Article Text

Statistics from Altmetric.com
Chronic pulmonary thromboembolic disease is an important cause of severe pulmonary hypertension, and as such is associated with significant morbidity and mortality. The prognosis of this condition reflects the degree of associated right ventricular dysfunction, with predictable mortality related to the severity of the underlying pulmonary hypertension.1 In recent years the epidemiology of this condition has been revised considerably. Once considered a rare condition, chronic thromboembolic pulmonary hypertension (CTEPH) was recently documented to complicate 3.8% of acute pulmonary embolic events.2
CTEPH is the only cause of severe pulmonary hypertension which is potentially curable without the need to resort to lung transplantation. Pulmonary endarterectomy (PEA) is the surgical procedure which removes the obstructing thromboembolic material, resulting in significant improvements (and in many cases normalisation) in right ventricular haemodynamics and function. This procedure requires a high degree of anaesthetic and surgical skill, coupled with assiduous preoperative evaluation of potential patients. Surgery is generally considered only in patients with proximal chronic thromboembolic disease as assessed by radiological investigations.
Over the past 20 years, the number of these procedures being performed has steadily increased. This reflects increased physician recognition of both pulmonary hypertension in general, and CTEPH in particular. Despite this increasing interest, however, outside of individual case series and reported experience, there are scant data available to support many of the theories and suppositions concerning this condition.
CTEPH: WHY IS IT IMPORTANT FOR THE CARDIOLOGIST?
Over the past 5–10 years, there have been major advances in the field of pulmonary hypertension. Underpinning this is an increasing understanding of the cellular, molecular and genetic mechanisms underlying pulmonary arterial hypertension (PAH), coupled with an increased range of effective treatment (drug) options for patients with this condition. This has created renewed interest among health professionals in general, and a significant increase in the number of referrals of patients with suspected pulmonary hypertension for assessment and treatment.
As a result of this increased profile, the World Health Organization has sponsored a number of expert consensus conferences to clarify the classification and treatment of pulmonary hypertension.3 Within the revised classification, CTEPH is recognised as a separate entity on the basis of its unique aetiology, and the recognition that it is potentially amenable to surgical cure. This mandates CTEPH being specifically considered in the diagnostic algorithm for any patient presenting with pulmonary hypertension.
There have been several recent comprehensive reviews of both CTEPH and pulmonary endarterectomy.4–6 The aim of this article will therefore be to highlight recent advances in our understanding of this condition and areas of controversy and difficulty, particularly where there is a direct impact on clinical practice.
NATURAL HISTORY OF CTEPH
The natural history of CTEPH within the population remains unclear. What has become apparent, however, is that the number of patients suffering from this condition has been grossly underestimated.
It is estimated that 3.8% of patients suffering an acute pulmonary embolus will develop CTEPH.2 Furthermore, even in patients receiving appropriate treatment for an acute pulmonary embolism, incomplete resolution occurs in a significant proportion of patients, placing them at risk of developing CTEPH.7–9
Accurately charting the epidemiology of CTEPH is made difficult with the historical recognition that many patients afflicted with this condition have no clear history of an acute thromboembolic event (either deep venous thrombosis, pulmonary embolism, or both).5 Such patients typically present with worsening dyspnoea on exertion, and are subsequently diagnosed with CTEPH during the diagnostic work up for pulmonary hypertension (see below). Although recognition of both pulmonary hypertension in general and CTEPH in particular has increased, there remains an unknown number of patients in the community with this condition undiagnosed.
Some patients present for the first time with severe pulmonary hypertension directly following a pulmonary thromboembolic event. This raises the issue of how to distinguish those patients with an acute thromboembolic event complicating unrecognised CTEPH, from those with a de novo major central acute event. This is a very important distinction because the treatment algorithms for the two scenarios are different.
As a general guide, if a patient presents with a right ventricular systolic pressure (RVSP) on echocardiography >50 mm Hg, without clinical or echocardiographic signs of right ventricular failure, it is likely they have a chronically “conditioned” right ventricle. Patients presenting de novo with an acute pulmonary embolism are unable to acutely generate and maintain an RVSP of this magnitude and will present with right ventricular failure and significant haemodynamic compromise. For these latter patients, treatment with thrombolysis, catheter fragmentation or surgical embolectomy may be considered.10 In patients with underlying CTEPH, however, surgical embolectomy alone is ineffective and potentially dangerous as it will remove only the acute embolus, leaving the underlying pulmonary hypertension unchanged and adversely impacting on the postoperative course. This distinction remains fraught with difficulty and relies principally on the judgement and experience of the treating clinician. Absolute rules cannot be applied.
