Article Text

Abstract
Background Ivacaftor is the first cystic fibrosis transmembrane conductance regulator (CFTR) modulator demonstrating clinical benefit in patients with cystic fibrosis (CF). As ivacaftor is intended for chronic, lifelong use, understanding long-term effects is important for patients and healthcare providers.
Objective This ongoing, observational, postapproval safety study evaluates clinical outcomes and disease progression in ivacaftor-treated patients using data from the US and the UK CF registries following commercial availability.
Methods Annual analyses compare ivacaftor-treated and untreated matched comparator patients for: risks of death, transplantation, hospitalisation, pulmonary exacerbation; prevalence of CF-related complications and microorganisms and lung function changes in a subset of patients who initiated ivacaftor in the first year of commercial availability. Results from the 2014 analyses (2 and 3 years following commercial availability in the UK and USA, respectively) are presented here.
Results Analyses included 1256 ivacaftor-treated and 6200 comparator patients from the USA and 411 ivacaftor-treated and 2069 comparator patients from the UK. No new safety concerns were identified based on the evaluation of clinical outcomes included in the analyses. As part of safety evaluations, ivacaftor-treated US patients were observed to have significantly lower risks of death (0.6% vs 1.6%, p=0.0110), transplantation (0.2% vs 1.1%, p=0.0017), hospitalisation (27.5% vs 43.1%, p<0.0001) and pulmonary exacerbation (27.8% vs 43.3%, p<0.0001) relative to comparators; trends were similar in the UK. In both registries, ivacaftor-treated patients had a lower prevalence of CF-related complications and select microorganisms and had better preserved lung function.
Conclusions While general limitations of observational research apply, analyses revealed favourable results for clinically important outcomes among ivacaftor-treated patients, adding to the growing body of literature supporting disease modification by CFTR modulation with ivacaftor.
EU PAS registration number EUPAS4270
- cystic fibrosis
- systemic disease and lungs
- respiratory infection
- rare lung diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
What are the important clinical outcomes among patients with cystic fibrosis (CF) treated with ivacaftor, the first cystic fibrosis transmembrane conductance regulator (CFTR) modulator?
What is the bottom line?
These observational analyses of the US and UK CF registry patient cohorts found no new safety concerns and revealed favourable results in clinically important outcomes, including lower risk of death and organ transplantation among ivacaftor-treated patients, adding to the growing body of literature supporting disease modification by CFTR modulation with ivacaftor.
Why read on?
We report the interim results from the largest to date evaluation of outcomes among patients with CF treated with ivacaftor for an average of 2 years in the USA and 1.3 years in the UK, demonstrating consistent clinical outcome patterns across two independent CF registries; the results are relevant to the broader CF population with CFTR mutations indicated for ivacaftor therapy.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease with serious, chronically debilitating morbidities and high premature mortality. CF affects >70 000 individuals worldwide,1 including approximately 33 000 in the USA2 and approximately 42 000 in Europe.3
Ivacaftor (Kalydeco, Vertex Pharmaceuticals Incorporated, Boston, Massachusetts, USA) is an orally bioavailable small molecule that targets the underlying defect in CF and represents the first in a new class of drugs, CFTR potentiators, which provide a novel therapeutic approach to the treatment of CF by improving the function of the CFTR protein. Clinical studies demonstrated improved lung function and other relevant clinical benefits (eg, improvements in respiratory symptoms and pulmonary exacerbations (PEx)) among patients treated with ivacaftor.4–7 Ivacaftor was first approved in the USA and the European Union (EU) in 2012 for treatment of CF in patients aged ≥6 years who have a G551D mutation in the CFTR gene. The indication was subsequently progressively expanded to patients aged ≥2 years who have one of the following mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, R117H (in the EU, in R117H mutation patients aged ≥18 years), or residual function mutations (in the USA only) that either result from a splicing defect in the CFTR gene or were shown to be responsive to ivacaftor in vitro.
Ivacaftor is intended for chronic, lifelong use. Therefore, understanding the long-term effects of therapy is important for patients, caregivers, healthcare providers, payers and regulators. Furthermore, since the therapy addresses the underlying cause of CF, long-term data are complementary to data from the clinical development programmes.
