Article Text

Abstract
Background: Matrix metalloproteinases (MMPs) are involved in the remodelling and degradation of extracellular matrix and may play a role in pulmonary tissue destruction in cystic fibrosis (CF).
Methods: Bronchoalveolar lavage (BAL) fluid levels of MMP-8, MMP-9, and their natural inhibitor TIMP-1 were measured on two occasions within 18 months in 23 children with mild CF, 13 of whom were treated with DNase.
Results: MMP-8 (39.3 (6.8) v 0.12 (0.01) ng/ml), MMP-9 (58.0 (11.4) v 0.5 (0.02) ng/ml), and the molar ratio of MMP-9/TIMP-1 (0.36 (0.05) v 0.048 (0.01)) were significantly higher in patients with CF than in control children without lung disease. Gelatine zymography showed the typical banding pattern of neutrophil derived MMP-9, including 130 kDa NGAL-MMP-9 complex and 92 kDa latent MMP-9 bands; 85 kDa bands (corresponding to active MMP-9) were seen in all patients. There was a close correlation between BAL fluid concentrations of MMPs and α2-macroglobulin, a marker of alveolocapillary leakage. After 18 months MMP levels were increased in untreated patients and decreased in patients treated with DNase.
Conclusions: Uninhibited MMPs may contribute to pulmonary tissue destruction even in CF patients with mild lung disease that may be positively affected by treatment with DNase.
- bronchoalveolar lavage
- cystic fibrosis
- metalloprotease
Statistics from Altmetric.com
Cystic fibrosis (CF) lung disease is characterised by an intense neutrophil dominated airway inflammation that is observed even in clinically stable patients with limited lung disease as well as in newborn infants diagnosed by neonatal screening.1–3 Many factors are thought to contribute to this inflammatory process that ultimately leads to tissue destruction and lung damage.
Metalloproteases (MMPs) are a group of enzymes that play a central role in the turnover of extracellular matrix (ECM) components as well as tissue degradation, repair mechanisms, and cell migration.4,5 More than 20 MMPs have been described which can be subdivided according to their function into five classes: gelatinases (MMP-2, MMP-9), collagenases (MMP-1, MMP-8, MMP-13), stromelysins (MMP-3, MMP-10), membrane-type MMPs (MT1-MMP, MT1-MMP-2, MT1-MMP-3), and others such as matrilysin (MMP-7), stromelysin 3 (MMP-11), and metalloelastase (MMP-12). They are secreted in inactive forms (zymogens) from different kind of cells or expressed as plasma membrane bound forms. While many cells within the respiratory tract are known to produce MMPs, both MMP-8 (also known as neutrophil collagenase) and MMP-9 in the lower respiratory tract are mainly derived from neutrophils which play a central part in CF airway inflammation. MMP-9 belongs to a group of gelatinases that has received special interest because of their ability to degrade both elastin and type IV collagen, a major component of basement membranes.6,7 MMPs are inactivated in vivo mainly by specific tissue inhibitors (TIMPs) that bind with high affinity in a molar 1:1 ratio to the catalytic site of MMPs.6
An excessive amount of MMPs is thought to be potentially harmful since it may lead to pulmonary tissue destruction.4–6 Although preliminary data using sputum have suggested that MMPs are increased in the airways of patients with CF, their role in CF lung disease is currently unclear.7 We have measured MMP-8 and MMP-9 and their specific inhibitor TIMP-1 in bronchoalveolar lavage (BAL) fluid of children with CF and limited lung disease.
METHODS
Patients
The study population consisted of 23 patients with CF of mean (SD) age 11.2 (3.2) years (range 6–18) with normal lung function defined as forced expiratory volume in 1 second (FEV1) above 80% predicted. Bronchoalveolar lavage was performed as part of the BEAT study, a prospective multicentre study to evaluate the evolution of bronchial inflammation in CF patients with preserved lung function and the influence of DNase on this inflammatory process.8 To study the change in MMPs over time and the possible influence of DNase, all patients were studied on two occasions with an interval of 18 months between them. Thirteen patients received DNase (2.5 mg once daily inhaled via a Pari Master compressor with a LC Plus nebuliser) after the first BAL, while in the remaining 10 patients treatment was unchanged. None of the patients received anti-inflammatory treatment (systemic or inhaled steroids, ibuprofen) during the study period. All patients were free of acute respiratory infections and pulmonary exacerbations 6 weeks before each BAL study.