In the majority of patients, months or even years (the so-called “honeymoon period”) may pass before clinically significant pulmonary hypertension manifests. This presumed gradual progression of pulmonary hypertension occurs in the absence of documented recurrent pulmonary embolic events and is thought to reflect progressive remodelling of the unobstructed pulmonary vasculature, stimulated by increased blood flow through these vessels, analogous to the changes seen in patients with Eisenmenger syndrome.11
Complicating the uncertainties surrounding the epidemiology is an inability to define an underlying predisposition to, or risk factor for, developing venous thromboembolism in the majority of patients with CTEPH. In a minority of patients, a prothrombotic tendency such as the presence of lupus anticoagulant or an hereditary thrombophilia such as protein C, protein S or anti-thrombin III deficiency will be identified.5 The role of other thrombophillic tendencies such as those associated with factor V Leiden, prothrombin gene mutations and hyperhomocysteinaemia are yet to be adequately studied in this condition. In a small number of patients, defects in fibrinolysis may be present.12
The clinical course of CTEPH reflects the progressive increase in the pulmonary vascular resistance (PVR) which characterises this condition. Progressive pulmonary hypertension, right ventricular dysfunction and death ensue if the condition is left untreated. As in all forms of pulmonary hypertension, prognosis reflects the severity of right ventricular dysfunction, which is intrinsically related to the mean pulmonary artery pressure (mPAP). Riedel et al1 showed that the prognosis of this condition progressively worsens with increasingly severe pulmonary hypertension. In this study, the 2 year survival for CTEPH patients with mPAP >50 mm Hg was only 20%.
With the above background, a very important clinical issue remains unanswered—that is, which patients with an acute pulmonary embolism should be monitored for the subsequent development of CTEPH? The precise answer to this frequently asked question is not known, but it is possible to define a group that is likely to be at the highest risk. This group includes: all patients with major central thromboembolic events7; patients presenting with significant haemodynamic disturbance and/or evidence of right ventricular dysfunction8; those with a documented thrombophilia; and those with persistent abnormalities of lung perfusion on follow up testing.9 Patients in these categories should probably be followed up for a minimum of 2 years, or at any time thereafter if symptoms develop. The vast majority of patients with an effectively treated pulmonary embolism will return to normal, with only a small percentage (3.8% being the published figure2) developing CTEPH; however, given how common acute pulmonary embolism is, this will still amount to a significant number of patients in absolute terms.
PATHOPHYSIOLOGY
The pathophysiology of this condition is related to the increased resistance to flow through the pulmonary arteries, that results initially from obstruction of pulmonary arterial vessels (from main to subsegmental levels) by organised thromboembolic material, and subsequently from vascular remodelling in small unobstructed vessels.11 Histological examination of resected PEA specimens showed organised thrombi of uniform age in 72% of cases, implying that a single, incompletely/inadequately resolved thromboembolic event was responsible for the disease. Intimal thickening was seen in all cases, consisting variously of collagen deposition, haemosiderin, inflammation, atherosclerosis and calcification.13 This intimal disease highlights the need for an endarterectomy to treat this condition effectively.
The histopathology raises a number of questions in terms of the underlying abnormalities which result in this condition. As mentioned above, identifiable abnormalities of coagulation (thrombophilia), clotting and/or defects in fibrinolysis are present in a minority of patients, although in a recent series of 122 patients, 41% were found to have raised factor VIII concentrations.14 Occasional patients will have underlying autoimmune or haematological disorders.
The pulmonary artery displays increased fibrinolytic potential compared with the aorta, and it is likely this plays a key role in the endogenous fibrinolysis that leads to the resolution of the vast majority of acute pulmonary emboli. In CTEPH an inherent endothelial cell mediated defect in fibrinolysis has not been shown, however.15
One curious (and now largely historical) risk for developing CTEPH was the use of ventriculo-atrial shunts in patients with hydrocephalus, with the cerebrospinal fluid itself being procoagulant.