In general, identifying an appropriate source of data for long-term outcome analyses in rare diseases can represent a challenge. In the field of CF, researchers have a unique opportunity to leverage relevant data collected via existing national CF registries for evaluations of effects of therapies. The US CF Foundation Patient Registry (US CFFPR) and UK CF Registry (UK CFR) are two of the largest registries worldwide and allow access for research purposes to the comprehensive demographic and clinical data from the vast majority of patients in the respective regions.2 8
This observational study was designed to evaluate key clinical outcomes and disease progression in ivacaftor-treated patients using data from the US CFFPR and the UK CFR for 5 years following commercial availability to fulfil general postapproval safety surveillance needs. The study is designated as a postauthorisation safety study (PASS) for a postmarketing commitment to the European Medicines Agency (EMA), and is disclosed on the EU electronic Register of Post-Authorisation Studies (EUPAS4270). Here, we present results from the 2014 data analyses (2 and 3 years following commercial availability in the UK and USA, respectively). The study was ongoing at the time of development of this manuscript.
Methods
Data sources
The US CFFPR tracks the treatments and health outcomes of patients with CF across >115 CFF-accredited care centres. Approximately 84% of all the US patients provide consent to participate in the registry.2
The UK CFR is an anonymised database of patients with CF across >32 paediatric and 28 adult CF care centres in England, Scotland, Wales and Northern Ireland who have consented to data entry (>99% of all the UK patients), which has approval from the National Research Ethics Committee and is maintained by the UK CF Trust.9
In both registries, investigators are physicians at accredited CF care centres, and patients participate in standard data collection for assessments of vital status (including death), hospitalisations, CF-related complications, PEx (or surrogate measures of PEx), respiratory microbiology, pregnancy, pulmonary function and organ transplantation.
Both registries use an online application for entry of pertinent data related to patients and their medical care by staff at CF care centres.
Study population
The source population for these analyses was all patients included in the US CFFPR and the UK CFR in 2014.
Ivacaftor cohorts in each registry included all patients with at least one record of having received ivacaftor in 2014 (both newly initiated in 2014 and on treatment before 2014). Ivacaftor-treated patients were matched in a 1:5 ratio with comparators who had never received ivacaftor.
Comparator patients were matched to ivacaftor-treated patients on age, sex and CFTR genotype severity based on established CFTR mutation class. For matching by genotype severity, comparators had to have both mutant alleles of similar severity to the mutant alleles of the ivacaftor-treated patient, ensuring that milder mutation class (IV–VI) patients were not matched to more severe mutation class (I–III) patients. For example, ivacaftor-treated patients with a G551D mutation on one allele and a class II mutation on the second allele would be matched with comparators with an F508del mutation on one allele and a class II mutation on the second. Although this matching strategy resulted in the comparator cohort including predominantly class I–II genotype patients versus predominantly class III in the ivacaftor cohort, it was the only feasible option to identify a concurrent cohort of untreated patients and was considered appropriate based on earlier research indicating generally comparable CF disease phenotypes between patients with genotype class III (eg, G551D) and those with genotype class II.9 10
In addition, subsets of ivacaftor-treated and comparator patients were defined for evaluation of lung function over time. These included patients who initiated ivacaftor in the first year of commercial availability (USA: 2012; UK: 2013), had no transplantation record and remained on treatment through 2014, and their matched comparators.
Data analysis
All of the evaluated outcomes were assessed as part of general safety surveillance, where any potential imbalances not favouring ivacaftor-treated patients were to be evaluated further to determine if they constitute a potential new safety signal. Analyses were restricted to the outcomes routinely collected by the registries; data on potential safety outcomes of cataract or drug-drug interactions are not available via registries and could not be evaluated in this study.
Key incident clinical outcomes of interest included death, organ transplantation, PEx and hospitalisation (USA: for any reason; UK: for intravenous therapy only). In both registries, PEx were defined as those requiring intravenous antibiotic use at home or in the hospital.
The risk of each incident outcome was calculated as the proportion of patients in each cohort with that outcome at any time during 2014. The risks were compared between cohorts by calculating relative risks (RRs) and 95% CIs, which were based on normal approximation. If the expected frequency was <5 in at least one substratum of the contingency table, Fisher’s exact p values were calculated in lieu of CIs. To address potential confounding, analyses were stratified by age, sex and per cent predicted FEV 1 (ppFEV1) (USA: average of the best-available values for each quarter; UK: annual recorded value). No further adjustment was carried out.
