Article Text

Abstract
A large part of the human genome is transcribed in non-coding RNAs, transcripts that do not code for protein, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). MiRNAs are short single-stranded RNA molecules that negatively regulate gene expression at the post-transcriptional level. They play an important regulatory role in many biological processes. Consequently, altered expression of these non-coding RNAs has been shown to lead to inflammation and disease. In contrast, lncRNAs, can both enhance or repress the expression of protein-coding genes. COPD is typically caused by tobacco smoking and leads to a progressive decline in lung function and a premature death. Exaggerated pulmonary inflammation is a hallmark feature in this disease, leading to obstructive bronchiolitis and emphysema. In this review, we discuss the miRNA expression patterns in lungs of patients with COPD and in mouse models and we highlight various miRNAs involved in COPD pathogenesis. In addition, we briefly discuss a specific lncRNA that is upregulated upon cigarette smoke exposure, providing a short introduction to this more recently discovered group of non-coding RNAs.
- COPD Pathology
Statistics from Altmetric.com
Search strategy
We searched PubMed with the search terms ‘COPD’ or ‘emphysema’ or ‘smoke’ in combination with one of the following terms: ‘microRNA’, ‘miRNA’, ‘micro-RNA’, ‘long non-coding RNA’, ‘lncRNA’ or ‘lincRNA’. We also specifically searched for the combination: ‘COPD/emphysema’, ‘miRNA/lncRNA’ and ‘biomarker’. We focused on lung and not on systemic disease manifestations of COPD and on ex vivo/in vivo studies of tissues or cells of patients with COPD and animal models instead of in vitro studies. The date of the last search was 10 April 2015. In addition, we consulted the reference lists of articles resulting from this search strategy and included those we judged relevant.
Introduction
COPD
COPD causes significant morbidity and mortality worldwide and is expected to become the third leading cause of death by 2020.1 COPD is characterised by a progressive and irreversible airflow limitation, associated with an abnormal inflammatory response of the lungs to noxious particles and gasses.1 Next to cigarette smoking, which is the main risk factor for COPD, exposure to smoke from biomass fuels and occupational exposure to chemicals or dust are also regarded as substantial threats.2 ,3
Exaggerated inflammation in the airways and lung parenchyma is critical in the development and progression of COPD, causing small airway obstruction (obstructive bronchiolitis) and destruction of lung parenchyma (emphysema).1 By triggering pattern recognition receptors (PRRs), noxious particles or gasses activate innate immune cells such as alveolar macrophages and airway epithelial cells. These cells subsequently release pro-inflammatory mediators such as chemokines and cytokines, mediating the recruitment of neutrophils and inflammatory monocytes from the blood circulation to the lung. The specialised antigen-presenting dendritic cells initiate adaptive immunity by promoting T helper 1 (Th1) and T helper 17 (Th17) cell differentiation and CD8+ cytotoxicity.
Obstructive bronchiolitis is caused by the accumulation of these immune cells in the airway lumen, airway wall and lung parenchyma and by structural alterations of the small airways (airway wall remodelling). Next to obstructive bronchiolitis, emphysema is the second pathological hallmark contributing to COPD. It compromises lung function and reduces the surface area available for gas exchange. Oxygen radicals and proteolytic enzymes are responsible for this lung destruction. Activated neutrophils and macrophages release such proteolytic enzymes, for example neutrophil elastase and matrix metalloproteinases (MMPs).4
The normal decline of lung function with age is accelerated in patients with COPD and slowly leads to disability and death.5 Furthermore, current pharmacological therapy is limited and is directed against the clinical manifestations, mainly leaving the underlying disease-causing process unaffected. The fact that only 20% of smokers develop COPD6 suggests that genetic and epigenetic factors contribute to the susceptibility for this disease.
Non-coding RNAs
Protein-coding genes represent only a small portion of the human genome (20 687 genes, <2%),7 whereas a large part is transcribed into non-coding RNAs (ncRNAs), including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). The purpose of this article is to establish a comprehensive overview of the literature on non-coding RNAs in COPD, focusing on miRNAs and lncRNAs. Literature on other non-coding RNAs in COPD (such as small nuclear RNAs, small nucleolar RNAs and piwi-interacting RNAs) is limited and beyond the scope of this review. For definition of terms see Table 1.