For comparison, 11 children without pulmonary diseases undergoing elective surgery for non-pulmonary illnesses were studied as described previously.9
The study was approved by the ethical committee of the University Hospital of Essen and written informed consent was obtained in all cases.
Bronchoalveolar lavage
Bronchoalveolar lavage was performed as described previously8 under sedation with midazolam (0.1–0.3 mg/kg) and local anaesthesia with lidocaine. Because it has been suggested that pulmonary inflammation is more pronounced in the upper lobes of patients with CF, BAL was performed in the lingula in all patients using aliquots of 20 ml up to a total volume of 3 ml/kg. The first sample was considered primarily a bronchial sample and was processed separately; subsequent samples were pooled for analysis. The reported measurement of BAL fluid proteins refers to the analysis of the pooled sample only.
After filtration through one layer of sterile gauze, BAL fluid was centrifuged at 500g for 10 minutes to separate the supernatants from the cell pellet. BAL fluid was stored as aliquots at –70°C for further analysis. The total cell count was performed on a haemocytometer (Coulter Electronics Ltd). The cell suspension of both the first and the pooled BAL sample was washed three times in Eagle’s minimal essential medium (MEM) containing 0.2% bovine serum albumin and 0.1% EDTA or in Hank’s solution and resuspended in MEM. Bronchoalveolar cells were counted with a cell counter and cell viability was assessed by trypan blue exclusion. Differential cell counts were obtained from smears stained with May-Gruenwald-Giemsa. At least 600 cells were counted in each subject.
Measurement of MMP-8, MMP-9, and TIMP-1 by ELISA
The concentrations of MMP-8, MMP-9, and TIMP-1 in BAL fluid were determined by ELISA (Biotrak, Amersham Life Science, UK) according to the manufacturer’s instructions. The total concentration of each enzyme was measured, whether latent, active, free, or complexed. In brief, samples were diluted in respective assay buffer and a standard curve was developed using known amounts of recombinant human MMP and TIMP. Colorimetric absorption was read at 450 nm using an SLT 340 ATTC microtitre plate reader. Concentrations were determined by relating the absorbance obtained to the standard curve. Each sample was assayed in duplicate and the values were within the linear portion of the standard curve. Sensitivity, specificity, and reproducibility were sufficiently reliable as previously reported.10
Gelatine zymography
Gelatine zymography was performed as previously described.11,12 Briefly, sodium dodecyl sulfate (SDS) gels containing 0.1% gelatine were used to identify gelatinolytic proteases from BAL fluid samples. Each lane was loaded with 30 μl of a 1:20 diluted sample. Electrophoresis was run at a constant current of 40 mA for 90 minutes. After electrophoresis, the gels were washed in 2.5% Triton-X-100 for 1 hour to remove SDS. The gel was then incubated at 37°C for 18 hours in enzyme buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, 5 mM CaCl2, and 0.02% Brij-35. Negative staining was performed for 2.5 hours with Coomassie brilliant blue to visualise the bands of gelatinolytic activity. To identify the enzymes, purified MMP-9 was loaded onto each gel. The character of the bands was analysed by incubating identical gels with 10 mM EDTA (a selective MMP inhibitor) and PMSF (a serine protease inhibitor).
Determination of α2-macroglobulin and lactoferrin
Levels of α2-macroglobulin and lactoferrin in BAL fluid were measured using highly sensitive immunoluminometric assays. Assays were performed as previously described.13 Light emission was determined using an Autolumat LB 953 (Berthold, Germany) at 37°C.
Statistical analysis
The data were analysed using non-parametric tests. The Mann-Whitney U test was used for unpaired samples, the Wilcoxon signed rank test was used for paired samples, and correlations were analysed with Spearman’s rank correlation. A p value of <0.05 was accepted as significant. Parametric data are presented as mean (SD) and non-parametric data as median with 95% confidence interval or range. Calculations were carried out using Statistica for Windows, Version 5, 1997.
