Article Text

Abstract
BACKGROUND Existing murine models of asthma lack many of the inflammatory and epithelial changes that are typical of the human disease. Moreover, these models are frequently complicated by allergic alveolitis.
METHODS High IgE responder BALB/c mice were systemically sensitised to ovalbumin and chronically challenged with low particle mass concentrations of aerosolised ovalbumin. Titres of anti-ovalbumin IgE in serum were measured at two weekly intervals by enzyme immunoassay, accumulation of inflammatory cells and histopathological abnormalities of the epithelium were quantified morphometrically in the trachea and the lungs, and airway reactivity was assessed by measuring bronchoconstriction following intravenous administration of methacholine.
RESULTS Mice sensitised by two intraperitoneal injections of ovalbumin developed high titres of IgE antibodies to ovalbumin. Following exposure to low concentrations of aerosolised antigen for up to eight weeks these animals developed a progressive inflammatory response in the airways, characterised by the presence of intraepithelial eosinophils and by infiltration of the lamina propria with lymphoid/mononuclear cells, without associated alveolitis. Goblet cell hyperplasia/metaplasia was induced in the intrapulmonary airways, while epithelial thickening and subepithelial fibrosis were evident following chronic exposure. In parallel, the mice developed increased sensitivity to induction of bronchospasm, as well as increased maximal reactivity. Non-immunised mice exposed to aerosolised ovalbumin had low or absent anti-ovalbumin IgE and did not exhibit inflammatory or epithelial changes, but developed airway hyperreactivity.
CONCLUSIONS This experimental model replicates many of the features of human asthma and should facilitate studies of pathogenetic mechanisms and of potential therapeutic agents.
- asthma
- mucosal inflammation
- epithelial abnormalities
- murine model
Statistics from Altmetric.com
Asthma is clinically characterised by bronchial hyperreactivity and symptoms of airway obstruction.1 Underlying the clinical manifestations is acute-on-chronic inflammation of the airway mucosa, with degranulation of mast cells, recruitment of eosinophils and neutrophils, as well as accumulation of activated T lymphocytes and other chronic inflammatory cells.2-4 Pulmonary parenchymal inflammation is not a significant feature of asthma,4 although limited numbers of eosinophils may be present in the alveoli immediately adjacent to involved airways.5 Abnormalities of the airway epithelium are prominent, including shedding and regeneration of ciliated cells, goblet cell hyperplasia and/or metaplasia, as well as a distinctive pattern of subepithelial fibrosis.4 ,6 Inflammatory cells are also observed within the epithelial layer, with intraepithelial eosinophils reported to be characteristic of asthma.7 The mucosal changes are consistently associated with hypertrophy and/or hyperplasia of the airway smooth muscle.8
While the inflammatory response within the airways has an atopic/allergic basis in at least two thirds of asthmatic patients,9 the mechanisms involved in the development of airway obstruction remain poorly understood. Accumulation of inflammatory cells and exudate, increased airway smooth muscle mass, deposition of connective tissue, and epithelial hyperplasia may all contribute to thickening of the airway wall, which appears to be the basis for the excessive diminution of airflow that accompanies bronchoconstriction in asthmatics.10 The pathogenesis of hyperreactivity is also unclear, although human studies suggest that it may be related to increased production of cytokines such as interleukin-511 ,12 or to epithelial injury by products of eosinophils13 ,14 with consequential loss of the epithelial barrier.15
Investigation of the pathophysiological mechanisms of chronic asthma has been limited by the lack of a satisfactory animal experimental model. Animals do not develop spontaneous asthma, although syndromes that have some similarity to human asthma have been described in horses16 and in cats.17 Attempts have been made to model the disease in a range of species including primates, sheep, rabbits, dogs, cats, guinea pigs, rats and mice.17-20 Models of allergic asthma in primates and sheep are considered to be the most reliably predictive of human responses to therapeutic agents,18 but these outbred animals are expensive and it is often difficult to ensure that they are free of subclinical infections. Many investigators have used guinea pigs for experimental studies, but these animals may yield exaggerated responses which are species-dependent and mediated by IgG rather than IgE, limiting their relevance to human allergic asthma.19Various models of allergic bronchopulmonary inflammation in laboratory rats and mice have recently been described. The relevance of experimental systems which employ antigens that bear little relationship to triggers of human asthma or which use pathophysiologically inappropriate methods for delivery of these antigens is debatable. However, models based upon sensitisation by systemic administration of protein antigens such as ovalbumin and subsequent inhalational challenge have gained widespread acceptance. In such models, concomitant development of both eosinophil dominated inflammation and airway hyperreactivity to cholinergic stimuli have repeatedly been demonstrated.21-24
Nevertheless, existing murine models of asthma have important limitations. Firstly, they do not usually exhibit the mucosal inflammation and recruitment of eosinophils into the epithelial layer that are characteristically associated with human asthma.25 Secondly, the majority involve relatively short term exposure to aerosolised antigen (usually no more than 1–2 weeks) and are thus devoid of the chronic inflammatory and epithelial changes that typify human asthma. Thirdly, most of the models are associated with an allergic alveolitis or hypersensitivity pneumonitis,26 which may overshadow the inflammatory lesions of the airways.