Definition of terms
MiRNAs are the most widely studied class of ncRNAs. They are small RNA molecules (approximately 21–25 nucleotides long) that negatively regulate gene expression post-transcriptionally, by means of mRNA degradation, inhibition of protein translation, or by a combination of both mechanisms.8 MiRNAs are predicted to regulate approximately 60% of all human protein-coding genes.9 Dysregulation of miRNAs is reported to be involved in the pathogenesis of many diseases, including pulmonary diseases such as COPD.10
Many forms of small ncRNAs are relatively strongly conserved across species.11 LncRNAs, in contrast, do not show such conservation and were originally classified as non-functional.12 They comprise a heterogeneous class of RNAs, varying in nucleotide length (200 nucleotides to over 100 kilobases), cellular location (nucleus, cytoplasm or both), splicing pattern and functional and structural role in various biological processes.
The tertiary structure of lncRNAs allows them to bind to and interact with proteins, explaining their involvement in many regulatory functions. They can prevent or facilitate the recruitment of RNA polymerase II, transcription factors and/or cofactors to gene promoters, thereby controlling transcription of target genes. They also function as molecular scaffolds, bringing together multiple protein partners. Hundreds of lncRNAs have been found to form ribonucleic–protein interactions with multiple protein partners, supporting the concept of a molecular scaffold.13 They can also guide specific regulatory protein complexes to the correct chromosomal locations in cis or in trans providing target specificity to these complexes.
Besides their predominantly transcriptional modes of actions described so far, lncRNAs can also base-pair with mRNA molecules and affect their stability or translation.14 Finally, lncRNAs can compete for miRNA binding, and in that way prevent the miRNA from carrying out its function.15 This is their so-called sponge function as competing endogenous RNA.
MiRNA expression upon cigarette smoke exposure and in COPD
As mentioned above, cigarette smoke (CS) is the uttermost important risk factor for the development of COPD, making it a relevant risk factor when studying COPD pathogenesis. It remains unknown by which mechanism CS is able to dysregulate miRNAs. Most likely, the high toxicity and mutagenicity of CS plays an important role, since CS contains radicals that can interact with DNA in vitro.16 Next to directly affecting the miRNA, disrupting miRNA regulatory mechanisms might be an alternative mechanism by which CS interferes with miRNA expression. An overview of the biological functions of miRNAs in the pathogenesis of COPD is illustrated in figure 1.

The biological functions of microRNAs and long non-coding RNAs in the pathogenesis of COPD. Illustration of the hallmarks of COPD, with chronic inflammation, airway remodelling and emphysema as pathological hallmarks and oxidative stress and imbalance in fluid homeostasis as pathogenetic mechanisms. CFTR, cystic fibrosis transmembrane conductance regulator; Cox-2, cyclooxygenase-2; IL-1R1, interleukin 1 receptor 1; let-7c, lethal-7c; miR, microRNA; MMP12, matrix metallopeptidase 12; MPK-1, mitogen-activated protein kinase phosphatase 1; SCAL1, smoke and cancer-associated lncRNA-1; SMAD7, mothers against decapentaplegic homologue 7; TNFR-II, tumour necrosis factor receptor II.
MiRNAs as potent regulators of gene and protein expression
Given the relative redundancy of complementary sequences between miRNAs and their target mRNAs, a single miRNA is capable of targeting several genes simultaneously. Moreover, by targeting transcription factors or genes involved in epigenetic regulation, miRNAs can indirectly alter expression of hundreds of genes at once. In addition, miRNAs have a tissue- and cell type-specific expression pattern, making them excellent fine-tuners of gene regulation. Consequently, dysregulation of miRNAs can have widespread effects on gene and protein expression, affecting a multitude of biological processes.
For example, miR-218 was downregulated in smokers relative to never-smokers in human bronchial epithelium,17 in induced sputum of current smokers with and without COPD in comparison to never-smokers10 and in the bronchoalveolar lavage (BAL) fluid of patients with COPD compared with control subjects.18 Amongst its targets, the transcription factor MAFG (v-maf musculoaponeurotic fibrosarcoma oncogene homologue G, avian) represents a gene of particular interest, since its binding sites were significantly overrepresented among genes altered in smokers.17 In this way, miR-218 is capable of indirectly regulating a multitude of genes in response to CS by modulating the expression of the transcription factor MAFG.
Although proteins typically have half-lives in the range of a couple of hours to a few weeks,19 changes in gene expression were found to persist in the bronchial epithelium of ex-smokers years after smoking cessation, indicating the presence of persisting alterations in gene regulation upon CS exposure.20 Likewise, genes differentially expressed between current and never-smokers were categorised based on their expression in former smokers. They were classified as rapidly reversible, slowly reversible and irreversible. Although the mRNA expression of most genes (79%) in this study was rapidly reversible upon smoking cessation, those genes classified as slowly reversible or irreversible still presented with a 1.5-fold change between former and never-smokers for at least 78 months after smoking cessation.21 In line with this, CS-induced changes in miRNA expression in murine lung are irreversible if a certain threshold is reached, depending on the duration and dose of smoke exposure.22 Taken together, these findings support the idea that persistent dysregulated gene expression, found in the lungs of smokers and ex-smokers, might be attributable to irreversible changes in miRNA expression induced by exposure to a sufficiently high dose of smoke, for a sufficiently long period of time.
