Article Text

Abstract
Background Parasympathetic pulmonary nerves release acetylcholine that induces smooth muscle constriction. Disruption of parasympathetic pulmonary nerves improves lung function and COPD symptoms.
Aims To evaluate ‘targeted lung denervation’ (TLD), a novel bronchoscopic therapy based on ablation of parasympathetic pulmonary nerves surrounding the main bronchi, as a potential therapy for COPD.
Methods This 1-year, prospective, multicentre study evaluated TLD in patients with COPD forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) (FEV1/FVC <0.70; FEV1 30%–60% predicted). Patients underwent staged TLD at 20 watts (W) or 15 W following baseline assessment off bronchodilators. Assessments were repeated on tiotropium before treatment and off bronchodilators at 30, 90, 180, 270 and 365 days after TLD. The primary endpoint was freedom from documented and sustained worsening of COPD directly attributable to TLD to 1 year. Secondary endpoints included technical feasibility, change in pulmonary function, exercise capacity, and quality of life.
Results Twenty-two patients were included (n=12 at 20 W, n=10 at 15 W). The procedures were technically feasible 93% of the time. Primary safety endpoint was achieved in 95%. Asymptomatic bronchial wall effects were observed in 3 patients at 20 W. The clinical safety profiles were similar between the two energy doses. At 1 year, changes from baseline in the 20 W dose compared to the 15 W dose were: FEV1 (+11.6%±32.3 vs +0.02%±15.1, p=0.324), submaximal cycle endurance (+6.8 min±12.8 vs 2.6 min±8.7, p=0.277), and St George's Respiratory Questionnaire (−11.1 points ±9.1 vs −0.9 points ±8.6, p=0.044).
Conclusions Bronchoscopic TLD, based on the concept of ablating parasympathetic pulmonary nerves, was feasible, safe, and well tolerated. Further investigation of this novel therapy is warranted.
Trial registration number NCT01483534.
- COPD ÀÜ Mechanisms
- Bronchoscopy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
-
Parasympathetic pulmonary nerves release acetylcholine that causes smooth muscle contraction and increased mucus production contributing to airway obstruction and symptoms in COPD. Is bronchoscopic radiofrequency (RF) ablation of these pulmonary nerves safe and feasible in patients with mild to moderate COPD?
What is the bottom line?
-
This first-in-human study evaluated bronchoscopic RF ablation of parasympathetic pulmonary nerves running along the main bronchi in patients with COPD. This approach is shown to be feasible and safe.
Why read on?
-
This new bronchoscopic therapy called ‘targeted lung denervation’, is a potential future treatment option for patients with advanced COPD.
Introduction
Cholinergic parasympathetic nerves innervate both large and small airways and provide the dominant innervation to human lungs.1 Acetylcholine released from these nerves regulates airway smooth muscle tone, mucus secretion, and potentially local inflammation through interaction with muscarinic receptors found throughout the bronchial tree.2 ,3 Furthermore, pulmonary parasympathetic activity is enhanced in COPD, and is the dominant reversible component of airway obstruction in this disease.2 As such, disruption of parasympathetic activity in the lungs is a logical, well characterised, and an effective approach to the treatment of COPD.
Historically, parasympathetic nerve disruption via surgical vagotomy has consistently demonstrated the bronchodilator effect of vagotomy in animals.4 ,5 Unfortunately, the majority of human research of vagotomy as a treatment for COPD is anecdotal or highly subjective.6 In one notable study, surgical denervation of the lungs in 19 patients with intractable bronchial asthma increased vital capacity from 2.36 L to 2.79 L, and maximal voluntary ventilation from 43 L to 50 L/min.7 Sputum production was essentially stopped in eight of the 11 patients with a previous history of heavy sputum production.