The prevalent outcomes evaluated as part of general safety surveillance included CF complications (CF-related diabetes, depression and hepatobiliary, pulmonary, gastrointestinal and bone/joint complications) and select pulmonary microorganisms. The US CFFPR defined the presence of pulmonary microorganisms based on at least one positive sputum culture within an evaluated 12-month period. The UK CFR defined chronic infections (such as chronic Pseudomonas) based on the presence of at least three positive cultures over a 12-month period.
Evaluations of lung function included tabulations of summary statistics (mean, SE and 95% CI) for ppFEV1 for each analysis year, as well as for changes from pretreatment baseline year (USA: 2011; UK: 2012) to 2014.
Analyses were carried out for each registry separately (ie, no pooled analyses were conducted) due to the differences in data capture between the USA and UK.
Results
Demographics
The analyses included 1256 ivacaftor-treated and 6200 comparator patients from the 2014 US CFFPR and 411 ivacaftor and 2069 comparator patients from the 2014 UK CFR. The average length of ivacaftor exposure (ascertained using registry records through 2014) was 2.0 years in the USA and 1.3 years in the UK ivacaftor cohort. Descriptive data are provided in table 1.
Patient characteristics, 2014 ivacaftor and comparator cohorts, USA and UK
In both registries, the ivacaftor and comparator cohorts were well matched with regard to age and sex. The matching by genotype severity, as intended, achieved a balanced representation of patients with more severe genotypes (classes I–III) versus all other genotypes (classes IV–VI and unknown) between the ivacaftor and comparator cohorts in both registries (table 1). Due to the nature of the ivacaftor indication and that of the matching algorithm, within the class I–III group, the majority of ivacaftor-treated patients had an overall class III CFTR genotype (USA: 81.2%; UK: 93.4%) versus class I or II genotype (USA: 87.6%; UK: 94.0%) in the majority of comparators. In the USA, 935 out of 1256 (74.4%) ivacaftor-treated patients had at least one copy of the G551D mutation, all of the 411 ivacaftor-treated patients in the UK had a G551D mutation.
In both registries, in 2014, ivacaftor-treated patients had statistically significantly better lung function than did comparators, as demonstrated by the higher proportion of those with ppFEV1 ≥70 (expected based on ivacaftor treatment benefits). Notably, during the pretreatment baseline year (USA: 2011; UK: 2012) within each registry, ivacaftor and comparator cohorts did not differ in terms of lung function or risks of PEx or hospitalisations (table 1).
Key clinical outcome analyses
The results of analyses comparing all of the clinical outcomes in this study between cohorts are summarised in figures 1–5 and table 2. The review of these results showed that none of the evaluated outcomes was significantly more common among ivacaftor-treated patients, indicating no potential new safety concerns deserving further evaluation. Instead, a number of observations consistently favouring ivacaftor-treated patients were made, as summarised in figures 1-5 and table 2.

Hospitalisations and pulmonary exacerbations, 2014 ivacaftor and comparator cohorts, (A) USA and (B) UK. ªHospitalisation due to any reason in the US CFFPR and for PEx in the UK CFR. bIn the UK CFR, PEx were defined as the requirement of intravenous antibiotic use at home or in the hospital. CFFPR, Cystic Fibrosis Foundation Patient Registry; CFR, Cystic Fibrosis Registry; PEx, pulmonary exacerbations; RR, relative risk.

(A) Hospitalisations and (B) PEx by age and lung function, 2014 ivacaftor and comparator cohorts, USA and UK. Error bars indicate 95% CIs. *P=0.23; †p=0.12. Hospitalisation due to any reason in the US CFFPR and for PEx in the UK CFR; PEx were defined as the requirement of intravenous antibiotic use at home or in the hospital. CFFPR, Cystic Fibrosis Foundation Patient Registry; CFR, Cystic Fibrosis Registry; PEx, pulmonary exacerbations; ppFEV1, per cent predicted FEV 1. Refer to online supplementary tables S1-S4 for tables corresponding to figure 2.