MiRNA expression profiling upon CS exposure and in COPD
In general, most studies report a global downregulation of miRNA abundance in response to cigarette smoking.
Graff et al23 found that cigarette smoking decreases global miRNA expression in human alveolar macrophages, possibly by modification of DICER, an RNA endonuclease involved in the maturation of miRNAs.24 Interestingly, the decrease in abundance was more pronounced in individuals with a heavy smoking history in comparison to light smokers, suggesting a correlation between the number of downregulated miRNAs and smoking history.23 Likewise, Izzotti et al25 also reported a downregulation of miRNA expression in the lungs of smoke-exposed rats. Notably, further research by the same group demonstrated that both age and gender influenced smoke-related miRNA dysregulation in murine lungs.26 In line with the results of smoke-exposed rat lungs25 and of human alveolar macrophages,23 the majority of differentially expressed miRNAs (23 out of 28) in human bronchial airway epithelium were downregulated in smokers compared with never-smokers.17
A global decrease in miRNA expression upon smoking can be explained by different possible mechanisms, such as a deficiency in miRNA processing (eg, due to Dicer inhibition), changes in primary miRNA transcription, an increase in miRNA degradation or localisation of miRNAs to stress granules (aggregates of mRNAs in conjunction with proteins that appear when the cell is under stress) and processing bodies (cytoplasmic foci containing aggregates of specific proteins and mRNAs), or relative upregulation of other RNA fractions, such as ribosomal RNAs or mRNAs. In the process of COPD pathogenesis, downregulation of miRNAs can provide a plausible mechanism for the observed upregulation of inflammatory mediators.
In contrast, Ezzie et al27 analysed lung tissue of smokers with or without COPD and found the expression of 57 miRNAs to be increased in patients with COPD, while only 13 miRNAs showed a decreased expression.
The discrepancy between this study and previous studies reporting a global downregulation of miRNAs10 ,17 ,23 might be due to the fact that miRNAs have a cell type-specific expression pattern and the global analysis of miRNA expression in whole lung tissue, where multiple cell types are present, might not reflect the situation in those cell types important for disease pathogenesis. Alternatively, differences in gene expression analysis workflow in general or normalisation procedure more specifically might also account for the observed discrepancies. Microarrays, reverse transcription quantitative PCR (RT-qPCR) and small RNA sequencing are widely used miRNA expression evaluation platforms. Nonetheless, these methods do not always deliver the same results.28 ,29 Microarray analysis, for example, can result in false positives (non-specific hybridisation) or false negatives (when hybridisation signals do not exceed the background level).
In addition, patient characteristics (such as COPD stage, age, gender, number of pack-years smoked, duration of smoking cessation) and sample size of the study population or differences in study design may also have a substantial influence on the results (for an overview of these parameters, see online supplemental table S1). Given that miRNA expression values are highly context dependent, an adequate experimental design and a sound statistical analysis are of high importance.
MiRNAs modulate the chronic pulmonary inflammation in COPD
Exaggerated and imbalanced inflammatory responses play an important role in COPD pathogenesis. These inflammatory responses are mediated by inflammatory proteins that are released upon CS exposure by structural cells of the lungs and by inflammatory cells. Pro-inflammatory transcription factors, such as nuclear factor кB (NF-кB), regulate the expression of these inflammatory proteins. Amplification of this inflammatory response upon CS exposure might lead to the development of COPD. For an overview of the expression pattern of the discussed miRNAs in patients with COPD, smokers and never-smokers see table 2. In addition, table 2 contains the accession number and the most recent annotation of each miRNA, as checked by miRBase Tracker.30 For an overview of miRNA profiling studies in rodent models discussed in this review see online supplemental table S2.