In more recent history, pharmacologic blockade of acetylcholine binding to muscarinic receptors has been shown to produce bronchodilation and decrease mucous production.8 One trial in COPD demonstrated tiotropium to have a 9.6% improvement in trough-forced expiratory volume in 1 s (FEV1) at 1 year accompanied by a −4.5-point change in St George's Respiratory Questionnaire (SGRQ).9 Similar improvements in lung function and health-related quality-of-life (HRQL) have been demonstrated by others.10 ,11 Tiotropium, when compared with placebo, also improves cycle endurance time by 3.9 min, 2.5 h after inhalation, and 2.9 min, 8 h after inhalation.12 A significant focus on drug development has resulted in numerous pharmaceutical therapies to help manage COPD, and the majority of today's treatment guidelines recommend anticholinergics as first-line therapy for patients with mild to advanced stages of COPD.13 ,14
Targeted lung denervation
Targeted lung denervation (TLD) is a novel bronchoscopic therapy that ablates the parasympathetic innervation of the lungs, and has a similar proposed mechanism of action to anticholinergic drugs.2 TLD therapy is delivered via a dual-cooled radiofrequency (RF) catheter (Holaira, Minneapolis, Minnesota, USA) (figure 1) designed to target tissue heating at depth, thereby producing a narrow band of ablation around the main bronchi while minimising effects to the inner surface of the airway. As RF current passes from the electrode through the airway and surrounding tissues, these tissues are heated. Coolant continuously circulated through the electrode and balloon removes heat from the surface of the airway wall. The net effect is targeted tissue ablation at depth with minimal heating and damage of the inner surface of the airway.

Description of the key components of the targeted lung denervation (TLD) catheter.
This targeted tissue ablation is intended to disrupt motor axons within bronchial nerve branches running along the outside of the main bronchi, thereby blocking parasympathetic signalling to the lungs and decreasing neuronal release of acetylcholine. This decrease in acetylcholine reduces airway obstruction in the whole lung by decreasing smooth muscle tone and mucous production.
Extensive animal and human cadaver testing was undertaken to develop the system and determine initial therapeutic RF energy levels (unpublished data). This first-in-human study was designed to investigate the safety and feasibility of TLD therapy in patients with moderate to severe COPD.
Methods
Study design and participants
This non-randomised, prospective, sequential, two-dose study was conducted at two sites in South Africa and one in The Netherlands between 31 October 2011 and 21 November 2013. Eligible patients were ≥40 years of age with COPD; defined as the ratio of the forced expiratory volume in 1 s (FEV1) to the forced vital capacity (FVC) of ≤0.70 and postbronchodilator FEV1 of 30%–60% of predicted normal values. Only patients with a 15% or greater relative increase in FEV1 following inhalation of 80 μg ipratropium bromide were included.
Key exclusion criteria included: COPD exacerbation or active respiratory infection within the past 4 weeks, more than 3 respiratory-related hospitalisations within 1 year of enrolment, previous lung surgery, suspicious pulmonary nodule, pulmonary hypertension, congestive heart failure, polycythaemia, SaO2 ≤88% or a PaO2 ≤7.3 kPa, and PaCO2 >8.0 kPa. Formal pulmonary rehabilitation was not a requirement for inclusion. Refer to the box 1 for a full list of inclusion and exclusion criteria.
Protocol Inclusion and Exclusion Criteria
Inclusion criteria
-
FEV1 30%–60%
-
FEV1/ FVC <70%
-
Patient is diagnosed with COPD (FEV1/ FVC <70%)
-
Positive relative change in FEV1 >15% following administration of ipratropium
-
≥40 years of age or older
-
Smoking history of at least 10 pack years
-
Non-smoking for a minimum of 6 months before consent and agreed to continue not smoking for the duration of the study
-
Patient has provided written informed consent
-
The patient is willing, able and agrees to complete all protocol-required baseline and follow-up assessments including taking and abstaining from medications
-
The patient has no child-bearing potential or a negative pregnancy test
-
Patient is a candidate for bronchoscopy in the opinion of the physician or per hospital guidelines
-
Current influenza vaccination and/or pneumococcus vaccination consistent with local recommendations and/or policy
Exclusion criteria
-
Pulmonary hypertension, peripheral oedema suggesting CHF or polycythaemia
-
Patient has an SaO2 ≤ 88% or a PaO2 ≤ 7.3 kPa (55 mm Hg)
-
Patient has a PaCO2 > 8.0 kPa (60 mm Hg)
-
Previous lung transplant, LVRS, median sternotomy, bullectomy or lobectomy
-
Pulmonary nodule requiring surgery