Supplementary file 1
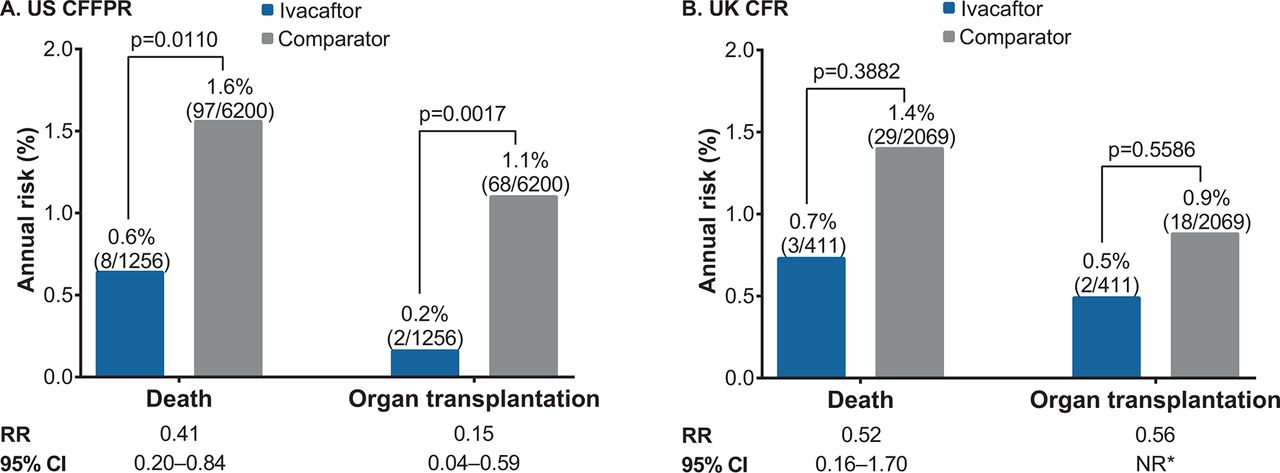
Death and organ transplantation, 2014 ivacaftor and comparator cohorts, (A) US CFFPR and (B) UK CFR. *Fisher’s exact p values are shown when the expected value is <5 in at least one cell of the contingency table. CFFPR, Cystic Fibrosis Foundation Patient Registry; CFR, Cystic Fibrosis Registry; NR, not reported; RR, relative risk.
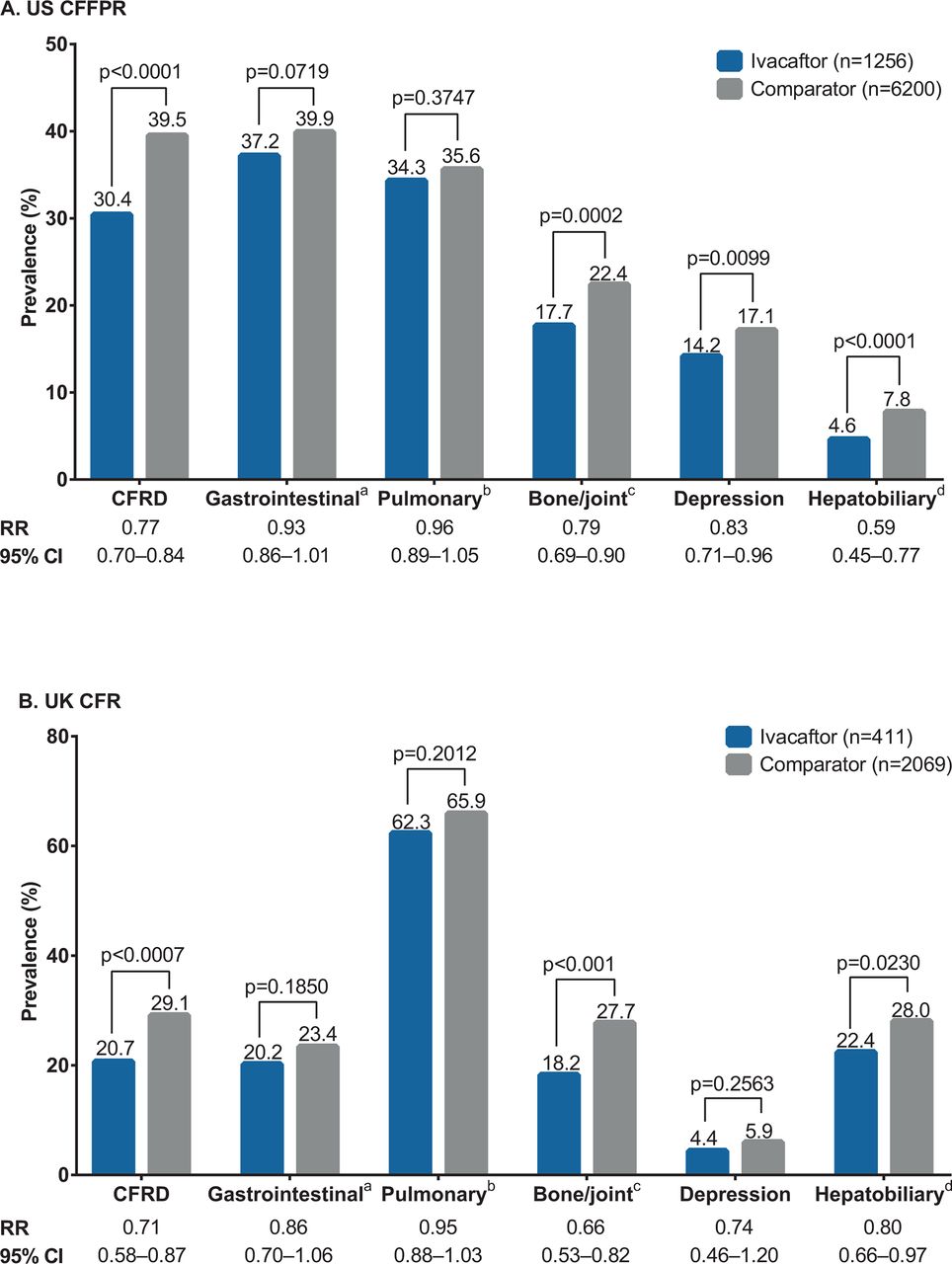
CF complications, 2014 ivacaftor and comparator cohorts, (A) US CFFPR and (B) UK CFR Direct comparisons between the US and the UK data on the prevalence of complications cannot be made because of the differences in complication screening and data capture between countries. aIncludes distal intestinal obstruction syndrome, fibrosing colonopathy/colonic stricture, gastro-oesophageal reflux disease, gastrointestinal bleed (non-variceal) requiring hospitalisation, peptic ulcer disease and rectal prolapse. bIncludes allergic bronchopulmonary aspergillosis, asthma, massive haemoptysis and pneumothorax requiring a chest tube. cIncludes arthritis/arthropathy, bone fracture, osteopaenia and osteoporosis. dThe US CFFPR includes gallstones, gallstones requiring surgery/procedure, liver disease (cirrhosis), cirrhosis complications (oesophageal varices, gastric varices, gastrointestinal bleed, splenomegaly, hypersplenism and ascites), liver disease (non-cirrhosis), hepatic steatosis and liver disease (other); the UK CFR also includes abnormal liver enzymes. CF, cystic fibrosis; CFFPR, Cystic Fibrosis Foundation Patient Registry; CFR, Cystic Fibrosis Registry; CFRD, cystic fibrosis-related diabetes; RR, relative risk.