Article Text

Statistics from Altmetric.com
Iron metabolism is of crucial importance in the biology and pathophysiology of the lower respiratory tract. As with many other factors involved in inflammation, it is very important that an appropriate iron balance is maintained. Local deficiency could impair growth and proliferation of cells responsible for the inflammatory response and tissue repair (lymphocytes and fibroblasts) and the synthesis of mediators (for example, arachidonic acid derivatives).1 In contrast, excessive accumulation of iron, especially in free form—that is, not bound to one of the specific iron-binding proteins—facilitates the generation of potentially toxic hydroxyl radicals2 and increases the ability of intracellular bacteria such as mycobacteria to grow.3-5
Research into iron metabolism in the lower respiratory tract has taken advantage of bronchoalveolar lavage, a technique by which it is possible to obtain the cells and fluid lining the alveoli. Determination of iron content in the alveolo-interstitial region shows that 80% is present in the cells and 20% in the epithelial lining fluid of the lung.6 Since alveolar macrophages constitute the major cell population in this region, and because the iron content of lymphocytes, neutrophils, and monocytes is very low in comparison with alveolar macrophages,6 ,7 these cells are of special interest.
In this paper we review current knowledge of iron and iron-binding proteins in bronchoalveolar lavage fluid, and the special features of iron metabolism in alveolar macrophages.
Background
Iron is principally required for haemoglobin synthesis, and its uptake, utilisation and storage are carefully regulated to ensure an adequate supply without excessive accumulation which could lead to toxicity. Intestinal iron absorption is related to erythropoietic requirements, although the regulatory mechanism(s) remain unknown. The usual source of iron in the lung is serum iron which is derived from catabolised erythrocytes and absorbed iron. However, in pathological states, iron can enter the lung by inhalation—for example, cigarette smoking or metallic dusts—or by catabolism of haemoglobin after alveolar haemorrhage. Furthermore, the hypoxia found in chronic lung diseases can stimulate intestinal iron absorption.8 ,9
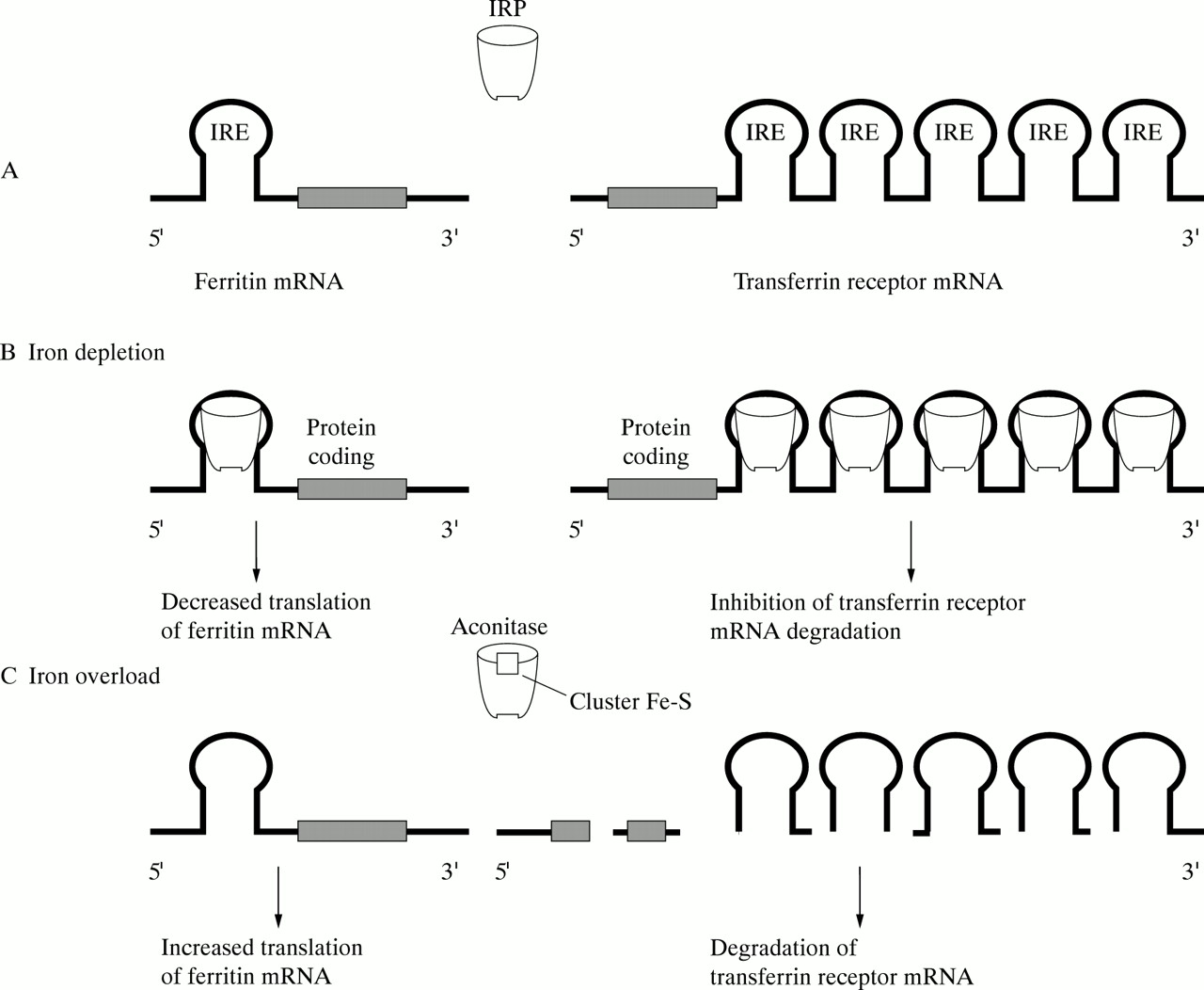