In this paper we describe a chronic inhalational challenge model of asthma in ovalbumin sensitised BALB/c mice. Using controlled exposure to low mass concentrations of aerosolised antigen, we have succeeded in eliciting acute-on-chronic allergic inflammation of the airways without an accompanying alveolitis. Furthermore, these mice exhibit abnormalities of airway epithelium similar to those observed in human asthma, as well as evidence of airway hyperreactivity to methacholine.
Methods
ANIMALS AND SENSITISATION
Specific pathogen-free female BALB/c mice aged 8–10 weeks were obtained from CULAS, Sydney. Animals were maintained in a laminar flow holding unit (Gelman Sciences, Sydney) and housed in autoclaved cages on autoclaved bedding in an air conditioned room on a 12 hour light/dark cycle. Autoclaved food and acidified water were provided ad libitum throughout. These mice, which are high IgE responders to ovalbumin,20 were either sensitised by inhalational exposure to ovalbumin without prior systemic immunisation or received an intraperitoneal injection of 10 μg of alum precipitated27 chicken egg ovalbumin (grade V, ⩾98% pure, Sigma, St Louis, Missouri, USA; unless otherwise specified, all chemicals were obtained from this source) 21 days before inhalational exposure and a booster injection seven days before inhalational exposure (hereafter referred to as boosted mice). All experimental procedures complied with the requirements of the Animal Care and Ethics Committee of the University of New South Wales (ref. no. 95/18).
INHALATIONAL EXPOSURE
Mice were exposed to aerosolised ovalbumin for 30 min/day on three days/week for up to eight weeks with assessment of responses usually at intervals of two weeks. Experimental groups comprised six mice at each time point. Exposures were carried out in a whole body inhalation exposure system (Unifab Corporation, Kalamazoo, Michigan, USA). During the exposure the animals were held in wire flow-through cage racks and filtered air was drawn through the 0.5 m3inhalation chamber at a flow rate of 250 l/min. Temperature and relative humidity were maintained at 20–25°C and 40–60% respectively. A solution of 2.5% ovalbumin in normal saline was aerosolised by delivery of compressed air to a sidestream jet nebuliser (Trimed, Sydney) and injected into the airstream prior to entering the chamber. The aerosol generated by this nebuliser comprised ⩾80% particles with a diameter of ⩽4 μm (and ⩾40% particles with a diameter of ⩽1 μm) which therefore had a ⩾90% probability of tracheobronchial deposition.28 The concentration and size distribution of particles within the breathing zone of the mice were continuously and cumulatively monitored using a light scatter device (PCAM-TX, PPM, Knoxville, Tennessee, USA). Particle concentration was maintained in the range of 10–20 mg/m3 by manual adjustment of a pressure regulator which controlled the air flow into the nebuliser. This approach was effective in achieving controlled exposure: the coefficient of variation of the mean daily aerosol concentration was <10% while the mean (SE) concentration over 24 exposures of an eight week experiment was 12.7 (0.2) mg/m3.
BLOOD AND TISSUE COLLECTION
Immediately prior to commencement of inhalational exposures, and thereafter at intervals of two weeks, mice were bled by retro-orbital puncture using heparinised capillary tubes. Plasma was diluted 1:20 in phosphate buffered saline (PBS) and subsequently assayed for ovalbumin specific IgE antibodies.
For each time point sampled, tissues for histopathological examination were collected 24 hours after the last inhalational exposure. Animals were killed by exsanguination following an overdose of pentobarbital and the lungs were perfused with 0.9% saline under a pressure of 40 cm H2O for 60 seconds to remove blood from the pulmonary capillary bed. The trachea was then cannulated with a blunted #19 needle and the lungs were inflated with 10% buffered formalin. After fixation overnight the longitudinally oriented trachea and a horizontal slice from the mid zone of the single lobed left lung were embedded in paraffin. Ribbons of 2–3 sections cut at 5 μm were stained with haematoxylin and eosin (for routine histopathological examination), with Gordon and Sweet’s reticulin stain (for assessment of collagen deposition), or with Alcian Blue-PAS (for enumeration of mucin secreting cells). Inflammatory and epithelial changes were assessed both in the trachea and in the intrapulmonary conducting airways.