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of lung function changes over time for ivacaftor and comparator patients, (A) US CFFPR and (B) UK CFR. *Within-group; †Between-group. aIncludes all patients followed from 2011 through 2014 (regardless of availability of ppFEV1 values in a given analysis year). bCalculations of change from baseline performed on patients with non-missing ppFEV1 values in 2011 and 2014 (n=636 for ivacaftor; n=2854 for comparator). cIncludes all patients followed from 2012 through 2014. dCalculations of change from baseline performed on patients with non-missing ppFEV1 values in 2012 and 2014 (n=250 for ivacaftor; n=1211 for comparator). CFFPR, Cystic Fibrosis Foundation Patient Registry; CFR, Cystic Fibrosis Registry; ppFEV1, per cent predicted FEV1.
Pulmonary microbiology, 2014 ivacaftor and comparator cohorts, US CFFPR and UK CFR
Hospitalisations and pulmonary exacerbations
In the 2014 US CFFPR, there were statistically significant lower risks of hospitalisation for any reason and PEx overall in the ivacaftor cohort versus comparator cohort (figure 1A). This was consistently observed within all age and ppFEV1 strata (figure 2).
Similarly, in the 2014 UK CFR, risks of hospitalisation for intravenous therapy and PEx were significantly lower in the ivacaftor versus comparator cohort, overall (figure 1B) and within all age and ppFEV1 strata (figure 2).
In both the USA and UK, similar trends in PEx and hospitalisations were observed when analyses were stratified by sex (data not shown).
Death and organ transplantation
In the USA in 2014, there were statistically significant lower risks of death and organ transplantations in the ivacaftor versus comparator cohort (figure 3A). The most common cause of death in the ivacaftor cohort was respiratory/cardiorespiratory (six out of eight patients), consistent with the distribution of causes of death in the general CF population.
In the UK in 2014, there were non-significant lower risks of death and organ transplantations in the ivacaftor versus comparator cohort (figure 3B). The cause of death was respiratory/cardiorespiratory for all (n=3) of the deceased patients in the ivacaftor cohort.