Iron is transported in the plasma to sites of utilisation and storage by the iron transport protein transferrin, an 80 kd molecule capable of reversibly binding two ferric irons (fig 1). Cellular uptake of iron is mediated by the transferrin receptor (CD71), which is expressed by all iron-requiring cells, and occurs by a process of receptor-mediated endocytosis in which the transferrin bound iron is released and retained by the cell, but transferrin itself is recycled (fig2).10 Excess iron is stored in ferritin, a protein consisting of 24 subunits in the form of a spherical cell, inside which up to 4000 iron atoms can be accommodated (fig 1). Two types of ferritin subunit exist, a heavy (H) subunit which predominates in ferritin molecules concerned with detoxification and short term storage, and a light (L) subunit found mainly in ferritin molecules involved in long term iron storage.11 Iron homeostasis is maintained mainly through post-transcriptional regulation of ferritin and transferrin receptor synthesis, which involves one or more cytoplasmic RNA binding iron regulatory proteins (IRPs) (fig 3) whose activity is itself determined by iron availability.12However, inflammatory mediators such as hydrogen peroxide and nitric oxide can also regulate IRP activity,13 and ferritin is subject to transcriptional upregulation during inflammation,14 thus providing mechanisms by which iron metabolism can be altered during inflammatory processes. Since the lower respiratory tract is constantly exposed to potential inflammatory stimuli, these mechanisms are of particular relevance, and it is not surprising that iron metabolism in the lower respiratory tract presents some special features.

Structure of iron-binding proteins. (A) Transferrin and lactoferrin. Both are glycoproteins with molecular masses in the range of 80 kDa and comprise two domains (N-terminal domain and C-terminal domain), each containing an iron binding site. Carbohydrate composition and glycosylation sites (not shown in the figure) differ between both molecules. (B) Ferritin. The ferritin molecule is a hollow protein shell enclosing a core of polynuclear iron oxide (up to 4500 iron atoms) (black). The protein shell is composed of 24 subunits of two structurally distinct types designed H (for heavy or heart) (white) and L (for light or liver) (grey), respectively. Different proportions of each subunit in a ferritin molecule give rise to isoferritins which have specific tissue distributions.

Iron uptake through transferrin receptor. (A) Binding of transferrin to transferrin receptors in coated pits. (B) Internalisation of transferrin-receptor complex. (C) Loss of clathrin, decrease of local pH, and release of iron. (D) Apotransferrin remains bound to the receptor because of its high affinity for the receptor at acid pH. (E) Recycling of apotransferrin-transferrin receptor complex to the plasma membrane. (F) Dissociation and release of apotransferrin.