ENZYME IMMUNOASSAY FOR IgE ANTIBODIES TO OVALBUMIN
IgE antibodies to ovalbumin in plasma samples from individual animals were quantified using an enzyme immunoassay. Microplate wells coated with 10 μg/ml of ovalbumin were incubated for one hour with 100 μl each of doubling dilutions of the test samples in PBS containing 0.05% Tween-20. Detection of bound anti-ovalbumin IgE was by sequential incubation with monoclonal rat anti-mouse IgE antibody (R35–92, PharMingen, San Diego, California, USA) and biotinylated rabbit anti-rat immunoglobulins (Dako, Glostrup, Denmark). To enhance the sensitivity of the assay, enzyme complex amplification29 was used as the reporter system. The reporter reagent was prepared by assembling complexes of streptavidin (Boehringer Mannheim) and biotinylated horseradish peroxidase in PBS-1% BSA, then cross linking these with biotinylated goat anti-horseradish peroxidase (Jackson Immunoresearch, West Grove, Pennsylvania, USA) and additional streptavidin. Colour development was by incubation with ABTS substrate in citrate phosphate buffer (pH 4.0) and the reaction product was read using a Titertek Multiskan photometer (ICN Biomedicals, Sydney) with a 405 nm filter. Each assay included six samples of pre-immune mouse plasma which were tested at a dilution of 1:20. The titre of a test sample was determined as the highest dilution at which the absorbance was greater than two standard deviations above the mean of the pre-immune samples. Titres of IgE anti-ovalbumin were logarithmically transformed to calculate the geometric mean for each group of experimental animals.
MORPHOMETRY
For quantitative analysis of the inflammatory response in the trachea, haematoxylin and eosin stained sections were examined using a Leica Q500MC image analysis system (Leica, Cambridge, UK) calibrated with a reference measurement slide. For each animal a section and an initial field of examination at one end of the section were selected at random. Using a ×40 objective, nuclear profiles in the lamina propria (defined as the region bounded by the epithelial basement membrane and the luminal border of the tracheal cartilage plates) were counted in the field and the length of the epithelial basement membrane was measured. The microscope stage was then advanced a linear distance of two microscopic fields and the process was repeated, continuing in this manner until the full length of the tracheal section had been examined. On average, this meant that five fields were analysed per section for these measurements. The data were used to calculate the mean number of cells per 100 μm of epithelial basement membrane in the lamina propria for individual animals.
The thickness of the epithelial layer was also measured in these sections, taking care to exclude fields in which the plane of section was tangential. Two standardised sampling points were used for measurement in each microscopic field and six measurements were made per section. The mean thickness for each animal was calculated.
In parallel, the length of the epithelial basement membrane for the entire section was measured using a ×10 objective. The full length of epithelial surface of the appropriately identified section was then examined using a ×100 oil immersion objective to enumerate intraepithelial eosinophils, which were readily identified on the basis of their strongly staining cytoplasmic granules, as well as neutrophils. The data were expressed as number of cells per mm of epithelial basement membrane for individual animals.
Reticulin stained sections were used for assessment of subepithelial collagenisation. The image analysis system was used to measure the thickness of the reticulin stained zone directly underneath the tracheal epithelium using a ×40 objective. Three standardised sampling points were used for measurement in each microscopic field with an average of 18 measurements per section. The mean thickness for each animal was calculated.
Metaplasia and/or hyperplasia of mucus secreting goblet cells was assessed in sections of lung tissue stained with Alcian Blue-PAS. Goblet cells are rarely present in the epithelium of the intrapulmonary airways of normal mice and their presence was thus readily apparent in these sections. The percentage of cells staining positive for mucins was enumerated in the largest visible airway which, in these horizontal sections through the hilum, was usually the left main bronchus. To minimise the effects of sampling error, each animal was categorised according to a semilogarithmic scale as grade 0 = <1% positive cells, grade 1 = 1–3%, grade 2 = 4–10%, grade 3 = 11–30%, and grade 4 = ⩾31%.