Expression pattern of miRNAs in patients with COPD, smokers and never-smokers
MiR-146a
Increased expression of cyclooxygenase-2 (COX-2) protein is a hallmark of chronic inflammation in CS-related diseases such as COPD and lung cancer.31 ,32 Sato et al showed that while interleukin (IL)-1β and tumour necrosis factor (TNF)-α, two potent inducers of the NF-кB pathway, induced the expression of miR-146a in both COPD and control fibroblasts, this was less pronounced in COPD fibroblasts. They suggested that this diminished miR-146a expression in COPD fibroblasts resulted in a reduced degradation and a prolonged half-life of COX-2 mRNA, a predicted target of miR-146a (TargetScan, miRBase), ultimately leading to overexpression of COX-2 protein.33 COX-2 is a key enzyme in prostaglandin E2 (PGE2) biosynthesis, a promoter of neutrophil recruitment.34 PGE2 also inhibits the repair functions of lung fibroblasts35 and is increased in the lungs of patients with COPD and in COPD fibroblasts in culture.33 ,36
Likewise, miR-146a was downregulated in sputum of patients with COPD and smokers compared with never-smokers and in smokers with COPD in comparison to smokers without COPD.10 In contrast, miR-146a expression was increased in lung tissue from patients with COPD in comparison to smokers.27
MiR-135b
MiR-135b seems to be involved in a negative feedback mechanism attempting to curtail excessive inflammatory responses (figure 2). MiR-135b was found to be significantly upregulated in the BAL fluid of CS-exposed mice and upon in vitro activation of the IL-1 receptor 1 (IL-1R1) by its agonist IL-1α.37 Further experiments indicated that CS induces an increase in pulmonary expression of miR-135b and that this increase is dependent on IL-1α/IL-1R1 signalling.37

Proposed negative feedback mechanism involving miR-135b. Cigarette smoke induces an increase in pulmonary expression of miR-135b in mice and this increase is dependent on IL-1α/IL-1R1 signalling.37 Activated caspase-1 generates the mature form of IL-1β by cleaving its precursor, pro-IL-1β. Both mature IL-1β and IL-1α activate the IL-1R1, which transduces its signal through myeloid differentiation primary response 88 protein (MyD88), ultimately resulting in the activation of the transcription factors NF-кB and activator protein 1 (AP-1, not shown here). NF-κB activation induces pro-IL-1β transcription and might also regulate the transcription of miR-135b (dashed arrow, observed in lung cancer cells82). IL-1R1 and caspase-1 are likely targeted by miR-135b.37 In the post-acute phase, miR-135b might thus function as a negative feedback regulator by binding on its own regulator (IL-1R1) and its downstream effector, caspase-1, in an attempt to halt the progression of inflammation into the chronic phase. Figure adapted from Halappanavar et al.37 IL-1α, interleukin 1α; IL-1RI, interleukin 1 receptor 1; IL-1RAcP, IL-1R accessory protein; miRNA, microRNA; mRNA, messenger RNA; MyD88, myeloid differentiation primary response protein 88; NF-кB, nuclear factor кB.
Furthermore, the authors demonstrated that IL-1R1 and caspase-1 are likely targeted by miR-135.37 Activated caspase-1 generates the mature form of IL-1β by cleaving its precursor, pro-IL-1β.38 Both mature IL-1β and IL-1α activate the IL-1R1. In the post-acute phase, miR-135b might thus function as a negative feedback regulator by binding on its own regulator (IL-1R1) and its downstream effector, caspase-1, in an attempt to halt the progression of inflammation into the chronic phase (figure 2). In accordance with this hypothesis, IL-1β has, in general, been described to be upregulated and implicated in early responses upon CS exposure,39 but not upon chronic CS exposure.40
An appropriate immune response is required to protect an organism against harmful attacks from various pathogens and other damaging events such as CS. However, when the activated immune response is too excessive, this can lead to pathological manifestations causing inflammatory disorders such as COPD. In this respect, negative feedback regulation is an important mechanism keeping inflammation in check. As certain miRNAs might be important negative feedback regulators, the feedback control may be lost due to reduced expression and concomitant exacerbated inflammation may occur.
Let-7c
Analysis of miRNA expression in induced sputum supernatant in the study of Van Pottelberge et al10 revealed that the expression of let-7c (lethal-7c) was significantly lower in patients with COPD in comparison with never-smokers and smokers without COPD. Moreover, let-7c was significantly associated with FEV1 (% predicted) and was previously reported to be downregulated in rat and mouse lungs upon CS exposure.22 ,25 The protein level of TNF receptor II (TNFR-II) in sputum, a predicted target gene of let-7c (MiRgen database), was inversely correlated with the level of let-7c. Gene set enrichment analysis revealed that target genes of let-7c are significantly enriched in patients with severe and very severe COPD in comparison to patients with moderate COPD. Furthermore, TNFR-II was enriched in the sputum of patients with severe and very severe COPD.10 TNFR-II plays an important role in COPD pathogenesis, evidenced by the fact that TNFR-II knockout mice are protected against CS-induced inflammation and emphysema.41 Let-7 mimics have been investigated as cancer therapeutics in mice42 and, given the demonstrated importance of let-7c in COPD pathogenesis, it might also be an interesting therapeutic target to investigate in preclinical COPD models.