-
History of recurrent respiratory infections (>3 hospitalisations within 1 year of consent)
-
Presence of a pacemaker, internal defibrillator or other implantable electronic devices
-
Active respiratory infection within the past 4 weeks
-
COPD exacerbation within the past 4 weeks
-
Myocardial infarction within the last 6 months
-
Unstable or life-threatening arrhythmia within the last year
-
Malignancy treated with radiation or chemotherapy within the last 2 years
-
Presence of other respiratory diseases (cystic fibrosis, tuberculosis, vocal cord dysfunction, Churg-Strauss syndrome, allergic bronchopulmonary aspergillosis)
-
Known hypersensitivity to anticholinergic drugs or components
-
Known allergy to medications required for bronchoscopy (such as lidocaine, atropine) that cannot be medically controlled
-
Clinical diagnosis of sleep apnoea
-
Clinical diagnosis of asthma or other respiratory disease other than COPD
-
Known coagulopathy
-
Patient is taking clopidogrel, coumadin or other blood-thinning medication
-
The patient has any disease or condition that might interfere with completion of this study (eg, life expectancy less than 1 year)
-
Patient is currently enrolled in another clinical trial
CHF, congestive heart failure; LVRS, lung volume reduction surgery; PaO2, partial pressure of oxygen; SaO2, saturation of oxygen.
This study was approved by local ethics committees and in accordance with the Declaration of Helsinki (1996), Good Clinical Practice guidelines, and local requirements. An operations committee, and a data monitoring committee, oversaw protocol management and safety for the study. An independent clinical event reviewer adjudicated all serious adverse events (SAE). This trial is registered with ClinicalTrials.gov, number NCT01483534.
Study endpoints
The primary safety endpoint of freedom from documented and sustained worsening of COPD directly attributable to the investigational device or procedure to a 365-days post-TLD therapy, was defined as a decrease in the individual patient's FEV1 by any amount at all follow-up time points, along with a report of an adverse event that was reported to have a probable or definite relation to the device. Secondary endpoints included technical feasibility of the device and change from baseline in pulmonary function tests, exercise capacity assessments, and HRQL. Technical feasibility was defined as the ability to access the target treatment area main bronchus and deliver RF energy to the entire circumference of the bronchus at the target treatment site.
Procedures
After informed consent, patients underwent baseline testing after a wash-out period of 8 days for long-acting muscarinic antagonists (LAMA), 24 h for long-acting β agonists (LABA) and 12 h for short-acting β agonists (SABA) and short-acting muscarinic antagonists (SAMA). Baseline and follow-up testing included spirometry, body plethysmography, cycle ergometry, 6 min walk test (6MWT), COPD-specific SGRQ (score range 0–100 with a minimally clinically important difference (MCID) of ≥4 negative units15), the Clinical COPD Questionnaire (CCQ, 7-day version, score range 0–6, with an MCID of 0.4 units16), the common Borg and modified Medical Research Council17 scales. Cycle ergometry was first conducted as an incremental maximal test to determine baseline maximum work rate (Wmax) off drugs, and subsequently as an endurance test conducted at a constant work rate of 75% of the Wmax.18 Current American Thoracic Society (ATS)/European Respiratory Society (ERS) guidelines were followed for pulmonary function testing,19 ,20 and ATS/American College of Chest Physicians (ACCP) guidelines for cycle ergometry21 and the 6MWT.22 A baseline chest CT scan was required to confirm appropriate bronchial anatomy and rule out other pulmonary abnormalities.
After completion of the wash-out baseline testing, patients underwent a minimum 8-day run-in period while on tiotropium bromide, and similar testing was performed at drug trough 24 h after the last dose of tiotropium to establish tiotropium trough baseline values. LABAs were again held for 24 h and SABAs and SAMAs for 12 h before this testing. All patients underwent early safety evaluation by phone at 3 days and 10 days postprocedure. Testing was repeated at washout baseline, tiotropium trough baseline, and 30, 90, 180, 270 and 365 days post-treatment, except for cycle ergometry which was performed at 90, 180 and 365 days post-treatment. Patients were provided a daily memory aid to track overall changes in health status. A total of three bronchoscopies were performed per patient; two in order to complete the two treatments and the third one to assess potential airway surface effects during the 90-day follow-up visit. Adverse events were tracked and recorded throughout the entire study period.