RESULTS
MMP-8, MMP-9, and TIMP-1 levels in BAL fluid
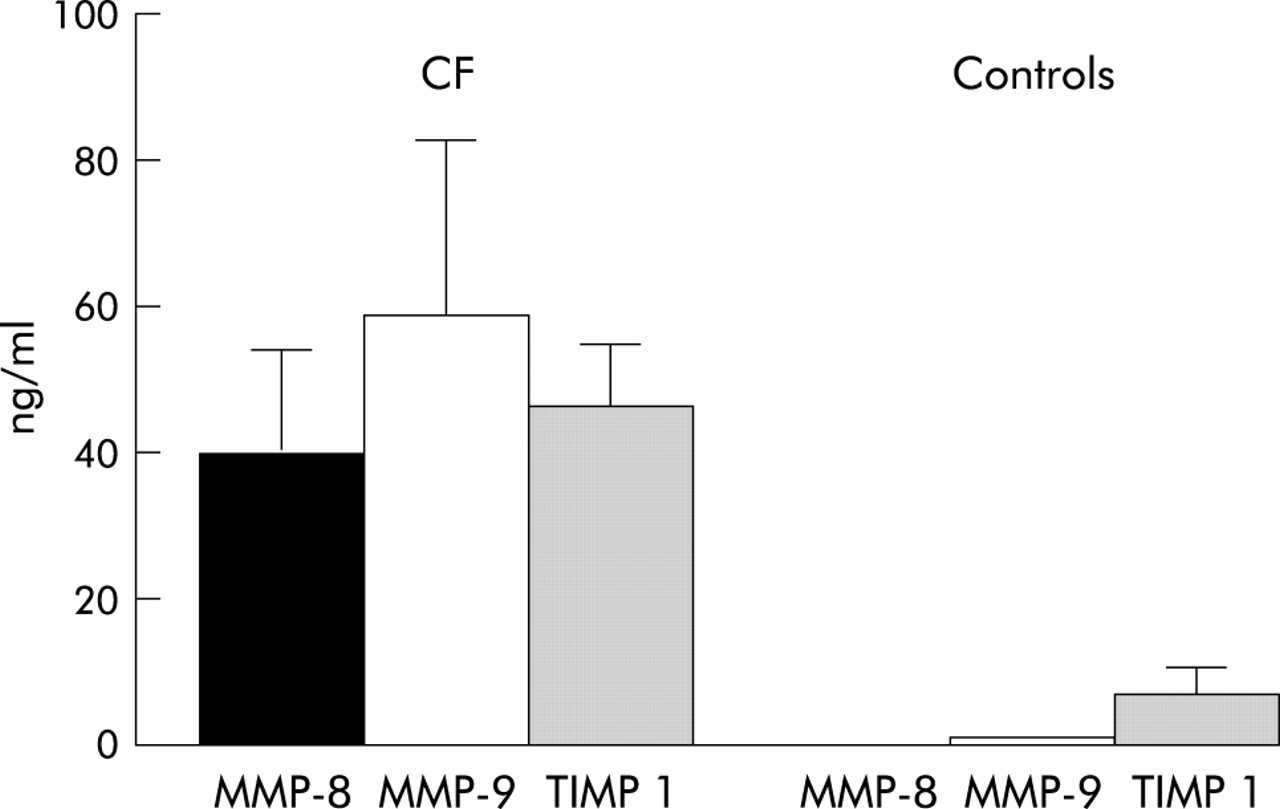
The concentrations of MMP-8 and MMP-9 in BAL fluid were significantly increased in patients with CF compared with healthy controls. MMP-8 was increased more than 300-fold and MMP-9 levels were 116-fold higher. In contrast, levels of TIMP-1 were only six times higher than in healthy controls (table 1, fig 1). There was a significant difference in the median (range) molar MMP-9/TIMP-1 ratio between patients (0.99 (0.22–3.85)) and controls 0.14 (0.03–4.00); p<0.01).