The most difficult issue currently encountered in defining the pathophysiology of CTEPH is the role of small vessel disease. Evidence for a critical role of small vessel arteriopathy in contributing to the pulmonary hypertension seen in this condition comes from two observations. Firstly, in some patients there is persisting pulmonary hypertension after PEA despite adequate macroscopic clearance of the chronic thromboembolic material. Secondly, there is increasing evidence of the effectiveness of the medical therapy used to treat PAH in some patients with inoperable CTEPH (see below).
The small vessel arteriopathy seen in CTEPH is not dissimilar to that responsible for idiopathic PAH, and is thought to reflect vascular remodelling which arises in response to increased flow and consequent shear stress in those parts of the distal pulmonary arterial vascular bed unobstructed by more proximal thrombotic material.11 This is analogous to the development of pulmonary vascular disease in Eisenmenger syndrome. The technique of partitioning pulmonary vascular resistance, using sophisticated software analysis of the pulmonary artery occlusion waveform, is a promising technique for the evaluation of the degree of small vessel disease present, allowing assessment of the risk of the PEA procedure and the risk of persistent pulmonary hypertension post-PEA.16
DIAGNOSIS OF CTEPH
CTEPH is diagnosed on the basis of radiological investigations performed during the assessment of a patient with pulmonary hypertension. A ventilation/perfusion (V/Q) lung scan is performed early in the diagnostic pathway to differentiate those patients with CTEPH from those with other forms of pulmonary hypertension. In CTEPH, the V/Q scan is almost always assessed as “high probability” with multiple mismatched segmental perfusion defects evident. A normal or low probability scan effectively rules out the diagnosis.
Following the initial diagnosis of CTEPH, subsequent investigations aim to define the nature and extent of thromboembolic disease and thus assess surgical suitability.
Traditionally, surgical suitability has been determined by the site of obstruction as seen on conventional pulmonary angiography (fig 1). More recently, magnetic resonance (pulmonary) angiography and computed tomographic pulmonary angiography (CTPA) have been used for this purpose. At the University of California San Diego (UCSD), pulmonary angioscopy has been used routinely to both confirm the diagnosis of CTEPH and assess surgical suitability. This, however, is a specialised technique and is not used to any degree outside this centre.
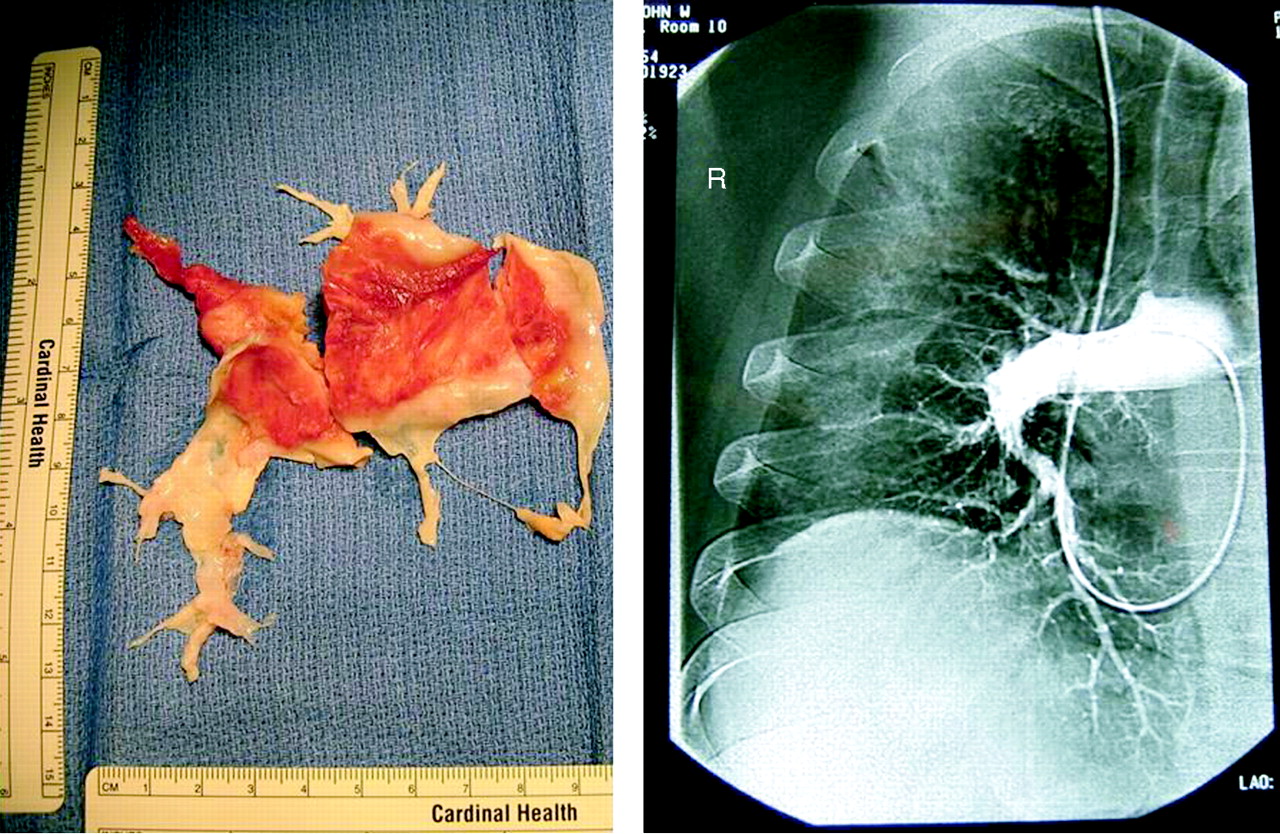
Pulmonary endarterectomy cast and corresponding pulmonary angiogram.
