND ANALYSIS
Data collected in multiple modalities will need to be integrated before analysis can proceed. In GS, these modalities include web-based forms that send encoded data directly into the database, and Windows forms that can feed into the database via a suitable network connection. All collection modalities use the barcode to pass data to the central phenotype database, where it is encoded using pre-defined look-up tables.
The GS informatics infrastructure provides a secure data resource to support the data needs of the GS projects. Servers are secured behind firewalls, with all data repositories placed on mirrored servers with raided drives ensuring maximum data redundancy and back-up maintained using offsite network-attached storage and local tape. All servers are physically located within secure managed environments without public access, and are attached to uninterruptible power supplies to ensure smooth shutdown in case of power outage.
The specific analyses performed will depend on the phenotypes collected, as considered in the initial study design. For example, a bi-allelic SNP has three possible genotypes, which can be stored as a simple categorical variable for use in association studies, or as an ordinal variable counting the number of copies of the minor allele, for use in, for example, logistic regression or score tests 21. Care must be taken in defining the classes used to collect the data, as illustrated by the ethnicity example in table 2⇑, and disease traits where precise phenotypic definition is also important, especially if data from more than one study are to be combined.
Continuous traits can be read directly into a database from the clinic, allowing instant validation, as well as rapid access to the data. Such traits may be analysed as phenotypes themselves in classic quantitative trait locus (QTL) analysis, or may be treated as risk factors/covariates that contribute towards disease risk. QTL analysis seeks to attribute some proportion of total trait variation to genetic effects, and identify the loci that contribute towards this variation. In more complex models of disease susceptibility, general linear models can be to evaluate the significance of marker genotype effects, while allowing the influence of other genetic/environmental covariates to be taken into consideration, for example, high blood pressure as a risk for developing heart disease. The large volumes of data that are generated by whole genome scans raise further statistical and computational issues that need to be considered.
RELEASE OF RESOURCES AND RETURN OF DATA
To use the resources of any biobank to the best possible advantage, it is important that provision is made for the samples and data to be accessed by researchers with expertise in areas outside the core team. Many of the GS scientists already work on identifying causes of illness in the Scottish population, but there are provisions in place to provide data to external researchers from academic or commercial organisations. It is important that pharmaceutical companies can access resources in order to research potential new drugs and treatments, and biobanks can only do this in partnership with industry. Detailed descriptions of the resources available can be found on the GS website 22.
Before making resources available, it essential that there are clear rules that govern how the samples and data can be used. The GS rules are contained in the Management, Access and Publications Policy, available in a first working version on the GS website. This is currently under consultation, with feedback welcomed from potential users and other interested parties. The policy explains the GS procedures for managing access to, and publication of data from, GS resources and is being used to guide the Resource Management and Development Committee. After a period of 12 months from signing a data or material transfer agreement to use GS resources, collaborators must return a copy of the final dataset used in their analyses, along with derived variables and descriptions of these variables. The overall purpose of the Management, Access and Publications Policy is to ensure that all applications are in keeping with the core aims of GS and comply with its ethical standards and strict rules on participant confidentiality.
CONCLUSIONS
In genetic analyses of complex traits, good sample and data management is necessary to ensure that data is passed accurately and efficiently from collection at the clinic to the researcher who will ultimately perform the analysis. The present review has outlined some of the problems that such projects may encounter and some of the principles that can be used to overcome them with relation to Generation Scotland. Complex disease susceptibility is influenced by many genes of small effect, and detecting these genes will require large population-based samples. With such large numbers, efficient sample storage and management are important. Similarly, to ensure valid conclusions can be drawn, genetic data needs to be well managed and accurate phenotypic data carefully collected, coded and validated. These issues require close collaboration between clinicians, researchers and information technologists, as the infrastructure would ideally be in place before any data are collected, and will become increasingly important as the size and scope of genetic studies increases.
Statement of interest
None declared.
Acknowledgments
The full members of the Generation Scotland Scientific Committee and their affiliations are: L. Hocking, G. Murray, E. Rattray, D. Reid, B. Smith, D. St Clair and L. Stott, all University of Aberdeen (Aberdeen, UK); J. Berg, G. Findlay, C. Jackson, K. Matthews, M.M. McGilchrist, A.D. Morris, C. Palmer, F. Sullivan and R. Wolf, all University of Dundee (Dundee, UK); D. Blackwood, H. Campbell, S. Cunningham-Burley, I. Deary, G. Haddow, S.M. Kerr, P. Linksted, P. McKeigue, D. MacIntyre, R. Morton, W. Muir, D.J. Porteous, S. Ralston, J. Ure and A. Wright, all University of Edinburgh (Edinburgh, UK); J. Cavanagh, J. Connell, M. Connor, A. Dominiczak, P. Ellis, B. Fitzpatrick, I. Ford, R. Gertz, P. Johnson, R. Lindsay, J. Pell and G. Watt, all University of Glasgow (Glasgow, UK); A. Philp and M. Sweetland, National Health Service (NHS) National Services Scotland (Edinburgh); and R.O. Sinnott, National eScience Centre (Glasgow). The authors are extremely grateful to all the participants and the whole Generation Scotland team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. The Chief Scientist Office of the Scottish Government and the Scottish Funding Council provide core support for Generation Scotland.
Footnotes
-
For editorial comments see page 233.
Earn CME accreditation by answering questions about this article. You will find these at the back of the printed copy of this issue or online at www.erj.ersjournals.com/current.shtml
- Received March 20, 2008.
- Accepted October 15, 2008.
- © ERS Journals Ltd