Levels of MMP–8, MMP–9, TIMP–1 and the percentage of neutrophils in BAL fluid of patients with cystic fibrosis and healthy controls at two different time points of the investigation
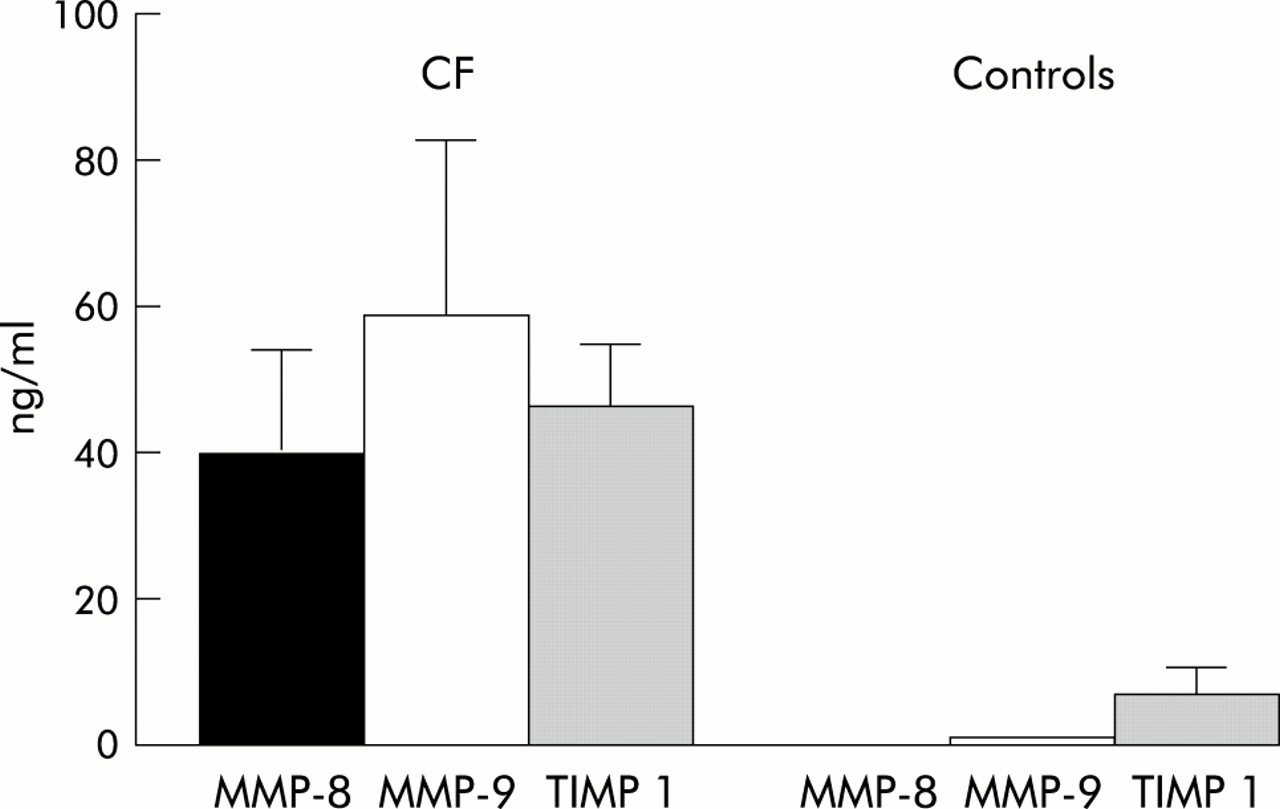
Levels of MMP-8, MMP-9, and TIMP-1 in BAL fluid of patients with cystic fibrosis and healthy controls (median and 95% confidence interval).
Correlation between percentage of cells and MMP levels
The mean (SD) percentage of neutrophils and macrophages in BAL fluid was 35 (4)% and 53 (4)%, respectively. In patients with CF the levels of MMP-8 and MMP-9 in BAL fluid was significantly correlated with the percentage of neutrophils in BAL fluid (MMP-8: p<0.05, r=0.5; MMP-9: p<0.05, r=0.5). Furthermore, levels of MMP-8 and MMP-9 were inversely correlated with the percentage of macrophages (MMP-8: p<0.005, r=–0.43; MMP-9: p=0.005, r=–0.42).
Determination of gelatinolytic activity
In all patients zymographic analysis showed three distinct bands with molecular weights of approximately 92, 130, and 220 kDa. They revealed the typical banding pattern of neutrophil derived MMP-9, consisting of pro-MMP-9 (92 kDa), HNL (human neutrophil lipocalin)-pro-MMP-9 complex (130 kDa), and the homodimeric form of MMP-9 (220 kDa). The pro-MMP-9 bands (92 kDa) were the most prominent bands in all patients. Furthermore, 85 kDa bands corresponding to the active form of MMP-9 were detected in most patients. In contrast, no gelatinolytic activity could be detected in controls. All bands disappeared in the presence of EDTA, an inhibitor of MMP, but the addition of PMSF, a serine protease inhibitor, had no effect. This indicates that the gelatinolytic activity was due to the presence of MMP.