Pulmonary hypertension per se is defined as a mean pulmonary artery pressure >25 mm Hg at rest, or >30 mm Hg on exercise. All patients with CTEPH should be assessed with a right heart catheter which provides both accurate prognostic information and the opportunity to perform pulmonary angiography.
It is not uncommon in the early stages of CTEPH for patients to have a near-normal resting mPAP, with significant pulmonary hypertension manifesting only with exercise. This is reflected in the clinical presentation, with the most common early symptom of all forms of pulmonary hypertension being exercise induced dyspnoea.
Pulmonary angiography
Pulmonary angiography is accepted as the definitive investigation for the diagnosis and assessment of surgically correctable CTEPH. When performed by experienced operators, conventional pulmonary angiography utilising modern contrast techniques is safe, even in patients with severe pulmonary hypertension.
A number of angiographic patterns are described in patients with CTEPH. Typical findings include vascular webs (which result from organisation of the thromboembolic material in the vessel lumen with subsequent scar formation), and abrupt vascular cut-offs representing complete occlusion of the vessel (fig 2). Filling defects per se are less common. Anteroposterior and lateral views should be taken to define the extent of disease fully.

Magnetic resonance angiography showing blunt cut off of right upper and middle lobes with absence of perfusion in right upper zone. Vascular web in left pulmonary artery (arrow).
Computed tomography (CT)
Modern scanners (particularly the new generation 64 slice helical scanners) can also accurately define the nature and extent of chronic thromboembolic disease. However, these scans do require interpretation by a radiologist familiar with this condition.
The hallmark of CTEPH on CTPA is the absence or sudden loss of contrast filled vessels, best appreciated by following individual vessels while scrolling through sequential slices. As in conventional pulmonary angiography, filling defects are not commonly seen. If filling defects are present, however, they tend to occur eccentrically within the vessel lumen, which is in contrast to the findings in acute pulmonary embolism where CTPA typically shows central filling defects in one or more pulmonary arterial vessels.
This is a very important distinction. Most radiologists will be very familiar with the typical changes of acute pulmonary embolism where CTPA is used widely for diagnosis, but the diagnosis of CTEPH will be missed unless the radiologist is specifically asked to look for the radiological signs of chronic thromboembolic disease.
Modern scanners and software also allow three-dimensional reconstruction of the pulmonary vascular tree, and as familiarity with this technique increases, conventional angiography is relied on less, particularly for diagnosis (as opposed to assessment of suitability for surgery).