Treatment
Pretreatment visual bronchoscopic inspection of the airways was conducted to reconfirm that appropriate airway anatomy existed. Due to the construction of this first-generation device, procedures were performed via rigid bronchoscopy under general anaesthesia. The dual-cooled catheter was placed through the rigid bronchoscope, and a flexible bronchoscope placed beside it for visualisation. The electrode was placed and activated in up to eight rotational positions per bronchus to achieve complete circumferential treatment. Total balloon inflation times were approximately 3 min per activation. The initial subjects were treated with 20 watts (W) in all positions except for the posterior-medial aspects of the left bronchus, where the power was reduced to 15 W due to the proximity of the oesophagus in those positions. Due to local airway effects, after a protocol amendment, additional patients underwent treatment with a more distal placement of the electrode along the medial wall to avoid the thin tissue of the carina, and a lower 15 W energy level in all position. Bronchoscopic and fluoroscopic visualisation was used to guide the electrode positioning (figure 2) throughout treatment. To maximise safety, the protocol mandated staged treatment (2 therapeutic procedures per patient) with the second bronchus being treated 30 days after the first. No special post procedure medications were required, and the use of tiotropium was stopped. Investigators were allowed to treat respiratory symptoms per standard-of-care and published guidelines, however, appropriate drug wash-out was required before follow-up testing.

(A and B) fluoroscopic view (A) and bronchoscopic view (B) of the electrode during the procedure. The electrode is indicated by the arrow.
Statistical methods
As this was a feasibility study, there was no primary study hypothesis with statistical inference. All p values were presented for informational purposes. According to the prespecified analysis plan, continuous data were summarised using means and SDs, or medians and quartiles, in the presence of non-normality. Categorical data were tabulated, with counts and percentages. All available data was summarised, with no imputation for missing data. Final analyses were conducted using SAS V.9.3 (SAS Institute, Cary, North Carolina, USA) by an independent statistical group (NAMSA, Minneapolis, Minnesota, USA).
Role of the funding source
The sponsor, D-JS, MM and CB designed the trial. Sites recruited patients and collected data on standardised case report forms. The sponsor managed the overall study and safety committees.
Results
Twelve patients were included in the 20 W cohort, and an additional 10 patients were included as part of the 15 W cohort at the same sites using the same study criteria (figure 3). Demographic and baseline characteristics were similar between the two cohorts, with the 15 W cohort enrolling patients with slightly worse baseline FEV1 (table 1). One-year follow-up data were available in 10 of 11 available patients in the 20 W cohort, and all 10 of the 15 W cohort. One patient in the 20 W cohort underwent coronary bypass surgery for a calcified coronary lesion during follow-up, and was withdrawn from the study by the site investigator before the second procedure. One patient in the 15 W cohort experienced a COPD exacerbation 10 days after the initial procedure and did not consent to the 2nd treatment, but continued follow-up.
Baseline characteristics. Data are mean (SD) unless stated otherwise

Clinical trial profile and patient flowchart.
The primary safety endpoint was achieved in 100% (11/11) in the 20 W cohort and 90% (9/10) in the 15 W cohort. The one subject that failed to meet the endpoint had a decrease from baseline in FEV1 at all follow-up time points, and a COPD exacerbation 1 day after the 2nd treatment that was reported as related to the lung denervation system and/or the bronchoscopy by the investigator.
No deaths occurred in this study. Seven postprocedure SAEs in the 30 days following treatment of either bronchus included COPD exacerbation (n=3), anaphylactic drug reaction, coronary artery bypass surgery, chest pain resulting in uneventful hospitalisation, and gastroparesis in a subject with a history of gastric issues. Four of these seven events occurred in a single patient. Nine longer-term SAEs out to 1 year included COPD exacerbation (n=5), pneumonia (n=2), influenza (H1N1) (n=1), and stomach cancer (n=1). Events were evenly distributed between both cohorts, with the majority of events reported during the periprocedural period (table 2).