Measurement of α2-macroglobulin and lactoferrin
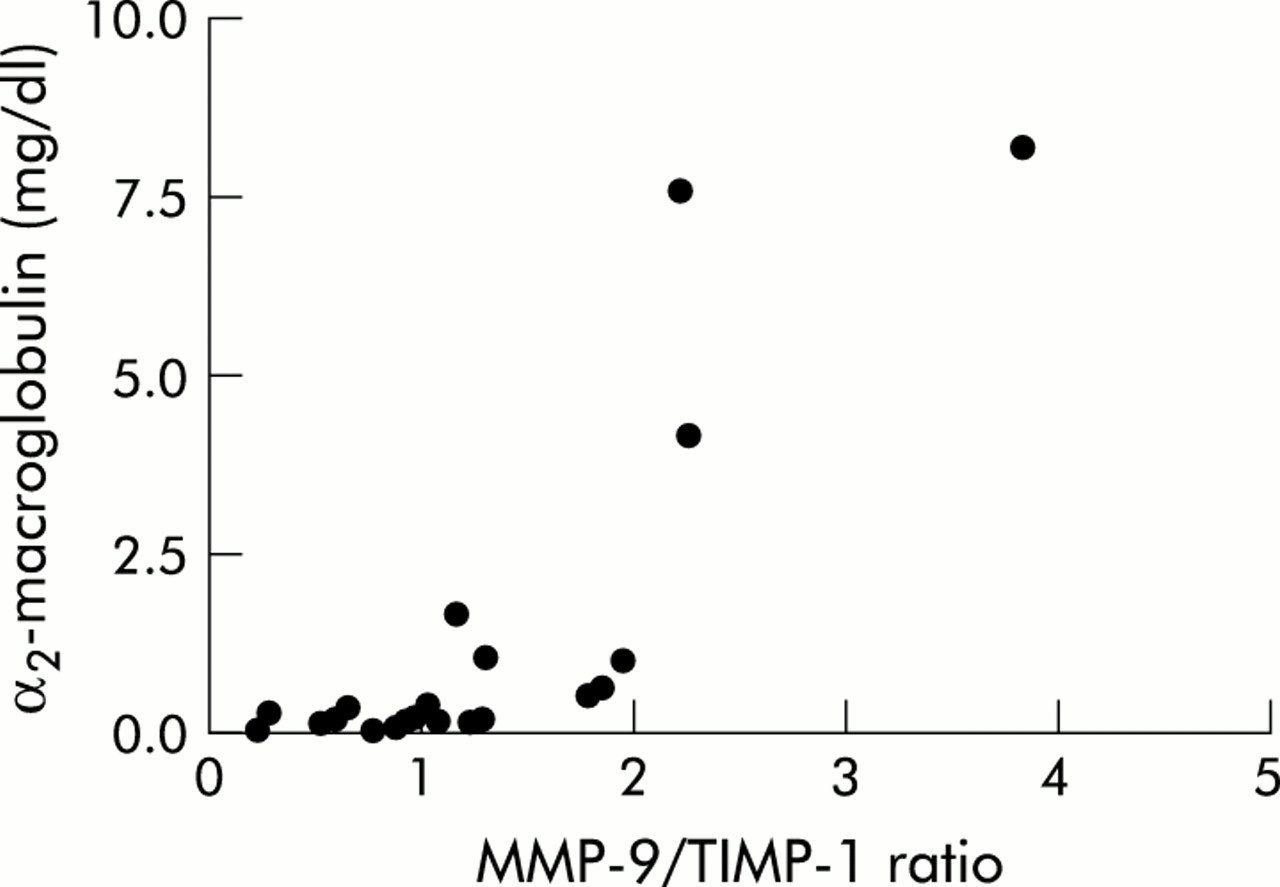
The median (range) level of α2-macroglobulin in BAL fluid was 0.21 (0.03–8.12) mg/l and the level of lactoferrin in BAL fluid was 2037 (1420–6070) μg/l which is significantly higher than an age matched reference group.13 A positive correlation was found between α2-macroglobulin and the molar MMP-9/TIMP-1 ratio (p<0.001, r=0.8; fig 2) and between lactoferrin and the molar MMP-9/TIMP-1 ratio (p=0.03, r=0.5). BAL fluid of patients with cystic fibrosis.