METHACHOLINE RESPONSIVENESS
Measurements of airway reactivity to intravenous methacholine were performed 48 hours after the last inhalational exposure. As previously described,23 a bronchospasm transducer (Ugo Basil 7020; Ugo Basile, Varese, Italy) coupled to a Lab Mac/8 analysis station (AdInstruments, Sydney) was used to determine airway constriction during cumulative intravenous administration of β-methacholine to anaesthetised mice ventilated under constant pressure. For each animal the increase in respiratory overflow volume provoked by each concentration of β-methacholine was represented as a percentage of maximal overflow obtained by occluding the tracheal cannula. For comparison between treatment groups a fourth order polynomial curve was fitted to the dose-response data and used to calculate the concentration that produced a 20% increase above baseline in airway occlusion. Control groups for these studies were mice that had been sham immunised with adjuvant alone as well as boosted mice, both exposed to aerosolised normal saline.
STATISTICAL ANALYSIS
Results of morphometry and assessment of airway reactivity are presented as arithmetic mean (SE) for each experimental group. In general, an initial one way analysis of variance followed by Dunnett’s multiple comparison test was used to examine differences between groups of animals exposed to ovalbumin and normal control animals. Comparison of the proportion of mucin positive cells in the intrapulmonary airways was based upon grading and therefore employed a non-parametric Kruskal-Wallis test followed by Dunn’s multiple comparison procedure. The software package GraphPad Prism (GraphPad Software, San Diego, California, USA) was used for all data analysis and preparation of graphs.
Results
None of the animals exhibited noticeable distress or significant tachypnoea during the period of study, nor was there any mortality.
ANTIBODY RESPONSE
Low levels of IgE antibodies to ovalbumin (geometric mean titre 1:40) were demonstrated in non-immunised mice after two weeks of inhalational exposure. However, antibody titres declined to undetectable levels thereafter. Boosted animals had high initial titres of anti-ovalbumin IgE (geometric mean 1:3630) which rose approximately fourfold following inhalational exposure.
AIRWAY INFLAMMATION
Trachea
The cellularity of the lamina propria of the trachea appeared unchanged after six weeks of exposure of non-immunised mice to aerosolised ovalbumin, although occasional eosinophils were observed within the epithelial layer. In contrast, boosted mice exhibited recruitment of eosinophils into the lamina propria of the trachea after exposure for only one week. This was followed by the progressive development of focal mononuclear cell infiltrates after two weeks of exposure, which became widespread by 6–8 weeks (fig 1A and B). Numerous intraepithelial eosinophils were also observed (fig2).

(A) Trachea from a boosted animal after six weeks of exposure. Aggregates of inflammatory cells are visible throughout the lamina propria. Note the subepithelial acellular zone of eosinophilic matrix material (arrows). Haematoxylin and eosin, bar = 200 μm. (B) Chronic inflammatory cells (lymphocytes, plasmacytoid lymphocytes and other mononuclear cells) in the lamina propria of the trachea from a boosted animal exposed for six weeks. Haematoxylin and eosin, bar = 50 μm.

Eosinophils within the tracheal epithelium of a boosted mouse after exposure to aerosolised ovalbumin for eight weeks. Haematoxylin and eosin, bar = 20 μm.
Intrapulmonary conducting airways
Accumulation of chronic inflammatory cells in the lamina propria was also evident in the intrapulmonary airways of boosted mice after six weeks of inhalation exposure to ovalbumin. This frequently took the form of focal aggregates of mononuclear cells (fig 3A) resembling bronchus associated lymphoid tissue which is normally not apparent in specific pathogen-free BALB/c mice. Inflammation in the intrapulmonary airways was not associated with any evidence of alveolitis (fig 3B). Of 54 mice examined histopathologically after varying periods of inhalational exposure to ovalbumin, only one exhibited a single focal area of parenchymal inflammation.

(A) Chronic inflammatory cells in the wall of the left main bronchus of a boosted animal after six weeks of exposure. Note the goblet cells in the airway epithelium (arrows). Haematoxylin and eosin, bar = 50 μm. (B) Absence of inflammatory changes in the lung parenchyma of a boosted animal after six weeks of exposure. Note the aggregates of inflammatory cells visible adjacent to the large intrapulmonary airways. Haematoxylin and eosin, bar = 400 μm.
Morphometric assessment of the cellular response in the airways was undertaken for non-immunised mice that were exposed to ovalbumin for six weeks and for boosted mice that were exposed for two, four, six or eight weeks. For convenience of sampling and measurement, quantification was performed in sections of trachea. Boosted mice exhibited statistically significant increases in the numbers of inflammatory cells in the lamina propria which were evident at all time points examined (table 1). Increased numbers of intraepithelial eosinophils were observed in the tracheal epithelium of all groups of exposed animals, although these were statistically significant only after six and eight weeks of exposure of boosted mice (table 1). Small increases in the numbers of intraepithelial neutrophils were observed in most groups of boosted animals after exposure to aerosolised ovalbumin, but these changes were not statistically significant.