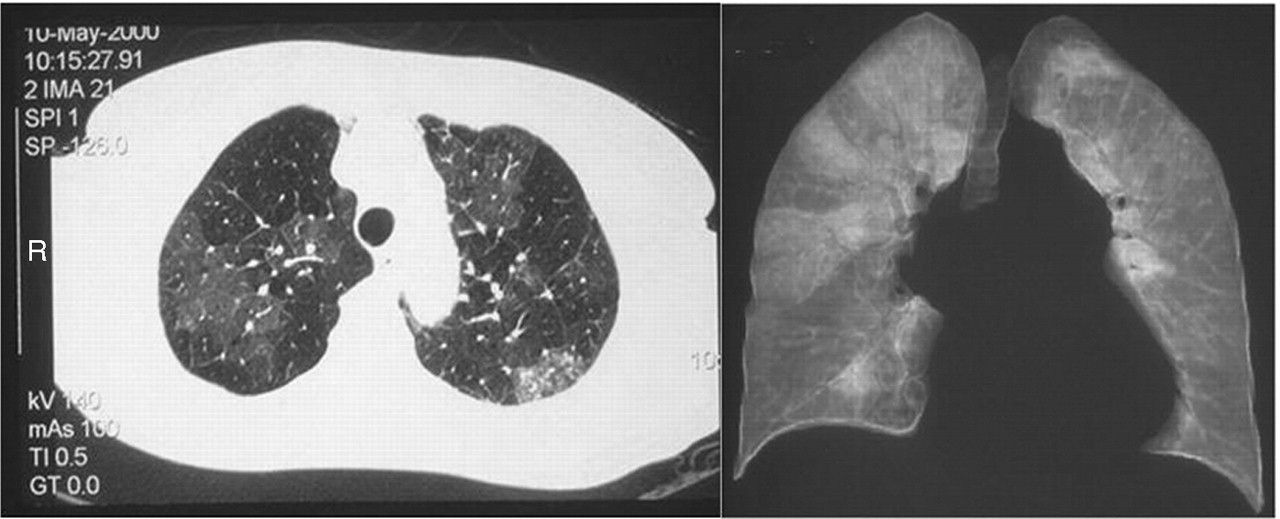
An additional technique available with chest CT is high resolution scanning (HRCT) of the lungs (fig 3). This allows detailed visualisation of the lung parenchyma, and in particular, the lung perfusion. The hallmark of CTEPH on HRCT is the presence of “mosaic perfusion”, which reflects the inhomogeneity of lung perfusion associated with this condition, analogous to the perfusion images seen with V/Q scanning.

Axial (left panel) and coronal high resolution computed tomographic (HRCT) slices demonstrating “mosaic perfusion”. Note on coronal view, the large wedge shaped perfusion defects in the right lung and almost total absence of perfusion in left lung.
Magnetic resonance angiography (MRA)
Both conventional and CT pulmonary angiography have the disadvantages of requiring exposure to both radiation and nephrotoxic contrast agents. Patients with CTEPH will often require repeated studies to confirm the diagnosis, assess surgical suitability and monitor outcome. Magnetic resonance angiography (MRA) has neither of these disadvantages, and as familiarity with this technique increases it is being relied on increasingly in the assessment of patients with this condition. In experienced hands, the images produced with MRA are comparable to those seen with conventional pulmonary angiography.
MRA also enables assessment of lung perfusion (in a similar way to HRCT) and provides a very useful non-invasive means of assessing right ventricular function.
TREATMENT OPTIONS
It is universally accepted that the definitive treatment for CTEPH is PEA (fig 4). On occasions, however, patients with CTEPH will have disease which is not accessible to surgery (distal obstructive changes), or the patients themselves will be unsuitable for surgery because of co-morbidity. In these situations, PAH-specific drug treatment and, in rare cases, balloon angioplasty may be considered. Lung (or heart–lung) transplantation can also be considered in selected cases where PEA is not indicated, or when significant pulmonary hypertension persists following PEA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dramatic improvement in calibre of vessel lumens following pulmonary endarterectomy (PEA). Left panel: pre-PEA. Right panel: post-PEA.
Pulmonary endarterectomy (PEA)
It is vitally important to recognise that the surgical treatment of CTEPH is a true endarterectomy, involving the stripping of the diseased intimal layer from the media. This procedure is not an embolectomy as may be applied in the acute situation.
PEA is performed via a median sternotomy on cardiopulmonary bypass. Deep hypothermic circulatory arrest is employed during the endarterectomy, to prevent back bleeding from bronchial collaterals obscuring the operating field. In the vast majority of cases the procedure is performed bilaterally, although there are reports of true unilateral disease.