Correlation between α2-macroglobulin and molar MMP-9/TIMP-1 ratio in BAL fluid of patients with cystic fibrosis
Sequential analysis of MMP concentrations in BAL fluid
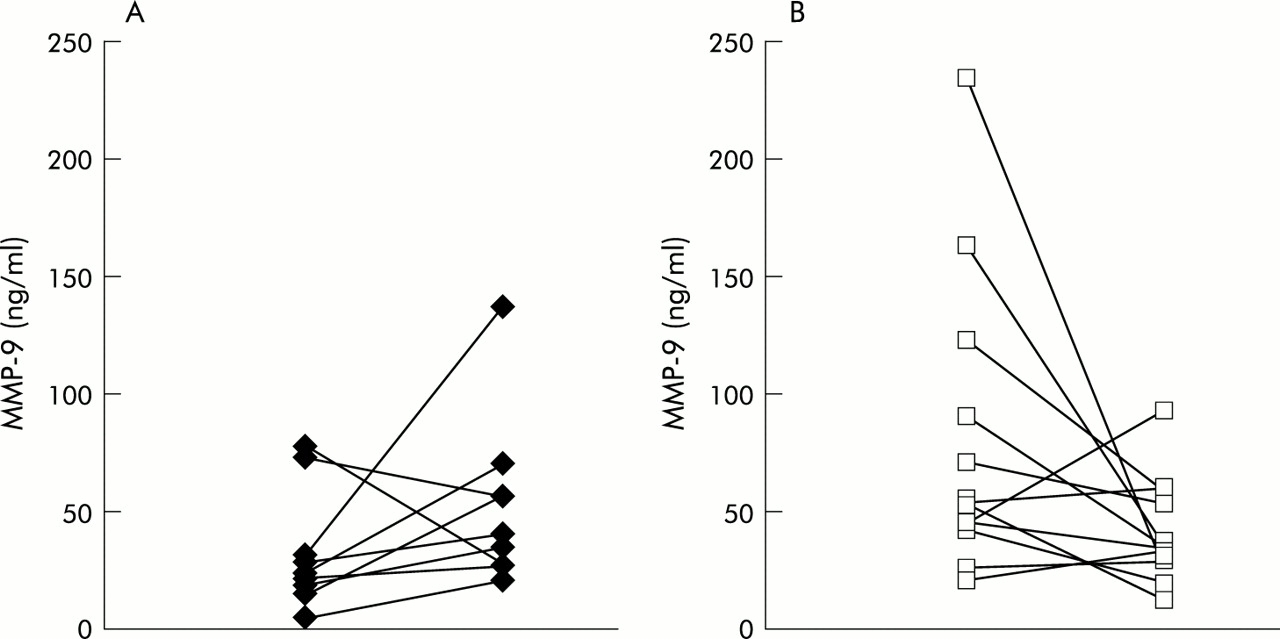
Baseline MMP levels were significantly higher in the DNase group (p=0.02 for MMP-8, p=0.01 for MMP-9). Investigation of the same patients 18 months after the first bronchoscopy showed an increase in MMP-8 and MMP-9 in patients not receiving DNase, whereas both MMP-8 and MMP-9 were decreased in DNase treated patients (MMP-8: change in median 15.5 v –23 ng/ml, p=0.007; MMP-9: change in median 16 v –20 ng/ml, p=0.02; table 1, fig 3). The differences in both the percentage of neutrophils and the TIMP levels failed to reach statistical significance (table 1). No significant changes in lung function were observed between the two groups, nor were there any significant differences in bacterial colonisation.
{kind=link}
{kind=link}
{kind=link}
Progression of MMP-9 concentrations of the same patients between the first and second BAL 18 months later in (A) untreated patients and (B) patients treated with DNase.
DISCUSSION
Neutrophil dominated airway inflammation is thought to play a central role in the development of bronchiectasis and destruction of lung tissue in patients with CF. We have shown that the neutrophil collagenase MMP-8 as well as MMP-9, which is involved in the degradation of both elastin and collagen IV, are increased in BAL fluid of CF patients with mild pulmonary disease. This increase is not counterbalanced by a similar increase in their inhibitor TIMP-1, resulting in a significantly increased MMP/TIMP ratio. Furthermore, the increase in the MMP-9/TIMP ratio correlated with the concentrations of α2-macroglobulin, a marker of leakage of plasma proteins through the alveolocapillary membrane. These data indicate that MMPs may contribute to the persistent inflammatory process in CF lung disease.