Summary of adverse events through 1 year
The RF energy applied to the airway wall resulted in local asymptomatic airway blanching, which resolved at the 3-month follow-up bronchoscopy in all 10 of the 15 W patients, and in eight of the 11 W–20 W patients with long-term follow-up (figure 4). One patient had a superficial tissue defect at the first treatment site seen at 30 days, without any airway abnormalities on CT. A second patient had a small 1.5 mm perforation through the thin tissue of the main carina, also discovered at 30 days. A third patient had a superficial tissue defect at the initial treatment site just distal to the main carina, as well as a 4 mm granuloma at the second treatment site, with a 20% stenosis observed during the follow-up bronchoscopy at 90 days (figure 5). The granuloma was electively removed by cauterisation. No other treatments or interventions were performed, and all showed complete or active healing during follow-up visual inspection.

Bronchoscopic confirmation of airway healing after radiofrequency energy delivery: (A) Left main bronchus pretreatment. (B) During treatment. (C) Immediately post-treatment. (D) 3-month follow-up.

CT and bronchoscopic findings seen before procedural enhancements: (A and B) A 4 mm granuloma indicated by white arrows on both transverse CT and bronchoscopic images. (C and D) Superficial airway effect indicated by white arrow on the bronchoscopic image with normal transverse CT image. (E and F) A 1.5 mm perforation through carina indicated by white arrows on both coronal CT and bronchoscopic images.
Technical feasibility was 93%, with 39 of 42 procedures delivering full circumferential treatment (up to eight individual activations) to the target treatment site of the main bronchus. Two patients had only one bronchus treated as outlined above. Three patients had incomplete (non-circumferential) treatment: two had airway geometries that limited electrode/balloon contact with the right mainstem bronchus wall, and one had an intraprocedural anaphylactic reaction to diclofenac that prohibited full left bronchus treatment.
No device-related adverse events occurred during the procedure. Mean (SD) procedure time (min) for the 20 W cohort was 47.75±8.13 for the right and 56.10±12.79 for the left bronchi. Mean procedure time for the 15 W cohort was 42.10±8.23 for the right and 51.33±25.76 for the left.
The results of FEV1 (% relative change), FVC, cycle ergometry endurance and SGRQ at each time point assessed are shown in figure 6. At the higher 20 W power level, significant differences from baseline were seen at the following assessment time points: FVC at 90 days (p=0.016) and 270 days (p=0.036); cycle endurance at 180 days (p=0.03), and SGRQ at 90 days (p=0.042), 180 days (p=0.019), 270 days (p=0.008) and 1 year (p=0.011). Variability across time is noted, and most likely due to the small sample size. No statistically significant differences in any of the variables measured were seen at the 15 W power level.

Secondary efficacy endpoints. Data represented as mean. Error bars represent SEM. *p <0.05 compared with baseline; FEV1: forced expiratory volume in 1 s; Cycle Erg. Endurance, Cycle Ergometry Endurance; SGRQ, St. George's Respiratory Questionnaire.
FEV1 per cent change from baseline at 1 year was 11.6±32.3% vs 0.02±15.1% (p=0.324) in the 20 W and 15 W cohorts, respectively (table 3). Similarly, FVC had a positive absolute positive change in FVC at 1 year of 198±516 mL in the 20 W cohort vs 61±485 mL (p=0.256) in the 15 W cohort (figure 6).
Comparison of outcomes at 1 year for patients with follow-up testing
Study results for exercise endurance and HRQL (figure 6) show improvement from baseline for endurance cycle ergometry was 6.8±12.8 vs 2.6±8.7 min (p=0.277) at 1 year. A significant difference was observed in HRQL as assessed by the SGRQ: −11.1±9.1 vs −0.9±8.6 (p=0.044) in the 20 W and 15 W cohorts, respectively.