Post-transcriptional regulation of intracellular iron metabolism. (A) Cells utilise trans-acting proteins (the IRPs or iron regulatory proteins) and a common cis-acting RNA element (the IRE or iron responsive element) to regulate the genes involved in cell iron homeostasis. IREs are stem-loop structures placed in the 5′ UTR of mRNA encoding ferritin and 5-aminolaevulinate synthase (not represented in the figure for simplicity) and in the 3′ UTR of mRNA encoding transferrin receptor. IRPs are bifunctional molecules which possess aconitase activity (in the case of IRP-1) or affinity for IREs, depending of iron availability. When intracellular iron is abundant, the IRPs are in the 4Fe-4S state which has high aconitase activity (IRP-1) but low affinity for IREs. Conversely, when iron is scarce the IRPs have negligible aconitase activity but high affinity for IREs. (B) When intracellular iron increases the IRPs interact with the IRE in the 5′ UTR of ferritin mRNA (thus preventing translation initiation) and with IREs in the 3′ UTR of transferrin receptor mRNA (thus inhibiting its degradation). (C) When intracellular iron decreases the absence of IRPs allows translation initiation of ferritin mRNA and degradation of transferrin receptor mRNA.
Iron and iron binding proteins in bronchoalveolar lavage fluid
Total iron in bronchoalveolar lavage (BAL) fluid has been determined in smokers and non-smokers using both the bronchial and the alveolar fraction.15 In neither group were differences found between the bronchial and alveolar fractions, thus validating the normal practice of using the total bronchoalveolar lavage for analysis. Secondly, the values obtained in the alveolar region have been reported to be significantly higher in smokers than in non-smokers, regardless of whether iron levels are expressed as the concentration per unit volume, the relationship with albumin, or the concentration in the epithelial lining fluid (table1).
Mean (SD) levels of iron in the bronchoalveolar lavage fluid in cigarette smokers and non-smokers
Although the available data are only semi-quantitative, it has nevertheless been possible to establish a positive correlation between total iron in the BAL fluid and iron concentrations in the alveolar macrophages.6 Iron in BAL fluid may be bound to specific iron binding proteins (transferrin, lactoferrin and ferritin) or “free” (in practice, usually bound to low molecular weight compounds or loosely bound by non-specific proteins such as albumin). Moreover, in occupational lung diseases iron can be found in ferruginous bodies (asbestos bodies) and in mineral particles. Since the form in which iron is present will depend upon the availability of binding sites on the specific binding proteins in the alveolar region, it is important to know the distribution of iron binding proteins in the lung.
TRANSFERRIN
Data in the literature on transferrin concentrations in BAL fluid are difficult to compare because of differences in assay methods (nephelometry, enzymatic immunoassay) and in the presentation of results in various reports. The number of studies actually carried out is also rather limited. Table 2 summarises the main findings in different groups of subjects.7 ,15-21 Despite these problems, the different studies tend to give similar values. One striking technical aspect is the absence of significant differences between alveolar (0.17 (0.03) μg/μg albumin) and bronchial samples (0.13 (0.04) μg/μg albumin).15
Mean (SE) concentrations of transferrin in bronchoalveolar lavage fluid (BALF) and epithelial lining fluid (ELF) in lung diseases
Transferrin levels as a percentage of total protein in BAL fluid are very high (4–5.6%) compared with values for plasma.7 ,22This suggests that transferrin in the lower respiratory tract originates not only through transudation from plasma, but also from local synthesis. A likely source is cells of the lymphomyeloid system, and this is discussed in more detail below.