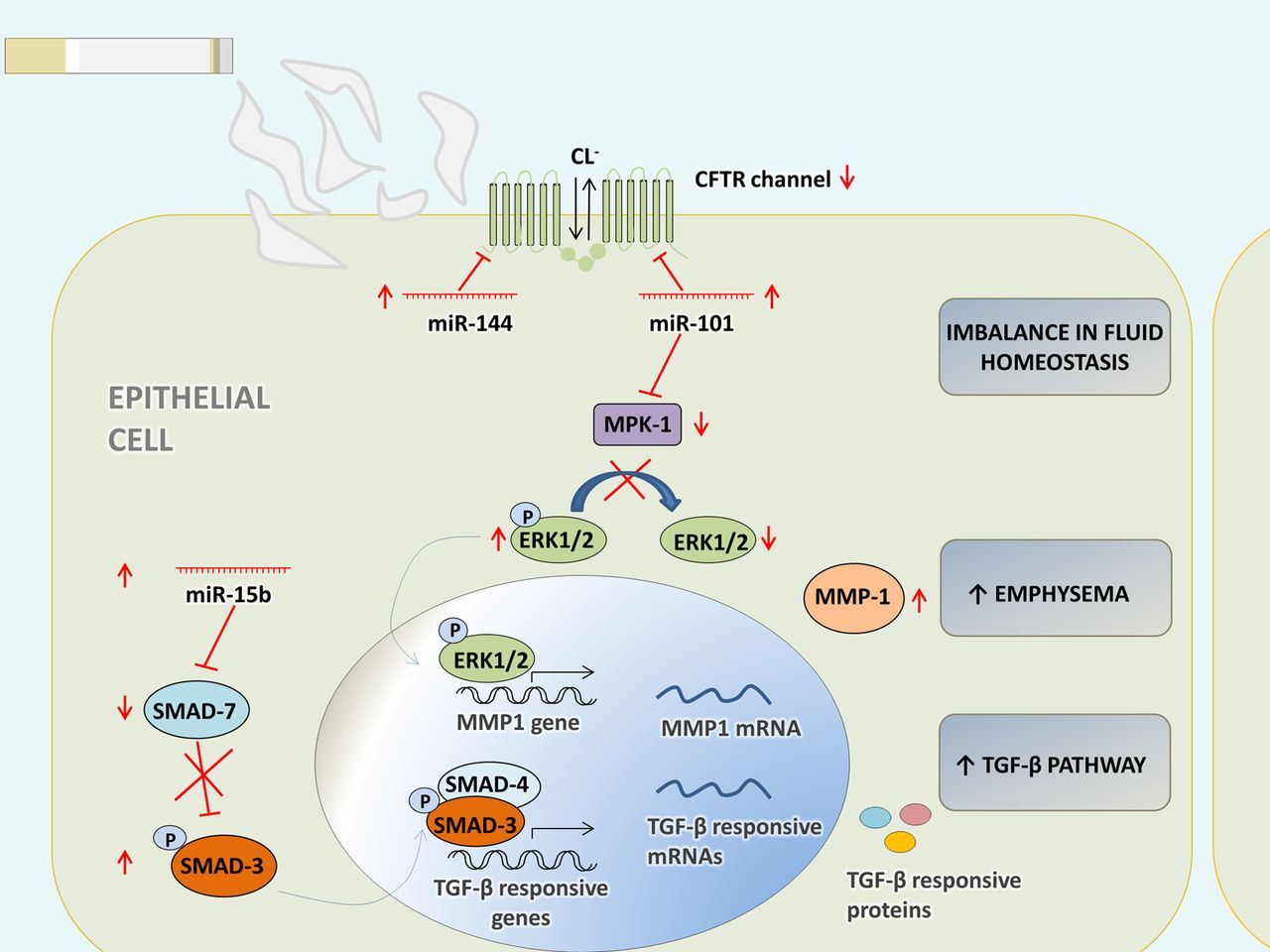
MiR-101 and miR-144
The cystic fibrosis transmembrane conductance regulator (CFTR) protein, a chloride channel involved in the maintenance of fluid homeostasis in the lung, is suppressed in the lungs of patients with COPD with a history of smoking.43 Hassan et al44 showed that expression of miR-144 and miR-101 suppresses CFTR protein in human bronchial epithelial cells and that they target the 3′ untranslated region of CFTR in vitro. Both miRNAs are upregulated by CS and cadmium.44
In addition, miR-144 was found to be upregulated in lung tissue from patients with COPD in comparison to smokers without COPD.27 Chronic cigarette smoking might thus upregulate miR-101 and miR-144, and this may result in the suppression of CFTR in the lungs of patients with COPD, causing an imbalance in fluid homeostasis (figure 3).