This operation is clearly a major undertaking, often performed in the setting of severe right ventricular dysfunction. The accurate assessment of surgical suitability is a vital aspect of achieving good outcomes for these patients. In experienced hands, the mortality associated with PEA is only 5–10%.17 For this reason, PEA should only be performed in centres with considerable experience in the assessment and management of patients with CTEPH.
There are a number of factors determining the outcome of PEA over and above the technical performance of the procedure. Increasing severity of right ventricular dysfunction (reflecting the severity of the underlying pulmonary hypertension) preoperatively is recognised as a risk for poorer outcome.17 Directly associated with this is the degree of residual pulmonary hypertension present postoperatively. In many cases, pulmonary and right ventricular haemodynamics will return to near normal within hours of the procedure. However, in some cases there is undoubtedly a significant contribution to the PVR from small vessel disease (see above). In this situation it may be some months before pulmonary and right ventricular remodelling occurs. Persistent pulmonary hypertension in the immediate postoperative period has an adverse effect on a severely compromised right ventricle and thus contributes significantly to postoperative morbidity and mortality.
The second factor influencing outcome is the occurrence of reperfusion lung injury. This represents a loss of endothelial integrity resulting in a spectrum of problems ranging from mild (non-cardiogenic) pulmonary oedema to a severe acute lung injury (diffuse alveolar damage). Following a successful PEA, this complication is the most important factor determining perioperative morbidity and mortality. Up to a third of patients undergoing PEA will require prolonged (>2 days) ventilatory support because of reperfusion injury, and this complication will be responsible for approximately half of the mortality associated with the procedure.5
Specific postoperative treatment strategies have been advocated to reduce the incidence of reperfusion injury and right ventricular dysfunction. These include the minimal use of catecholamines along with lung protective ventilation strategies utilising low tidal volumes and low peak inspiratory pressures.18
The other major factor influencing morbidity following PEA is cerebral injury associated with cardiopulmonary bypass and circulatory arrest. Minimal circulatory arrest times and reduced morbidity are achieved with increased surgical experience. A number of techniques involving antegrade cerebral perfusion have been advocated to reduce the incidence of this complication.19
Long term outcomes following PEA are generally excellent with most patients experiencing significant improvements in haemodynamics and functional capacity. Not surprisingly, the major determinate of a good outcome is the degree of residual pulmonary hypertension. In our experience, reverse pulmonary vascular remodelling can occur for up to a year post-PEA, but in most cases a plateau in improvement will occur between 3–6 months. It is thought that this reverse remodelling primarily involves the small vessel arteriopathy arising in that part of the pulmonary arterial vascular bed which was unobstructed. The role of PAH specific drugs targeting this small vessel disease post-PEA is unclear. There is a randomised controlled trial of an endothelin receptor antagonist currently in progress addressing this specific question.
Chronic thromboembolic pulmonary hypertension (CTEPH): key points
-
CTEPH should be specifically considered in the diagnostic workup of all patients with pulmonary hypertension
-
The prevalence and incidence of CTEPH have been grossly underestimated
-
Following an acute pulmonary embolism, patients at risk of developing CTEPH include those with any of the following: a major central thromboembolic event, significant haemodynamic disturbance on presentation, a documented thrombophilia, or persistent abnormalities of lung perfusion on follow up
-
If present to a significant degree, small vessel arteriopathy contributes substantially to the outcome of treatment for CTEPH
-
Ventilation/perfusion (V/Q) scanning is the screening investigation of choice for CTEPH
-
Patients with suspected CTEPH should be referred to a centre with significant experience in the assessment and treatment of this condition
-
In patients with operable CTEPH, pulmonary endarterectomy is the treatment of choice
Alternative therapeutic options
Balloon angioplasty has been reported as a treatment option in highly selected patients with CTEPH not amenable to surgical intervention. This technique is only applicable in cases where discrete stenoses are present in the main or segmental pulmonary arteries, and is ineffective if significant distal or small vessel disease existed.