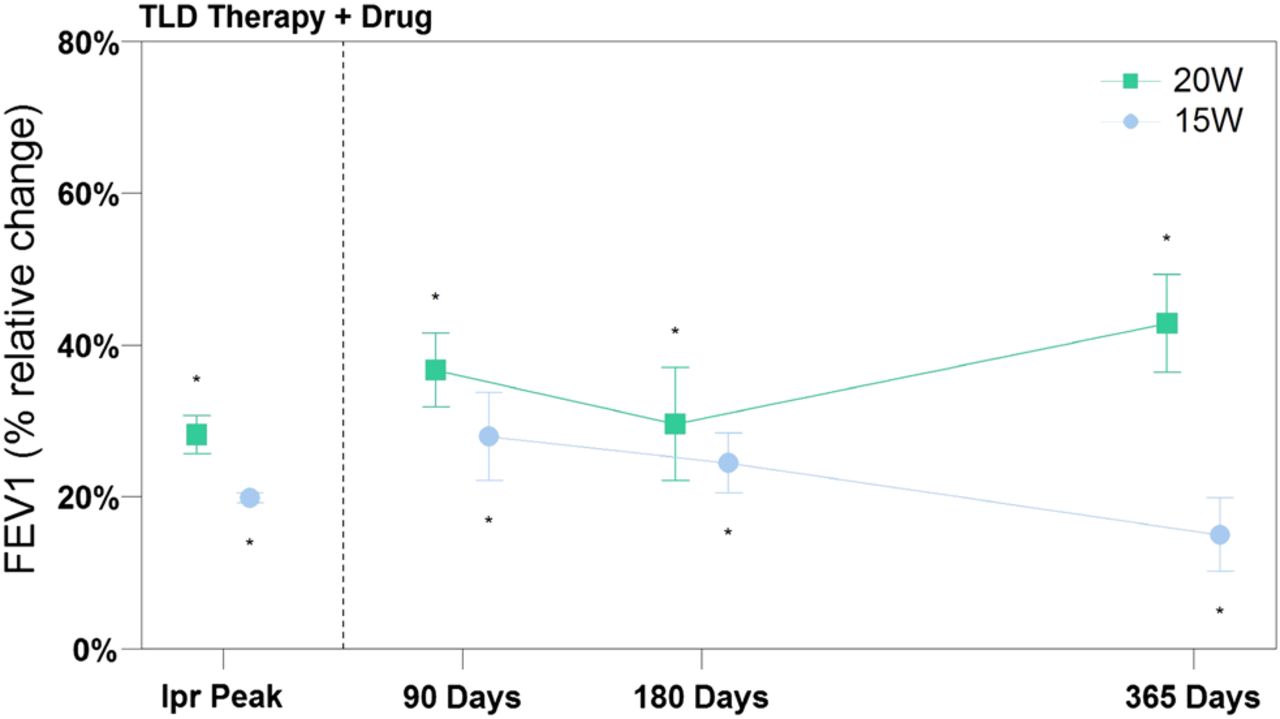
At 90, 180 and 365 days, the effect on FEV1 of TLD therapy plus 80 μg ipratropium bromide at peak was assessed. As illustrated in figure 7, the combination therapy might result in an increase in FEV1 over those seen with an inhaled anticholinergic drug alone. Significant differences between TLD +drug vs TLD alone were seen at each matching time points assessed for both energy cohorts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Efficacy measures of targeted lung denervation (TLD) therapy +inhaled ipratropium bromide. Data represented as mean. Error bars represent SEM. *p<0.05 compared with baseline. Follow-up data points were compared with results of TLD alone, and all were statistically significant.
Discussion
This first-in-human clinical trial evaluated the novel TLD therapy, designed to ablate the parasympathetic pulmonary nerves surrounding the main bronchi, thereby decreasing bronchomotor tone in patients with COPD. As a feasibility evaluation, this study included a drug ‘wash-out’ period to establish baseline values in order to understand the effect of this new therapy alone. This study demonstrated TLD to be feasible, safe and well tolerated. The primary endpoint was met in 95% of patients, and technical feasibility was 93%. A tendency toward improvements in lung function, exercise capacity, and HRQL were observed in the 20 W cohort with statistical significance achieved for FVC at 90 days (p=0.016) and 270 days (p=0.036); cycle endurance at 180 days (p=0.03) and SGRQ at 90 days (p=0.042), 180 days (p=0.019), 270 days (p=0.008) and 1 year (p=0.011). These improvements tended to be larger than those seen in the 15 W cohort with statistically significant difference in SGRQ at 1 year (p=0.044). Additionally, early data suggest an additive effect when TLD is combined with inhaled anticholinergic drugs.