{kind=link}
{kind=link}
{kind=link}
Schematic overview of hypothetic functions of miR-144, miR-101 and miR-15b in bronchial epithelial cells. The CFTR protein is suppressed in the lungs of patients with COPD with a history of smoking.43 MiR-144 and miR-101 are upregulated by cigarette smoke and in patients with COPD.27 ,44 Expression of miR-144 and miR-101 suppresses CFTR protein in HBE cells and both miRNAs target the 3′ untranslated region of CFTR in vitro.44 Chronic cigarette smoking might upregulate miR-101 and miR-144, and this may result in the suppression of CFTR in the lungs of patients with COPD, causing an imbalance in fluid homeostasis. MiR-101 targets MPK-1.53 High expression of this miRNA could lead to diminished dephosphorylation by MPK-1 and a sustained activation of ERK 1/2, seen in patients with COPD.54 Notably, ERK1/2 signalling is also required for the increased expression of MMP-1 in emphysematous lung tissue of chronic smokers.55 SMAD7 is a predicted target of miR-15b. It has an inhibitory function in transforming growth factor β (TGF-β) signalling and was markedly downregulated in bronchial biopsies of patients with COPD.27 Overexpression of miR-15b in Beas2B bronchial epithelial cells resulted in decreased SMAD7 protein expression and, consequently, an increased early phosphorylation of SMAD3 in response to TGF-β.27 CFTR, cystic fibrosis transmembrane conductance regulator; Cl−, chlorine; ERK 1/2, extracellular signal-regulated kinase 1/2; miR, microRNA; MMP1, matrix metallopeptidase 1; MPK1, mitogen-activated protein kinase phosphatase 1; SMAD, mothers against decapentaplegic; TGF-β, transforming growth factor β.
MiR-15b
MiR-15b is significantly upregulated in COPD tissue and is expressed in bronchial epithelium and alveolar wall in type II pneumocytes.27 SMAD7 (mothers against decapentaplegic homologue 7) is involved in the pathogenesis of COPD45 and is a predicted target of miR-15b (TargetScan V.5.1, PicTar, miRanda) (figure 3). SMAD7 has an inhibitory function in transforming growth factor β (TGF-β) signalling and was markedly downregulated in bronchial biopsies of patients with COPD.27 Moreover, knockdown of miR-15b in Beas2B bronchial epithelial cells resulted in increased SMAD7 protein expression and, consequently, a reduced early phosphorylation of SMAD3 in response to TGF-β. In line with this, overexpression of miR-15b caused an increased early phosphorylation of SMAD3.27
MiRNA-199a-5p
CS causes proteins to misfold, leading to ER stress and the activation of the unfolded protein response.46 GRP78 (glucose response protein 78), IRE1 (inositol requiring enzyme 1α), ATF6 (activation of transcription factor 6), NF-кB1 and RELA (V-Rel avian reticuloendotheliosis viral oncogene homologue A) are all involved in the unfolded protein response and are confirmed targets of miR-199a-5p.47–49
By studying isolated peripheral blood monocytes from individuals with or without COPD, a mechanism was put forward whereby hypermethylation of the miR-199a-5p promoter induces miR-199a-5p silencing, followed by derepression of proteins involved in the unfolded protein response and consequent unfolded protein response activation in patients with COPD.49
Recently, Chatila et al50 demonstrated that miR-199a-5p is significantly higher expressed in T-regulatory (Treg) cells compared with T-effector (Teff) cells in peripheral blood. Moreover miR-199a-5p was approximately fourfold lower in Treg cells of patients with COPD compared with healthy smokers.50 The role of Treg cells in the pathogenesis in COPD is still undetermined. These cells are CD4+ T cells with an immunoregulatory function, mediating a suppressive effect on other T cells and antigen-presenting dendritic cells.
The role of miRNAs in emphysema
COPD is a heterogeneous disease, with patients suffering from varying degrees of obstructive bronchiolitis and emphysema. Importantly, there is only a weak correlation between these two disease manifestations, where the same FEV1 value (as measured by spirometry) can be accompanied by a low or high degree of emphysema (as evidenced by pathological examination of lung tissue resection specimens, CT scan of the lungs (imaging), or measurement of the diffusing capacity of the lungs) in different patients.51 However, most studies on miRNA expression in COPD use a disease classification solely based on the degree of airflow obstruction. For an overview of the expression pattern of the discussed miRNAs in patients with COPD, smokers and never-smokers see table 2.