Mean (SE) of inflammatory and epithelial changes in the airways as assessed by morphometry
EPITHELIAL LESIONS
Trachea
Exposure of non-immunised mice to aerosolised ovalbumin for six weeks was not associated with any evidence of epithelial abnormalities. Following chronic exposure of boosted mice there was a generalised increase in the thickness of the epithelium together with accumulation of eosinophilic extracellular matrix material beneath the epithelial layer (fig 1A). Reticulin staining confirmed the deposition of collagen within this zone (fig 4A and B). Occasional focal areas of epithelial discontinuity were also observed in relation to aggregates of inflammatory cells. Morphometric measurements showed that the mean thickness of the epithelium was significantly increased after eight weeks of exposure (table 1) and the mean thickness of the subepithelial zone of collagenisation was significantly increased from four weeks onwards (table 1).

(A) Trachea from a non-immunised mouse after exposure for six weeks showing normal thickness of the subepithelial collagenous zone (arrow). Reticulin stain, bar = 50 μm. (B) Trachea from a boosted mouse after exposure for eight weeks showing marked thickening of the subepithelial collagenous zone (arrow) and of the epithelial layer. Reticulin stain, bar = 50 μm.
Intrapulmonary conducting airways
Analogous to the changes observed in the trachea, subepithelial fibrosis was also noted in the intrapulmonary conducting airways of boosted mice after chronic exposure to ovalbumin. More striking, however, was the increase in the number of goblet cells in intrapulmonary airways of these mice. Goblet cells were virtually absent from the epithelium of the intrapulmonary airways of normal mice. Occasional Alcian blue or PAS positive cells were observed in the epithelium of the left main bronchus of non-immunised mice exposed to ovalbumin for six weeks (fig 5A). In contrast, marked hyperplasia/metaplasia of mucus-secreting cells was evident in the bronchial epithelium of boosted mice exposed in a similar fashion (fig5B). Morphometric quantification revealed a significant increase in the proportion of goblet cells as early as two weeks after commencement of inhalational exposure to ovalbumin (table 1). This was not associated with significant epithelial thickening.