Three procedural adjustments were made during the study in response to asymptomatic airway wall effects observed in three of 12 patients in the 20 W group. These effects were postulated to be attributable to a combination of energy delivery to the thin, thermally sensitive tissue of the main carina, imperfect balloon contact that limited surface cooling and, potentially, the higher energy level. Subsequent computer modelling and bench testing confirmed heat accumulation in the thin tissue of the carina, and post hoc analysis of procedural imaging indicated the balloon was not able to accommodate itself to sharply tapering or sudden geometric changes of the airway. As a result, the procedure was modified to more distal electrode placement away from the main carina, more detailed visual assessment of balloon contact before activation, and decrease in overall power to 15 W to further reduce the potential for undesired airway wall effects.
In this study, the 12 patients treated with 20 W tended to have greater changes from baseline in spirometry, exercise capacity and HRQL, when compared with the 10 patients treated with 15 W. The added effect in the 20 W group might be attributable to more effective denervation due to a deeper tissue effect at higher energy. On the other hand, the 20 W group patients were slightly more reversible to ipratropium at baseline, which theoretically, might mean that these patients are more sensitive to TLD therapy. However, we found no difference between the two groups in tiotropium through FEV1 levels. Apart from the airway effects observed, safety profiles for the two energy levels were similar. This might imply that future catheter designs that better accommodate human airway irregularities will ensure better surface cooling and, thus, allow higher energy levels to be used.
In this paper, we introduced TLD, a novel bronchoscopic treatment concept for symptomatic patients suffering from COPD. Based on the concept of ablating parasympathetic pulmonary nerves, TLD was shown to be feasible, safe and well tolerated. TLD has the potential to overcome many of the limitations of inhaled drugs for the treatment of COPD. First, TLD may eliminate inhaler compliance issues for the 63% of new tiotropium users who discontinue treatment after 1 year.23 Second, TLD would not be subject to the peak and trough variations seen with drugs.24 Third, TLD may eliminate variable regional drug delivery and deposition in patients with obstructive lung disease25 by ablating the nerves that travel throughout the bronchial tree independent of regional airflow obstruction. Fourth, by interfering with parasympathetic nerve-derived acetylcholine by two different mechanisms, the combination of TLD +inhaled anticholinergic drugs, as suggested in figure 7, may have a synergistic effect that results in a reduction in airway obstruction and mucus production, as well as inhibition of local airway inflammation induced by non-neural muscarinic action.8 ,26 Further investigation and progressive product development of this novel therapy is warranted, with focus on further refining energy delivery to ablate the nerves and optimise patient selection.
Acknowledgments
We would like to thank the investigators, their staff and teams, and the patients who consented to be part of this investigation. We acknowledge the work of the late Prof C Bolliger for his important contributions to this work, and to the field of pulmonary medicine.
References
Footnotes
-
Collaborators Panorama Medi Clinic & University of Stellenbosch, Cape Town, South Africa (Ethics Committee reference # M11/05/013): Chris Bolliger Coenraad Koegelenberg, Johan Theron, Firdows Noor, Elvis Irusen, Dorothy Steyn; University of Groningen, Groningen, The Netherlands (Ethics Committee reference # NL36608.042.11): Dirk-Jan Slebos, Nick Ten Hacken, Karin Klooster.
-
Contributors D-JS, KK, CFNK, JT, DS and CTB recruited and treated patients in this trial. MM is the inventor of the lung denervation system. AV was involved in data analysis and data interpretation. All authors helped to write the report. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit it for publication.
-
Funding Holaira, Inc., Minneapolis, Minnesota, USA.
-
Competing interests All clinical trial expenses were reimbursed by the study sponsor (Holaira, Inc.). D-JS is the overall study principal investigator. AV is the principle investigator of a second study conducted by the sponsor. MM is founder and chief technology officer of the study sponsor.
-
Ethics approval The medical ethics committees of the study sites involved, in South Africa and in The Netherlands.
-
Provenance and peer review Not commissioned; internally peer reviewed.
Linked Articles
- Airwaves