MiR-638
Notably, miRNA expression seems to be correlated with regional emphysema severity.52 MiR-638, for example, was positively correlated with emphysema severity and had more than 50 positively or negatively regulated targets. Inhibition of this miRNA in lung fibroblasts resulted in overexpression of its predicted targets, including genes involved in the oxidative stress response and extracellular matrix remodelling.52
MiR-101
Hassan and colleagues found miR-101 to be highly expressed in the lungs of patients with COPD and in the lungs of mice subjected to CS.44 MiR-101 targets mitogen-activated protein kinase (MAPK) phosphatase 1 (MPK-1), an inhibitor of MAPKs.53 High expression of this miRNA could lead to reduced dephosphorylation of extracellular signal-regulated kinase (ERK) 1/2 by MPK-1 and a sustained activation of ERK 1/2, seen in patients with COPD54 (figure 3). Notably, ERK1/2 signalling is also required for the increased expression of the protease MMP-1 in emphysematous lung tissue of chronic smokers.55
MiR-452
MiR-452 is highly downregulated in alveolar macrophages from active smokers compared with non-smokers.23 Upon transfection of a miR-452 inhibitor in human monocytic THP-1 cells that are induced to differentiate toward a macrophage phenotype, the expression of matrix metallopeptidase 12 (MMP12) mRNA (a predicted target, MicroCosm database) was increased. MMP12 is secreted by macrophages and plays an important role in the pathogenesis of COPD by degrading elastin, thereby causing emphysema.56
MiR-199a-5p
In contrast to Hassan et al,49 Mizuno and colleagues reported an increased miR-199a-5p expression in lung tissue samples of patients with COPD in comparison to control samples of never-smokers with normal lung function.57
The authors propose a mechanism whereby oxidative stress mediates an upregulation of TP53, which in turn upregulates miR-34a, followed by regulation of miR-199a-5p, possibly through AKT inactivation.57
Transfection of a miR-199a-5p precursor in human pulmonary microvascular endothelial cells resulted in a decreased hypoxia inducible factor 1α (HIF-1α) expression,57 in line with decreased expression in lungs from patients with COPD.58
Furthermore, HIF-1α positively regulates vascular endothelial growth factor (VEGF) gene expression,59 which is of importance, since conditional knockout of the VEGF ligand60 and inhibition of its receptor61 in mouse lungs resulted in emphysema. In addition, in a genome-wide gene expression profiling study of lung tissue samples from current and ex-smokers with and without COPD, VEGF was one of the most downregulated genes in COPD samples.62
MiR-34c and miR-34b
In a recent study miRNAs differentially expressed between mild and moderate emphysematous lung were identified.63 Five miRNAs were significantly downregulated in lungs from patients with moderate emphysema, with miR-34c exhibiting the largest fold change. Supporting these findings, a more than threefold downregulation of miR-34c was observed in sputum from smokers with or without COPD compared with never-smokers.10 Moreover, miR-34c was significantly downregulated in the lungs of rats exposed to CS.25
MiR-34b originates from the same primary transcript as miR-34c and its expression is also reduced in the sputum from smokers with or without COPD compared with never-smokers10 and in the lungs of mice22 and rats25 exposed to CS.
MiR-34c acts as a tumour suppressor and is downregulated in many types of cancer, including lung cancer.64 A synthetic mimic of miR-34 (MRX34) is the first miRNA mimic that has reached a phase I clinical trial status,65 aiming to restore its function in a range of cancers.
Further, given the downregulation of miR-34c in serum of patients with COPD compared with healthy controls, it was suggested that this miRNA might serve in the clinic as a biomarker in COPD.66 These studies open a window to the potential for certain miRNAs to serve as diagnostic and therapeutic agents in the combat against COPD.
Use of non-coding RNAs as biomarkers in COPD
Since miRNAs are remarkably stable in biofluids67 and can be easily and reproducibly detected, they make excellent candidates for risk prediction and minimally invasive diagnosis of COPD.
For example, levels of serum miR-21 and miR-181a and their ratio are suggested to have potential biomarker value for predicting the development of COPD in heavy asymptomatic smokers.68
Also in serum, five miRNAs (miR-34c-5p, miR-20a, miR-28-3p, miR-100 and miR-7) were differentially expressed between patients with COPD and controls.66 This suggests that these miRNAs might have a biological function in the pathogenesis of COPD and in the future might serve as biomarkers of COPD.