To address whether there were any underlying differences in mortality and transplantation patterns between the ivacaftor-eligible population and comparator population prior to the availability of ivacaftor, we evaluated the 2010 US CFFPR mortality and transplantation statistics for patients older than 6 years of age with at least one copy of G551D (the most common class III mutation) and those homozygous for F508del (the most common class II mutation). There were no differences in mortality or transplantation between those two populations (mortality: 1.9% vs 1.8%, respectively; transplantation: 1.1% vs 1.0%, respectively).
The small numbers of deaths and transplantations among ivacaftor-treated patients precluded meaningful stratified analyses of these outcomes; these events were observed in patients aged >18 years who had moderate-to-severe lung function impairment, consistent with CF disease epidemiology.
Cystic fibrosis complications
The majority of the evaluated CF complications were less common in the ivacaftor than in the comparator cohorts in both registries (figure 4).
In the USA, relative to comparators, ivacaftor-treated patients had a statistically significant lower prevalence of CF-related diabetes, hepatobiliary complications, bone/joint complications and depression. There was no statistically significant difference in the prevalence of two other complication categories evaluated (gastrointestinal and pulmonary complications), although both were less commonly reported among ivacaftor-treated patients (figure 4A).
In the UK, the observed patterns were similar to those in the USA (figure 4B).
Pulmonary microbiology
The majority of patients in both registries had bacterial culture results available for the pulmonary microbiology analyses (USA: 97.6% of ivacaftor-treated patients and 96.1% of comparators; UK: 99.5% of ivacaftor-treated patients and 98.3% of comparators).
Evaluation of the prevalence of pulmonary microorganisms among patients with available culture results showed that most pathogens were less commonly reported in ivacaftor cohorts in both registries (table 2). Pseudomonas aeruginosa was consistently less prevalent among ivacaftor-treated than among comparator patients in both the USA and UK. Prevalence of Aspergillus spp was also notably lower among ivacaftor-treated than among comparator patients in the USA and UK. Several other microorganisms, including Staphylococcus aureus, showed a trend towards lower prevalence among ivacaftor-treated than among comparator patients in both registries (table 2).
Lung function
In the USA, analyses of lung function were performed in the subsets of 708 patients who initiated ivacaftor in 2012 and had no record of organ transplantation and 3249 matched untreated comparators. In the UK, the lung function analyses included 277 patients who initiated therapy in 2013 and had no record of lung transplantation and 1365 comparators.
In the USA, the mean ppFEV1 increased in ivacaftor-treated patients and decreased in comparators from the baseline year (2011) to 2014 (figure 5A). For patients with lung function measurements available for both 2011 and 2014, the ppFEV1 improved significantly by 1.4 percentage points in ivacaftor-treated patients and decreased significantly by 5.3 percentage points in comparators (between-group p<0.0001).
In the UK, the mean ppFEV1 increased in ivacaftor-treated patients and decreased in comparators from the baseline year (2012) to 2014 (figure 5B). For patients with lung function measurements available for both 2012 and 2014, the ppFEV1 improved significantly by 6.6 percentage points in ivacaftor-treated patients and decreased significantly by 1.5 percentage points in comparator patients (between-group p<0.001).
Discussion
Ivacaftor has been demonstrated to improve lung function and nutritional status as well as to reduce the frequency of PEx in clinical studies, with the effect sustained for up to 144 weeks.11 Additionally, the rate of lung function decline in ivacaftor-treated patients was shown to be slower by nearly half compared with that in comparators over a 3-year analysis period.12 We aimed to monitor patients treated with ivacaftor for any potential new safety concerns via evaluation of important clinical outcomes captured in the national CF registries in the USA and UK. The analyses also allowed to further assess if the effects of ivacaftor on long-term clinical outcomes, including mortality, are consistent with data supporting the disease modification potential for CFTR modulator therapy.
Data from two large independent national CF patient registries evaluated in this study revealed consistently favourable findings with respect to clinically important outcomes among ivacaftor-treated patients, adding to the literature on the effects of ivacaftor therapy.