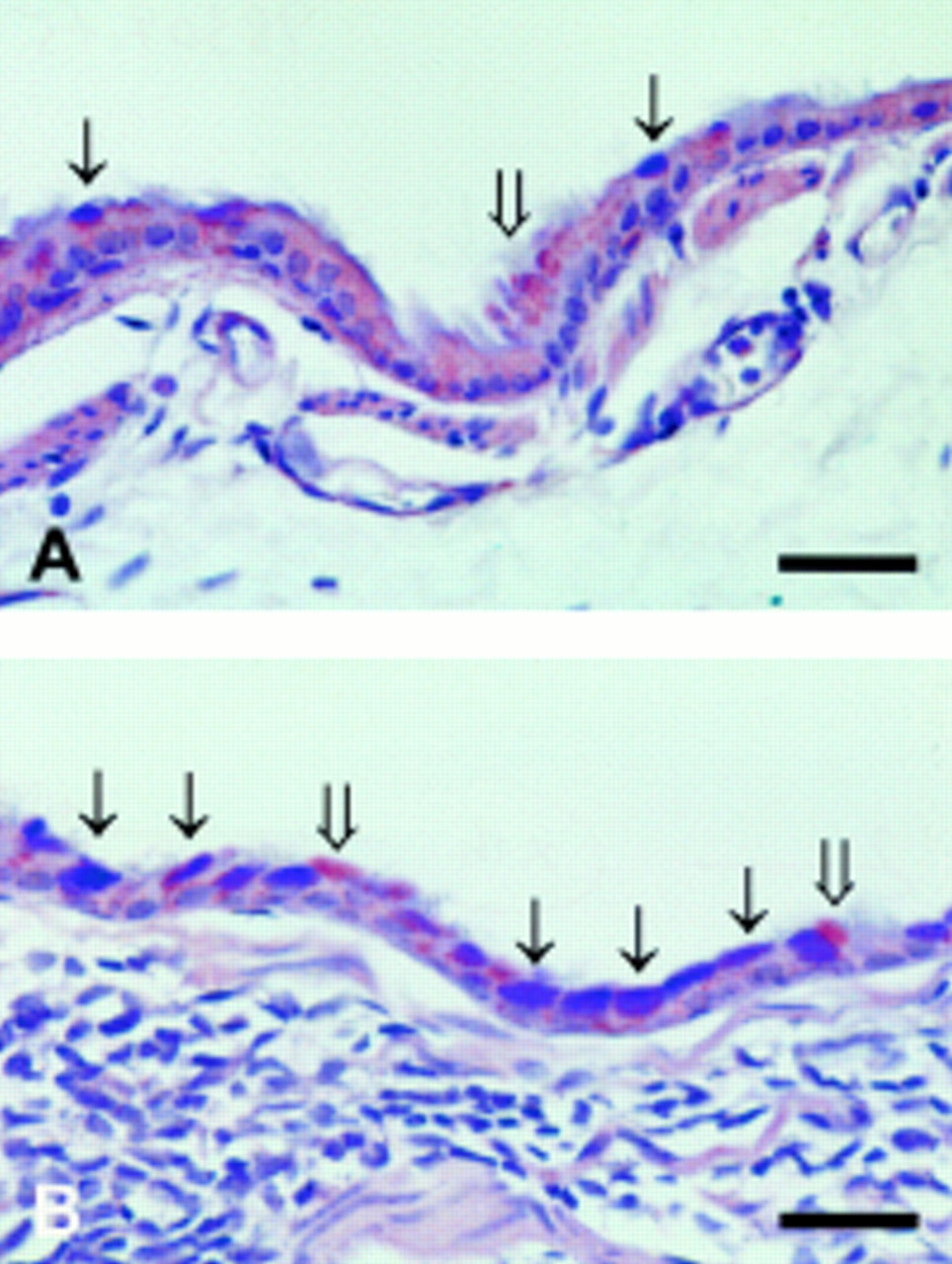
(A) Left main bronchus from a non-immunised mouse after exposure to aerosolised ovalbumin for six weeks showing occasional mucin-producing cells in the epithelium staining with Alcian Blue (thin arrows) or with PAS (wide arrow); bar = 50 μm. (B) Left main bronchus from a boosted mouse after exposure to aerosolised ovalbumin for six weeks showing many epithelial cells staining with Alcian Blue (thin arrows) as well as some cells staining with PAS (wide arrows). Note the numerous inflammatory cells in the airway wall; bar = 50 μm.
AIRWAY HYPERREACTIVITY
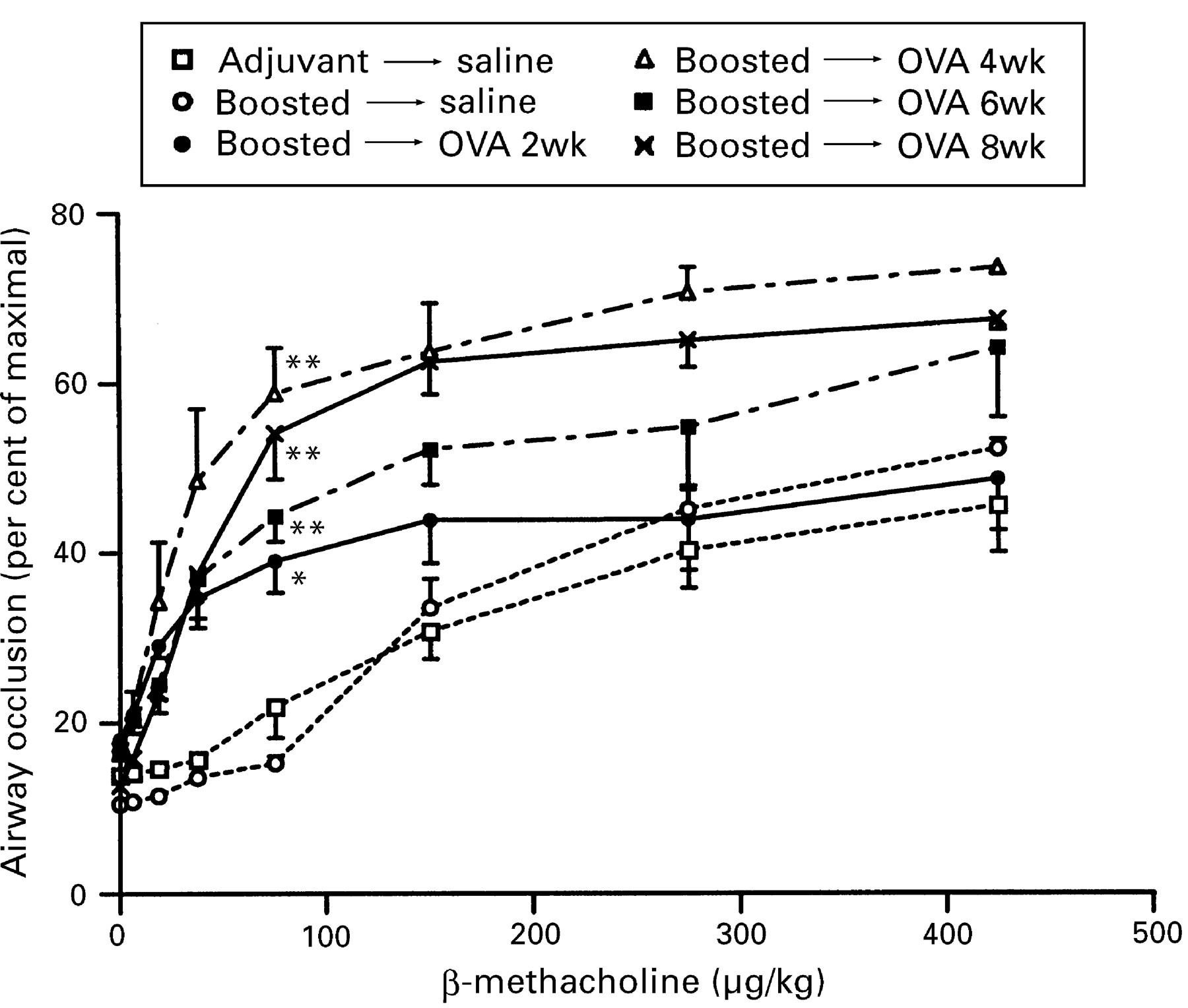
Following exposure to aerosolised ovalbumin, both non-immunised and boosted mice developed hyperreactivity to induction of bronchospasm by the non-specific cholinergic agonist β-methacholine. In non-immunised mice airway reactivity was unchanged after two weeks, but increased sensitivity to β-methacholine (a left-shifted dose-response curve) as well as increased maximal reactivity (a higher plateau) were apparent at all time points thereafter. In boosted mice increased sensitivity to β-methacholine was evident after two weeks of exposure, with an increased maximal response also being demonstrable at later time points (fig 6). The presence of airway hyperresponsiveness was further exemplified by calculating the concentration of β-methacholine that produced a 20% increase above baseline in airway occlusion, shown in table 2 for each experimental group.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Induction of bronchospasm by administration of increasing doses of intravenous β-methacholine to boosted mice following exposure to aerosolised ovalbumin (OVA) compared with mice that received adjuvant alone or boosted mice that were exposed to aerosolised saline. For the dose of 75 μg/kg significant differences compared to animals that received adjuvant and aerosolised saline are shown as * = p <0.05, ** = p <0.01.
Concentration of β-methacholine (μg/kg) producing 20% increase above baseline in airway occlusion
Discussion