Comparison of transferrin levels in different groups has revealed that smoking, despite increasing iron and ferritin levels (see below), appears not to affect transferrin concentrations in the BAL fluid,7 ,15 ,17 ,23 and levels are also normal in BAL fluid from individuals with Pneumocystis carinii infection.21 ,24 On the other hand, local transferrin concentrations are lower in patients with chronic obstructive pulmonary disease and toxic oil syndrome than in controls,17 ,25 while patients with sarcoidosis have higher BAL fluid transferrin levels than controls. These high levels correlate positively with the percentage of lymphocytes in the BAL fluid,16 ,24 supporting the proposal (see below) that local synthesis by activated lymphocytes makes a significant contribution. Patients with ARDS (acute respiratory distress syndrome) also have increased transferrin levels.25
Transferrin in the lower respiratory tract may perform a number of different functions. Its capacity to bind iron may allow it to exert an indirect antioxidant effect by preventing the occurrence of “free” iron. Quantitative studies have shown that the combination of transferrin, lactoferrin and ferritin prevents the occurrence of “free” iron in the respiratory tract in non-smokers but not in smokers.19 Quantitatively, transferrin is the main antioxidant in the lung; the role of caeruloplasmin is less important and vitamins C and E and albumin play only a minor part.19It is probably because of this indirect antioxidant action that the clinical evolution of adult ARDS patients is better when there are high levels of transferrin in the BAL fluid.26 In addition, pulmonary Pseudomonas aeruginosa infections are associated with transferrin cleavage which will liberate iron, allowing it to catalyse formation of free oxygen radicals.27 On the other hand, transferrin is able to provide iron to cells that express CD71, the transferrin receptor. In the alveolar region B and T lymphocytes and alveolar macrophages all express CD71,1 ,16 ,28 suggesting that transferrin plays an important role in delivering iron required for normal metabolic function in these cells.
FERRITIN