The effect of MMPs on airway remodelling and lung tissue destruction has been studied in various lung diseases. A deficiency of MMPs has been postulated to play a role in insufficient pulmonary remodelling in patients with interstitial lung diseases.14 In the acute respiratory distress syndrome (ARDS) increased MMP-9 concentrations are positively correlated with the concentration of 7S collagen, a marker of basement membrane disruption,15,16 and upregulation of collagenases and gelatinases is associated with increased parenchymal cell death in patients with COPD.17 MMP-8 concentrations in BAL fluid of patients with bronchiectasis were shown to reflect the severity of lung disease with highest concentrations in patients with active disease.18 Measurements of MMPs in patients with CF have been limited to sputum analysis in patients with more advanced disease.7 Uninhibited MMP-9 activity was positively correlated with the increase in type IV collagen degradation products.7 Our data show that an imbalance of the MMP/TIMP ratio and evidence of collagen breakdown is found even in CF patients with mild pulmonary involvement who are clinically stable with few symptoms of active lung disease.
α2-macroglobulin, a large (725 kDa) plasma glycoprotein, is thought to be a marker for alveolocapillary leakage since it is not physiologically found in the alveolar compartment of adults and is present in low concentrations in children without lung disease.13 It has previously been used to study plasma leakage in both parenchymal and airway diseases.19,20 In the present study we found a significant influx of α2-macroglobulin in the lungs of patients with CF which was positively correlated with the increased level of MMP-9 which is known to degrade collagen IV, a major component of basement membranes. Our results therefore provide indirect evidence for degradation of basement membranes in this patient population with functionally mild pulmonary involvement.
There are several possible explanations for the high concentrations of MMP-8 and MMP-9 in the BAL fluid of patients with CF. CF lung disease is characterised by an intense neutrophil dominated airway inflammation, and neutrophils adhere to the endothelium cell surface of postcapillary venules, cross the endothelium through tight junctions and penetrate the basement membrane, leaving the vascular system to accumulate at the site of inflammation.21 It is likely that neutrophils are the main source of MMP-8 and MMP-9 in the pulmonary compartment in our patient population. Whereas MMP-8 is exclusively stored and released by neutrophils, MMP-9 is produced by different cell types such as macrophages, epithelial cells, and fibroblasts.22,23 Zymography of MMP-9 clearly showed three distinct bands with molecular weights of 92, 130, and 220 kDa. This characteristic banding pattern of neutrophil derived MMP-9 consisted of pro-MMP-9 (92 kDa), human neutrophil lipocalin (HNL)-pro-MMP-9 complex (130 kDa), and a homodimeric form of MMP-9 (220 kDa).24 HNL is a 25 kDa lipocalin which is specifically stored in granules of neutrophils. It is associated with neutrophil gelatinases and is thus a marker for PMN released MMP-9 in zymography. All these data suggest that neutrophils are the main source of MMP-8 and MMP-9 in our CF patient population.
Although MMP-9 in our study was mainly derived from neutrophils, epithelial cells are also known to produce MMP-9.23 This process appears to be downregulated by nitric oxide (NO).25 The production of NO by epithelial cells has been found to be decreased in CF26 and reduced formation of NO in CF epithelial cells could therefore lead to unopposed production of MMP-9 contributing to the exaggerated inflammatory response in CF airways.
In our sequential BAL studies we observed a positive effect of DNase on MMP concentrations in BAL fluid. Patients treated with DNase had significantly decreased BAL fluid concentrations of MMP-8 and MMP-9 compared with untreated patients in whom the concentration of MMPs increased during the observation period of 18 months. Baseline levels of MMPs were higher in the DNase group and marked intraindividual differences were observed in the response to treatment, a finding that is similar to the variable clinical response observed with this drug. DNase is primarily a mucolytic agent as it breaks down polymerised DNA which is a major contributor to increased sputum viscosity in CF airway secretions.27 Previous studies assessing the effect of DNase on inflammatory markers in CF sputum and BAL fluid have indicated that it may increase neutrophil elastase and IL-8 levels in sputum as elastase and IL-8 in sputum are bound to DNA and may be released after inhalation of DNase.28 This finding is not supported by our longitudinal study of BAL fluid as recently reported.29 In contrast, the results of the present study suggest that, rather than increasing inflammatory cytokines in CF, DNase decreases airway inflammation by improving clearance of retained secretions.30,31 We speculate that early treatment with DNase may diminish structural lung damage in patients with CF.
REFERENCES
Footnotes
-
The study was in part supported by the German CF Foundation and Roche, Germany.