A significantly lower proportion of ivacaftor-treated versus comparator patients in both registries evaluated in this study had PEx, an important CF disease outcome associated with loss of lung function13 and shown to predict CF mortality.14 Similarly, a significantly lower proportion of ivacaftor versus comparator patients had hospitalisations. The fact that the risk of these events during pretreatment baseline year was similar between the ivacaftor and comparator cohorts suggests that the observed patterns are unlikely to be attributable to the underlying differences between mutation classes rather than to ivacaftor. The results of PEx analyses strengthen the findings of the previous smaller reports, such as the G551D Observational Study (GOAL) evaluating 133 ivacaftor-treated patients across 28 US centres and revealing a decrease in the rate of PEx 1 year after ivacaftor initiation.15 A single-centre study of 29 ivacaftor-naïve patients also found a significant reduction in the likelihood of requiring intravenous antibiotics in the 6 months following ivacaftor initiation compared with the prior 6 months.16 Other smaller studies have also suggested a decrease in PEx requiring intravenous antibiotics after ivacaftor initiation.17 18 The lower risk of hospitalisations was also consistent with the earlier reports from smaller studies, including GOAL,19 where the proportion of patients who were hospitalised declined by 19.1% during the 6 months following ivacaftor initiation compared with the immediately preceding 6 months.15
Although very small number of events of deaths and transplants observed in this study make the interpretation of the analyses challenging, ivacaftor-treated patients had statistically significant lower risks of these events than did untreated matched comparators in the USA, and a similar trend was observed in the UK. The fact that ivacaftor-treated patients predominantly had a class III genotype with the G551D mutation while comparators were predominantly genotype class I/II patients also raises a question if survival differences existed between these two populations prior to availability of ivacaftor. However, evaluation of transplantation and mortality patterns in patients older than 6 years of age with at least one copy of G551D mutation versus those who are homozygous for F508del mutation in the US CFFPR in 2010 showed that no such differences favouring patients with the G551D mutation existed. Previous studies have also shown that mortality patterns were similar between patients homozygous for F508del and those heterozygous for F508del and G551D before availability of ivacaftor.9 These historical data support the hypothesis that the patterns of significantly lower mortality and transplantation observed in this study could potentially be attributed to ivacaftor. Only one small study describing mortality and transplantation among 21 ivacaftor and 35 control patients has been published prior to these analyses, where ivacaftor appeared to improve transplantation-free survival, significantly reducing the combined outcome of death plus transplantation (HR 0.13; 95% CI 0.04 to 0.43).20
The prevalence of a number of complications across organ systems was significantly lower among ivacaftor-treated versus comparator patients. These included hepatobiliary and bone/joint complications, depression (significant in the USA only), and CF-related diabetes, which is the most frequently described comorbidity in studies of ivacaftor use. CF-related diabetes is associated with deterioration in health status and contributes to poor weight gain, decreased lung function and increased mortality21 and may also spur the emergence of diabetes-related microvascular complications, including microalbuminuria, retinopathy, gastrointestinal symptoms and autonomic neuropathy.22 In a number of earlier small studies, ivacaftor appeared to improve insulin response to oral glucose,23 reduce levels of haemoglobin A1c (suggesting improved glycaemic control)24 and reduce insulin requirement in a patient with existing CF-related diabetes.25
Additionally, several key microorganisms such as S. aureus, P. aeruginosa and Aspergillus spp were less prevalent among ivacaftor-treated patients in this study. Our findings on P. aeruginosa are consistent with the GOAL study where there were significant reductions in the percentage of patients with at least one documented positive P. aeruginosa culture 6 months after ivacaftor initiation relative to the preceding 6 months,19 and where 29% of patients who previously had a positive culture were found to be culture negative after 1 year of treatment.15
Furthermore, analyses of patients who initiated ivacaftor in the first year of commercial availability (primarily those with a G551D mutation) and remained on therapy through 2014 also demonstrated lung function improvements relative to baseline, as compared with sustained decline among the untreated comparators. Differences in patterns of lung function changes noted between the USA and UK (numerically greater improvements in ivacaftor-treated patients in the UK but more pronounced decline in untreated comparators in the USA) were likely due to multiple factors, including differences in data collection between the registries and acknowledged differences in lung function in the CF population between the regions (with the UK patients having lower ppFEV1 than their US counterparts). Despite these differences, the between-group difference in lung function change was comparable in the two registries. The results of lung function analyses are directionally consistent with those of the clinical studies11 and with prior analyses demonstrating the reduced rate of lung function decline12 but should not be directly compared due to the nature of the study, including unselected patient population and lack of standardised lung function measurements at predefined frequent intervals.