Ferritin determination in BAL fluid has been performed almost exclusively in humans (smokers and non-smokers). The results are shown in table 3.7 ,15 ,17 ,23 All the studies have shown a significant rise in BAL fluid ferritin levels in smokers, regardless of whether or not chronic bronchitis was present. The most probable origin of ferritin is moribund alveolar macrophages since these cells do not secrete ferritin actively17 ,23 and there is a correlation between release of ferritin and LDH (lactic dehydrogenase).
Mean (SE) concentration of ferritin in bronchoalveolar lavage fluid (BALF) in smokers and non-smokers
The biological role of ferritin in BAL fluid is debatable since, depending on conditions, it may either sequester iron (thus exerting an antioxidant effect) or serve as an iron source that can, in the presence of superoxide, catalyse the generation of toxic hydroxyl radicals.29 In this setting patients with smoker’s lung have higher levels of superoxide anion30 and ascorbic acid,31 agents that can mobilise iron from ferritin and thus promote the generation of hydroxyl radicals.
LACTOFERRIN
Lactoferrin is present in BAL fluid, the mean concentration being approximately 10 times lower than that of transferrin (47.7 (0.1) μg/ml of pulmonary epithelial fluid in controls).15 ,20Lactoferrin in BAL fluid comes mainly from the airway rather than the alveolar region,32 which explains why smokers have significantly increased lactoferrin levels in the BAL fluid (159.4 (0.37) μg/ml of epithelial fluid).15
Iron metabolism and the alveolar macrophage
TOTAL INTRACELLULAR IRON
The total amount of iron in alveolar macrophages has been evaluated using cytochemical (Perls), colorimetric (ferrozine), or particle induced x ray emission (PIXE) tech niques.6 ,7 ,15 ,17 ,33 Possible sources are inhalation of iron-rich dust, phagocytosis of moribund cells (including erythrocytes), and uptake of iron from the iron binding proteins transferrin or lactoferrin. These mechanisms are shown schematically in fig 4. The amounts of iron in alveolar macrophages reported by different authors (table 4) show that smokers (with or without chronic bronchitis or bronchopulmonary neoplasm) and individuals exposed to inorganic dusts or with Goodpasture’s syndrome all have higher levels than normal controls.6 ,7 ,15 ,17 ,31
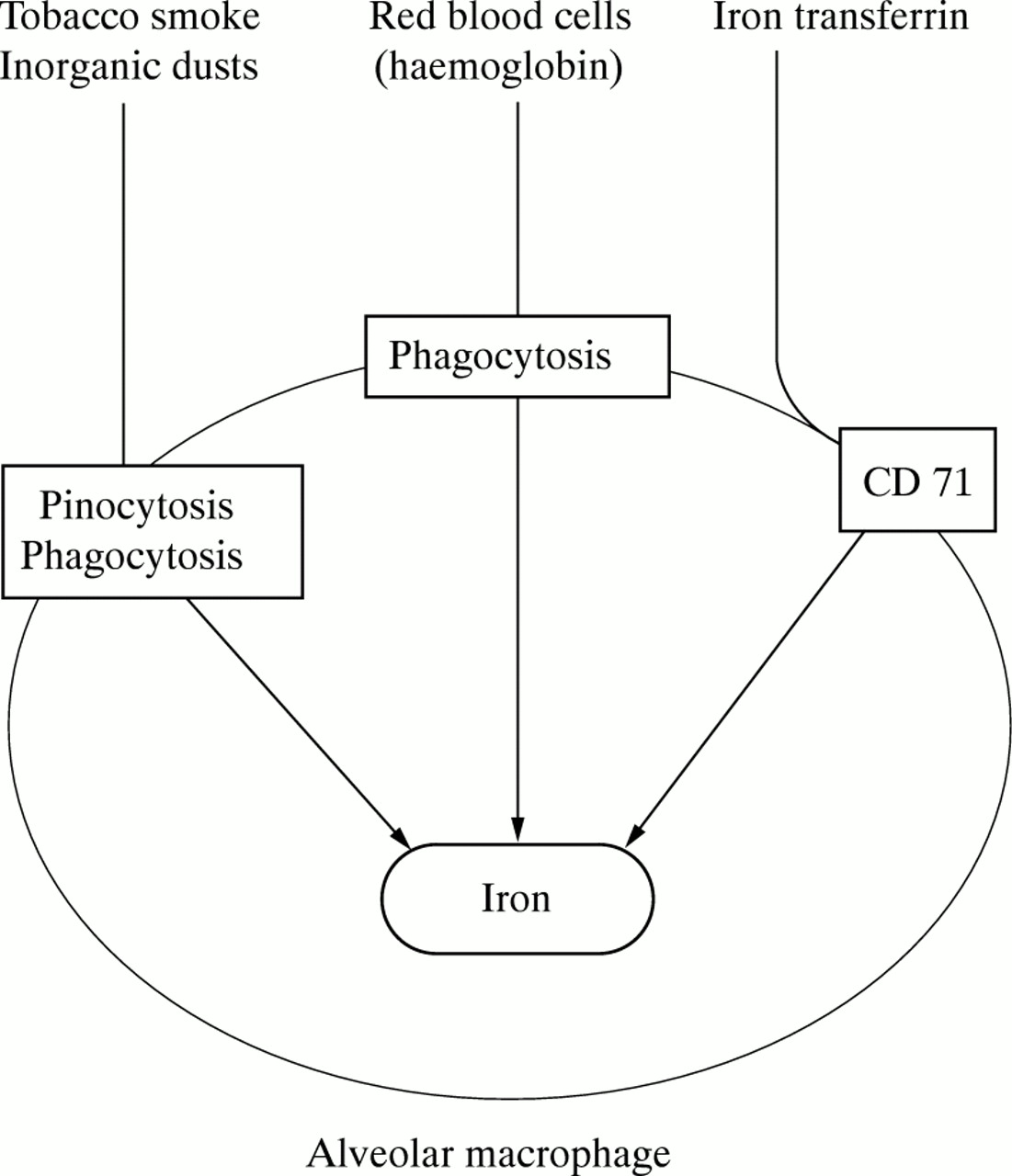
Sources of intramacrophagic iron. CD 71 = transferrin receptor.