Medical treatment utilising PAH specific drugs is reported as being efficacious in a number of scenarios involving CTEPH. The aim of drug treatment in this situation is to improve PVR by targeting secondary small vessel arteriopathy. Drugs such as bosentan, prostacyclin and iloprost have all been used with varying efficacy as a bridge to PEA in unstable patients, in patients with residual pulmonary hypertension post-PEA, and as definitive treatment in patients with CTEPH unsuitable for surgery. Bosentan treatment in patients with inoperable CTEPH resulted in improvements in haemodynamics, exercise tolerance (6 min walk test) and functional class.20 Intravenous prostacyclin used preoperatively improved haemodynamics. Inhaled iloprost used pre-PEA was ineffective but improved haemodynamics significantly when used postoperatively.
All patients receive lifelong anticoagulant treatment with warfarin or its equivalent. Antiplatelet agents are used as dictated by underlying disorders such as essential thrombocythaemia. It is common practice to insert an inferior vena cava (IVC) filter preoperatively although the evidence for this is based largely on historical practice. The presence of a filter is, however, reassuring if anticoagulation needs to be stopped at any time. Modern correctly placed IVC filters in the setting of ongoing anticoagulation are associated with few (if any) adverse effects.
CONDITIONS MIMICKING CTEPH
There are a number of disease states which can radiologically mimic CTEPH. These include pulmonary artery sarcoma, fibrosing mediastinitis, and large vessel arteritis (Takayasu disease). It is important to recognise these conditions as they are not amenable to PEA, although pulmonary artery sarcoma can be effectively palliated with surgical resection in some cases. The distinction between these conditions and CTEPH on diagnostic imaging is often subtle, further highlighting the need for these patients to be seen at centres with experience in assessing these patients.
SUMMARY
CTEPH is an important cause of severe pulmonary hypertension, which has been under recognised in the past. Appropriate diagnosis and assessment is vital as the majority of patients with this condition can be effectively cured with pulmonary endarterectomy. Assessment and surgical treatment of these patients should only be carried out in a centre with significant experience in treating this condition. For those patients unsuitable for surgery, or with significant pulmonary hypertension post-PEA, PAH specific drugs may be an effective therapeutic option.
Additional references appear on the Heart website— http://heart.bmj.com/supplemental
INTERACTIVE MULTIPLE CHOICE QUESTIONS
This Education in Heart article has an accompanying series of six EBAC accredited multiple choice questions (MCQs).
To access the questions, click on BMJ Learning: Take this module on BMJ Learning from the content box at the top right and bottom left of the online article. For more information please go to: http://heart.bmj.com/misc/education.dtl Please note: The MCQs are hosted on BMJ Learning—the best available learning website for medical professionals from the BMJ Group.
If prompted, subscribers must sign into Heart with their journal’s username and password. All users must also complete a one-time registration on BMJ Learning and subsequently log in (with a BMJ Learning username and password) on every visit.
MULTIPLE CHOICE QUESTIONS
Education in Heart Interactive (heart.bmj.com/misc/education.dtl)
Education in Heart (EiH) articles each have an accompanying series of six multiple choice questions. These are hosted on BMJ Learning—the best available learning website for medical professionals from the BMJ Group. Each article is submitted to EBAC (European Board for Accreditation in Cardiology; ebac-cme.org) for 1 hour of external CPD credit.
Free access for subscribers: For full details of the resources available to subscribers please see: heart.bmj.com/misc/education.dtl#access
How to access the questions: Click on BMJ Learning: Take this module on BMJ Learning from the online article content box, table of contents or EiH collection (heart.bmj.com/cgi/collection/heart_education).
If prompted, subscribers must sign into Heart with their journal’s username and password.
Please note: All users must also complete a one-time registration on BMJ Learning. Users will then subsequently log in (with a BMJ Learning username and password) on every visit in order to log activity and provide appropriate access.
Activating your subscription to Heart: If you have not yet activated your online subscription to Heart, please visit journals.bmj.com/cgi/activate/basic and enter your six digit (all numeric) customer number (found above your address label with your print copy). If you have any queries, please contact subscriptions{at}bmjgroup.com
Case based learning: You may also be interested in the cardiology interactive case histories published in association with Heart, for more information please see: heart.bmj.com/misc/education.dtl#ichs
REFERENCES
Supplementary materials
Files in this Data Supplement:
Footnotes
-
In compliance with EBAC/EACCME guidelines, all authors participating in Education in Heart have disclosed potential conflicts of interest that might cause a bias in the article