In this study we sought to develop a model of allergic asthma which replicated the chronic inflammatory and epithelial changes of the human disease. Chronic inhalational exposure of mice that had been sensitised by repeated systemic administration of antigen yielded a model that exhibited many of the characteristics of human asthma. Notably, the inflammatory response in these animals involved the accumulation of a mixture of acute and chronic inflammatory cells in the lamina propria of both the trachea and the intrapulmonary conducting airways, together with the presence of intraepithelial eosinophils. Various epithelial abnormalities were also observed including hypertrophy, goblet cell hyperplasia/metaplasia, and subepithelial fibrosis. In parallel, the mice demonstrated enhanced bronchoconstriction of the airways in response to a cholinergic agonist. To elicit changes comparable to those of human asthma, it was necessary to perform chronic inhalational exposure to the aerosolised antigen as most of the relevant abnormalities were only apparent after 4–6 weeks of exposure. This model is thus relatively labour intensive but does succeed in achieving a new level of fidelity of asthmatic lesions.
Chronic inflammation of the lamina propria of the airways and intraepithelial accumulation of eosinophils are both hallmarks of human asthmatic inflammation.7 ,30 To our knowledge acute-on-chronic inflammation of the airway wall has not hitherto been demonstrated in experimental asthma. Most reported inhalational exposure models are primarily characterised by acute parenchymal inflammation of the lung, with perivascular and peribronchiolar accumulation of large numbers of eosinophils.24 ,31-36However, there is usually a paucity of intraepithelial eosinophils.37 Thus these systems model allergic acute inflammation but are not representative of the asthmatic inflammatory response. In contrast, the inflammatory response in our experimental model was selectively localised to the airways, without any confounding acute alveolitis/hypersensitivity pneumonitis. Furthermore, whereas previous studies of chronic antigen exposure have consistently reported the development of pulmonary granulomas,38-40 no such lesions were observed in our model. We believe that the absence of parenchymal inflammation was probably a result of careful maintenance of a low mass concentration of aerosol in the exposure chamber. In preliminary experiments we found that a single uncontrolled exposure to aerosolised ovalbumin for one hour readily induced alveolitis in all boosted exposed animals (G M Velan and R K Kumar, unpublished results).