Limitations
A general concern in any observational study is the potential for confounding, which was partially addressed in our study by matching patients in the ivacaftor and comparator cohorts. Although the comparator cohort included predominantly class I–II patients versus predominantly class III in the ivacaftor cohort, two retrospective cohort studies using the US CFFPR data showed that CF disease phenotype is generally comparable between the class III (eg, G551D) patients and those with genotype class II.9 10 As a result of successful matching, the ivacaftor and comparator cohorts in each registry were similar in terms of sex and age distribution and showed similar average ppFEV1 distribution and PEx and hospitalisation risks in the year preceding ivacaftor market availability. Potential confounding was further addressed by performing stratified analyses by age, sex and ppFEV1; nevertheless, the possibility of residual confounding by unmeasured factors cannot be fully excluded. For instance, some underlying differences between the cohorts in the rate of developing chronic complications or acquiring pathogens may exist and could partially explain the lower prevalence of CF-related complications and select microorganisms observed in ivacaftor-treated patients in this study. In future, more detailed analyses of the patterns in chronic CF complications and bacterial pathogens in patients treated with CFTR modulators may be needed to elucidate the role of the therapy.
Other potential limitations of our analysis are inherent to observational research in general and include lack of scheduled visits and standardised assessments and no precise dates of therapy initiation (which may introduce exposure misclassification in the research studies). In the USA, while ivacaftor and comparator cohorts had similar average number of visits per patient in the year prior to ivacaftor availability (5.6 visits for each cohort), the average number of visits in 2014 was lower in ivacaftor versus comparator cohort (4.7 vs 5.7, p<0.0001), consistent with the observation of lower observed risk of hospitalisations and PEx. Annual data reporting in the UK CFR precluded a similar description of patient visits. Both the US and the UK registries report high degrees of data completeness. In the US CFFPR in 2015, complications were reported in 97% of the patients enrolled in the registry.26 Since 2012, the US CFF has conducted annual data audits of selected CF centres by comparing the registry data entered by those centres with the data from their electronic medical records. These audits show high accuracy of the registry data with matching rates up to 99% in some data elements.2 Similarly, the UK CFR dataset is considered 90% complete, particularly for complications. The UK CFR is used by the National Health Service to inform the payment system so that funding to CF centres is based on the severity of disease including complications. This provides an incentive for accurate and complete data entry.
Conclusion
While the general limitations of observational research apply, this study represents the largest analyses of patients treated with ivacaftor to date and adds to the growing body of literature supporting disease modification by CFTR modulation with ivacaftor. Since registries capture data from a majority of patients with CF in their respective geographic regions and since no exclusion criteria were applied to ivacaftor-treated patients in this study, the results can be considered relevant to the broader CF population with CFTR mutations indicated for ivacaftor therapy.
Acknowledgments
The authors would like to thank the Cystic Fibrosis Foundation and the Cystic Fibrosis Trust for the use of patient registry data to conduct this study. Additionally, the authors would like to thank the patients, care providers and clinic coordinators at CF centres throughout the USA and the UK for their contributions to the patient registries. The authors would also like to thank Yan Bai, MD, PhD, for substantial contributions to this study (YB is a former employee of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company). Editorial coordination and support were provided by Gauri Dixit, PhD (GD is an employee of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company). Medical writing and editorial support were provided by Stephanie Vadasz, PhD, Shannon Davis and Dena McWain of Ashfield Healthcare Communications, which received funding from Vertex Pharmaceuticals Incorporated.
References
Footnotes
Contributors LBes, NV, MH, LBen, ST and CS conceived the study. LBes, NV, MH, LBen, ST, CS, BCM and DB designed the study. AS, SN and AE acquired the data. LBes, NV, LBen, ST, CS, AS, SN and AE analysed the data. All authors interpreted the data. LBes and NV drafted the manuscript. All authors critically revised the manuscript for important intellectual content and approved the manuscript for publication.
Funding This study was sponsored by Vertex Pharmaceuticals Incorporated.
Competing interests MH, CS, ST and NV are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. LB and LB are former employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. GSS has served on advisory boards for Vertex Pharmaceuticals Incorporated and on the US CFFPR committee. AE is an employee and AS is a contractor for the US CF Foundation, which provided data for this study. BCM is an employee of the CF Foundation. DB and SN are members of the Steering Committee of the UK CF Registry, which provided data for this study. MWK is a consultant to Vertex Pharmaceuticals Incorporated.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note Leona Bessonova and Leif Bengtsson were employees of Vertex Pharmaceuticals Incorporated at the time of this study.