Mean (SE) concentration of iron in alveolar macrophages (AM) in relation to occupation and lung diseases
The high iron content in alveolar macrophages of smokers and individuals exposed to inorganic dusts is probably due to a large extent to inhalation of the metal. Thompson et al have reported15 that tobacco has a high iron load (440–1150 μg/g of tobacco and 420 μg/g paper). If one assumes that each cigarette has 0.7 mg of tobacco and that each inhalation furnishes 0.1%, a smoker of one pack per day would inhale 1.12 μg of iron. If one takes into account that alveolar macrophages of smokers retain more iron than those of control subjects, the intake of exogenous iron under these circumstances would be significant.17 The increased iron levels in alveolar macrophages of patients with Goodpasture’s disease probably arise from haemoglobin iron derived from phagocytosed erythrocytes. However, Custer et al 34 reported that alveolar macrophages had only a limited ability to metabolise haemoglobin, and McGowan et al 31 found that alveolar macrophages do not contain haem oxygenase which is required for the liberation of iron from haemoglobin. However, haem oxygenase is induced by haem,35 ,36 so it may be that the enzyme is induced in the alveolar macrophages of patients with Goodpasture’s disease following phagocytosis of alveolar erythrocytes.
Iron uptake by alveolar macrophages from specific transport proteins (lactoferrin and transferrin) will be discussed below.
DISTRIBUTION OF IRON WITHIN ALVEOLAR MACROPHAGES
The distribution of iron among the different intracellular compartments in alveolar macrophages shows some distinctive features. Theoretically, intracellular iron may be present in three different forms: bound to ferritin, as haemosiderin, or in the form of a transit pool. Early x ray dispersion studies in smokers and non-smokers showed that intra-alveolar macrophage iron is located in lysosomes, probably in the form of haemosiderin.37 In a study carried out with alveolar macrophages from subjects with different types of lung disease, Costabel et al 38 reported a mean ferritin content of between 355 and 8500 ng/106alveolar macrophages. Unlike monocytes, in which isoferritins containing H (heavy) subunits predominate, there is a clear predominance of L (light) subunit containing ferritin in alveolar macrophages, only 5% of the ferritin being of the H type. The L isoferritin is less effective than the H type in detoxification of iron and inhibition of peroxidation, so ferritin iron in alveolar macrophages may be a potential source of tissue damage. Furthermore, in smokers total ferritin in alveolar macrophages is considerably higher than in non-smokers (5059 (1486) versus 290 (51) ng/106) and, although the actual amount of H isoferritin also increases, its relative proportion decreases to as low as 1%.7Furthermore, cultured alveolar macrophages from smokers release higher amounts of ferritin than those from non-smokers.23
Excellent correlation exists between total intracellular iron and ferritin concentrations. However, whereas ferritin is found in the cytoplasm, most of the iron is insoluble and found in the mitochondrial and lysosomal factions.17 Only about 25% of intracellular iron is bound to ferritin, implying that the remaining 75% must be bound to other substances. Direct evidence using spin trapping and deoxyribose techniques2 suggests that the intracellular accumulation of iron has a protective rather than an aggressive function since exogenous iron overload followed by an immune stimulus does not result in generation of hydroxyl radicals. This effect is probably due to iron sequestration rather than a failure of alveolar macrophages to respond to the stimuli, since triggering of intracellular oxidant systems and expression of heat shock proteins were normal.2
Alveolar macrophages from smokers transfer internalised iron transferrin to cytoplasm more slowly than those of non-smokers, and although their capacity to take up iron is similar to that of non-smokers, subsequent release is slower.31
TRANSFERRIN RECEPTORS IN THE ALVEOLAR MACROPHAGE
The presence of transferrin receptors on alveolar macrophages was first reported by Hirata et al 1using Scatchard analysis and Northern blotting. They found a Ka (association constant) of 4.4 (0.7) × 10–8M–1 and 4.4 (1.2) × 104 receptors per cell, although McGowan et al,31 using similar techniques, reported a higher number of receptors (5 × 105 per cell). These results are similar to those cited by Wyllie et al in alveolar macrophages from rabbits.39
Expression of transferrin receptors by alveolar macrophages varies in different interstitial lung diseases. Haslam et al 40 reported an increase in transferrin receptor expression in granulomatous pulmonary disease, although there was considerable within-group variability (14–96%). In contrast, our group found a decreased transferrin receptor expression by the alveolar macrophages of patients with hypersensitivity pneumonitis,41 an observation similar to those of Costabelet al.42