In sputum, an easily accessible body fluid, detection of both miRNAs and lncRNAs has also been reported.10 ,69 In addition, by using sampling of exhaled breath condensates, another minimally invasive method, it is also possible to detect differential expression of miRNAs.70
Dysregulation of long non-coding RNAs by CS exposure and in patients with COPD
Unlike miRNAs, lncRNAs are much less characterised. The function of some individual lncRNAs (such as HOTAIR (LNCipedia ID: lnc-SMUG1-771 ,72)) has been elucidated, but the function of most lncRNAs remains unknown.
Recently, a novel lncRNA was characterised, named the smoke and cancer-associated lncRNA-1 (SCAL1, lnc-ARRDC3-1), which is upregulated by CS extract in vitro73 and is elevated in the airway epithelia of cigarette smokers versus non-smokers.74 ,75 As its name suggests, SCAL1 is also elevated in many lung cancer cell lines. Small interfering RNA knockdown experiments demonstrated that SCAL1 is transcriptionally regulated by nuclear factor erythroid 2 related factor (NRF2). NRF2 is involved in the regulation of protective antioxidant genes76 and NRF2 loss of function sensitises lung cells to COPD-like changes.77 Furthermore, NRF2 expression is reduced in pulmonary macrophages of patients with COPD.78
Knockdown of SCAL1 in bronchial epithelial cells resulted in a significant potentiation of CS-induced cytotoxicity, indicating that it might function as a protector against oxidative stress (see figure 1). It is suggested that SCAL1 mediates some of the cytoprotective functions of NRF2 toward CS-induced stress.73
The authors also analysed several publically available databases and found that CS is able to alter the expression of numerous lncRNAs in airway epithelial cells.73 LncRNAs, similar to miRNAs, might function as regulators of large sets of genes that protect against environmental stresses such as CS exposure.
Interestingly, lncRNAs are expressed in a more pronounced tissue-specific manner than protein-coding genes,79 making them attractive future diagnostic and therapeutic targets.
Recently, a study was published comparing the lncRNA expression profile in lung tissue from non-smokers and smokers with or without COPD.80 Upon comparison of patients with COPD and smokers without COPD, upregulation of 120 and downregulation of 43 lncRNAs was reported. Given the small sample size and the inclusion of only male subjects, additional larger studies with both genders are warranted.
Future directions
COPD is a heterogeneous disease, with patients suffering from varying degrees of obstructive bronchiolitis and emphysema. As it stands, most studies on non-coding RNA expression profiling in COPD characterise patients solely by measurements of airflow obstruction, not taking the degree of emphysema into account (see online supplemental table S1). Nonetheless, emphysema is independently associated with poor clinical outcome and might also be the predominant disease manifestation in certain patients with COPD.81 Given this limitation, future studies should make an accurate assessment of emphysema extent and severity to produce a more complete classification of patients with COPD, allowing a more precise comparison between different groups.
As discussed in this review, great difficulties arise when trying to find overlap between miRNA profiling studies. Next to taking differences in patient characteristics into account, it remains important that findings are replicated in independent validation cohorts, by different studies and by different laboratory measurement methods. Since RT-qPCR and microarray can only quantify known targets, (small) RNA sequencing forms an interesting method for discovery of novel tissue-and disease-specific ncRNAs.
Upon differential expression analysis of ncRNAs, the resulting (top) hits should be functionally validated. Exhaustive profiling experiments result in lists of statistically significant differentially expressed ncRNAs, but little is known about the biological significance of these findings. This can be achieved for example by in vivo experiments, using either transgenic/knockout animals or perturbation experiments.
Conclusion
Current therapy for COPD has only limited disease-modifying effects. Hence, there is a great need for novel therapeutic interventions and preventive strategies. Research concerning the pathogenetic mechanisms causing COPD would greatly contribute to an improved therapy for patients suffering from this disease. The above-mentioned studies highlight the involvement of ncRNAs in the host response towards environmental exposures. In doing so, they are capable of influencing the susceptibility towards lung diseases such as COPD, linking environmental exposures to disease development and progression. Identification of ncRNAs important in these processes could provide new clinical targets as well as diagnostic and prognostic tools for COPD.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online table 1
- Data supplement 2 - Online table 2
Footnotes
Contributors EGDS performed the literature search and wrote the manuscript. PM, JV, GGB and KRB reviewed and edited the manuscript.
Funding Presented work within the Department of Respiratory Medicine of Ghent University is funded by grants from the Fund for Scientific Research in Flanders (FWO Vlaanderen, G.0A99.13), the Concerted Research Action of the Ghent University (BOF/GOA, 01G02714) and the Interuniversity Attraction Poles Program (IUAP, P7/30).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.