Most of the previously described murine models of asthma do not demonstrate many of the chronic epithelial changes that are seen in the human disease. Although increased numbers of goblet cells are frequently observed,24 ,35 ,40 this appears to be a non-specific response to inflammation which may even develop following acute exposure to antigen.36 To date the only model in which subepithelial fibrosis has been described involved an artificial challenge protocol in which repeated intratracheal injections of antigen were administered.40 In our model hyperplasia and/or metaplasia of mucus secreting cells was evident but, in addition, the animals developed epithelial hypertrophy and subepithelial fibrosis.
In mice that were not systemically sensitised low titres of IgE evident following initial inhalational exposure to antigen were later suppressed, a finding that closely parallels the observations of Holt and coworkers.41 ,42 As in other such models,43 no statistically significant inflammatory response developed in these animals. In contrast, systemically sensitised and boosted mice which had high initial titres of anti-ovalbumin IgE developed chronic inflammation of the airway walls. Previously reported studies of the relationship between serum IgE titres and allergic bronchopulmonary inflammation have yielded contradictory findings.33 ,37 ,44 ,45 It should be noted, however, that the observed association does not establish any cause and effect relationship between IgE titres and the inflammatory response in this model, as titres of anti-ovalbumin IgG were also raised in these animals (data not shown).
Boosted mice chronically exposed to aerosolised ovalbumin exhibited marked airway hyperreactivity to methacholine. The sensitivity to bronchoconstriction and the maximal response to this agonist, both of which are relevant criteria of hyperreactivity,46 were concomitantly increased in these mice. Unlike the short term hyperresponsiveness observed in some experimental models,47 increased methacholine responsiveness was demonstrable 48 hours after the last exposure to antigen. In contrast to many previously reported experimental models, parenchymal inflammation would not have contributed to the development of hyperreactivity. However, we recognise that the technique used for measurement of bronchial reactivity in this study has no direct parallel to assessment of airway responsiveness as performed in human subjects.48 In future studies we intend to evaluate airway responses using whole body plethysmographic assessment of reactivity to inhaled methacholine in conscious unrestrained animals,49which more realistically approximates the technique used in humans. In addition, we hope to characterise further the relationship between airway lesions and methacholine responsiveness using techniques which permit distinction between reactivity of airway and parenchymal origin.50
Of considerable interest is the evidence that, in these experiments, airway hyperreactivity did not correlate with IgE titres or with the magnitude of the inflammatory response in the airway wall. The mechanism of hyperreactivity in the non-immunised mice is unclear. However, whereas repeated exposure to aerosolised ovalbumin elicited airway hyperreactivity in both non-immunised and boosted mice, this took longer to induce in the former group of animals which became hyperreactive only after four weeks of inhalational exposure to antigen. This time course is consistent with induction of an immune response, which might then have led to local production of cytokines that are important in hyperreactivity. In this context, both animal22 ,23 ,33 and human studies12 ,51 ,52indicate that interleukin-5, probably derived from T lymphocytes, plays a major role in the development and maintenance of airway hyperreactivity. Thus it is possible that, following chronic exposure in the non-immunised mice, local production of interleukin-5 by sensitised T lymphocytes was responsible for induction of hyperreactivity.
While mice can never wholly replicate the features of human asthma, given the significant differences in the microanatomy and physiology of the respiratory system, the experimental model described herein simulates the morphological and functional changes of asthma with a greater degree of accuracy than previously reported models. For experimental convenience ovalbumin was used as the antigen for these studies. However, our approach employing chronic inhalational exposure to low mass concentrations of aerosol can probably be extended to other antigens. We believe this improved model should permit meaningful studies of the pathogenesis of chronic inflammatory and epithelial lesions of asthma, as well as of potential therapeutic agents.
Acknowledgments
This work was supported by grants from the Asthma Foundation of New South Wales and the Government Employees Medical Research Fund. We thank Ms Angelina Enno for preparation of the tissue sections, Dr Mark Wilson for providing reagents for enzyme complex amplification, and Mr Michael Oakey for assistance with the preparation of figures. Jason Temelkovski is a recipient of an Australian Postgraduate Award.