TRANSFERRIN SYNTHESIS BY ALVEOLAR MACROPHAGES AND OTHER PULMONARY CELLS
The cells responsible for the greater part of serum transferrin synthesis are hepatocytes. However, other cells (Sertoli, muscle and brain cells) can also synthesise transferrin.43 In the lung, transferrin synthesis by inflammatory cells (macrophages and lymphocytes) is of special interest. It has been known for many years that macrophages (including alveolar macrophages) from experimental animals are able to synthesise transferrrin.3 In mouse alveolar macrophages basal transferrin synthesis is higher than in monocytes or peritoneal macrophages and is increased by inflammatory stimuli.44 However, the ability of human alveolar macrophages to synthesise transferrin is debatable. Using ELISA, Wesselius et al 7 detected transferrin within human alveolar macrophages, there being no significant differences between smokers and non-smokers. However, the methods used do not distinguish between endogenous synthesis and endocytic uptake of exogenous transferrin. Since alveolar macrophages from smokers contained less transferrin than those of controls, the latter origin seems more likely. Furthermore our group,45using metabolic labelling and immunoprecipitation techniques, found that the alveolar macrophages of control subjects did not synthesise transferrin. It may be that in man alveolar transferrin synthesis is due to activated T lymphocytes, in particular CD4 cells, which have been shown to synthesise transferrin in vitro.46 Moreover, we have found that lymphocytes of patients with diffuse alveolar-interstitial lung disease synthesise and release transferrin both in vitro47 and in vivo.24 Finally, it may be that other cells in the lung contribute to local transferrin synthesis, as certain types of small cell carcinoma are able to produce transferrin.48
Iron and lung disease
Iron deposition in the lung is associated with tissue injury and fibrosis. Accumulation of iron in the lung has been demonstrated in smokers and patients with various pulmonary diseases (see above). In smokers the increased iron deposition in BAL fluid and/or alveolar macrophages6 ,7 ,15 is higher in the upper lobes than in the lower lobes, so that iron accumulation is related to areas of emphysema or lung cancer.49 Iron also accumulates in the lungs of patients with occupational diseases6 ,50-52 and during alveolar haemorrhage6 or hyperoxic lung injury.53 In addition, iron in the lung increases with age54 and in pulmonary allografts.55 However, the first determinant factor for iron toxicity is not its concentration in the lung but its ability to remain in a “free” form. Thus, the mobilisation of iron from ferritin31 or mineral particles51 or the release of iron from transferrin27 ,56 is associated with lung injury.
Free iron in the lung exerts toxic effects through its ability to catalyse highly reactive hydroxyl radicals from less reactive superoxide and hydrogen peroxide via the Fenton and Haber-Weiss reaction,22 and/or through its ability to stimulate fibrogenesis57 (fig 5). Several findings support this interpretation: (a) the demonstration of pro-oxidant iron in the lungs of patients with ARDS58; (b) the finding of oxidised proteins in BAL fluid in interstitial lung diseases59; and (c) the increased levels of non-transferrin bound iron in preterm babies.60 In addition, the treatment with iron chelators prevents lung injury both in animal models61 and in human disease caused by toxins—that is, smoke inhalation62—or microorganisms (Pneumocystis carinii).63 Hydroxyl radicals exert damage on all types of organic molecules (carbohydrates, lipids, proteins and nucleic acids).64 ,65

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanisms of iron mediated lung diseases. NADPH = reduced nicotinamide adenine dinucleotide phosphate; H2O2 = hydrogen peroxide; OH • = hydroxyl radical; OH – = hydroxyl anion; O2 – • = superoxide radical.
Summary and perspectives
In summary, data provided in this review demonstrate the presence of iron and iron binding proteins in different lung compartments. Moreover, in different pulmonary diseases and/or after inhalation of toxins both the amount of iron and its distribution is modified.
However, many questions about iron metabolism in the lung remain unanswered. For example, the relationship between nitric oxide and iron management in alveolar macrophages, the interaction between differently saturated transferrin molecules and alveolar surfactant during acute alveolar damage,66 ,67 and the role of iron in the defence against infectious agents in the lung are not fully understood. A better knowledge of these questions will aid our understanding of the pathogenesis of pulmonary diseases and will help us to develop new approaches to their treatment.
Acknowledgments
This work has been supported in part by a grant from Comision Internacional de Ciencia y Tecnología (Spain) SAF 96–0167.