Article Text

Abstract
Background: Matrix metalloproteases (MMPs) are believed to be important in the pathogenesis of cigarette smoke-induced emphysema, but this hypothesis has only been proved in the mouse and its applicability to other species, particularly humans, is uncertain. The role of MMPs in smoke-induced small airway remodelling is unknown.
Methods: The effects of a dual MMP-9/MMP-12 inhibitor, AZ11557272, on the development of anatomical and functional changes of chronic obstructive pulmonary disease (COPD) in guinea pigs exposed daily to cigarette smoke for up to 6 months were examined.
Results: At all times, smoke-induced increases in lavage inflammatory cells, lavage desmosine (a marker of elastin breakdown) and serum tumour necrosis factor α (TNFα) were completely abolished by AZ11557272. At 6 months there was an increase in lung volumes and airspace size. AZ11557272 returned the pressure- volume curve to control levels, decreased smoke-induced increases in total lung capacity, residual volume and vital capacity by about 70%, and also reversed smoke-induced airspace enlargement by about 70%. There was a very strong correlation between surface to volume ratio and both lavage desmosine and serum TNFα levels. AZ11557272 protected against smoke-mediated increases in small airway wall thickness but did not prevent smoke-induced increases in mean pulmonary artery pressure.
Conclusions: An MMP-9/MMP-12 inhibitor can substantially ameliorate morphological emphysema, small airway remodelling and the functional consequences of these lesions in a non-murine species. These findings strengthen the idea that MMPs are important mediators of the anatomical changes behind COPD in humans, and suggest that MMP-9 and MMP-12 may be potential intervention targets.
- COPD, chronic obstructive pulmonary disease
- Lm, mean airspace size
- MMP, matrix metalloprotease
- Sv, surface to volume ratio
- TGFβ, transforming growth factor β
- TNFα, tumour necrosis factor α
Statistics from Altmetric.com
- COPD, chronic obstructive pulmonary disease
- Lm, mean airspace size
- MMP, matrix metalloprotease
- Sv, surface to volume ratio
- TGFβ, transforming growth factor β
- TNFα, tumour necrosis factor α
The pathogenesis of cigarette smoke-induced emphysema is an area of intense investigation and some confusion. Modern theories of the mechanisms behind emphysema originate from the reports of Gross and colleagues that instillation of proteolytic enzymes into the lungs of experimental animals produced emphysema,1 and the clinical observation that patients deficient in the serine elastase inhibitor α1-antitrypsin developed early onset emphysema, particularly if they smoked.2 These findings led to the protease-antiprotease hypothesis which states that cigarette smoke causes an influx of inflammatory cells into the lung and that these cells release proteases that overwhelm the local antiproteolytic defences, leading to matrix destruction and emphysema.
The protease-antiprotease hypothesis is generally accepted, but the exact cells and proteases that are the crucial mediators of matrix destruction remain to be determined. Recent data from mouse models suggest that neutrophils, neutrophil-derived serine proteases and matrix metalloproteases (MMPs) are all involved. Neutrophil elastase knockout mice are about 60% protected against increases in airspace size,3 whereas MMP-12 (macrophage metalloelastase) knockout mice are 100% protected.3,4 We have suggested that neutrophil recruitment is linked to MMP-12 by a mechanism in which MMP-12 causes release of active tumour necrosis factor α (TNFα), and TNFα subsequently elicits a neutrophil influx into the lung with matrix destruction mediated by neutrophil-derived proteases, primarily neutrophil elastase.5
Although there is a widespread belief that metalloproteases also play a role in the pathogenesis of emphysema in humans, virtually all published data come from mouse models and the role of metalloproteases in this context in other species is unknown. This is an important issue because differences in the types of metalloproteases found in different species, as well as differing roles for cognate metalloproteases across species, might render observations in mice of little relevance to humans.
Small airway remodelling is a separate but equally important cause of airflow obstruction in humans exposed to cigarette smoke. Very little is known about the pathogenesis of small airway remodelling; the usual assumption is that it is secondary to smoke-induced inflammation, although there is no proof of this idea (see later).6
To evaluate a potential role for metalloproteases in the development of COPD in a non-murine species, and to determine what specific metalloproteases might be involved, we examined the effects of AZ11557272, a dual MMP-9/MMP-12 inhibitor with high selectivity over other MMPs, in guinea pigs exposed to smoke for up to 6 months.
METHODS
Smoke exposure and treatment with AZ11557272
Groups of 6–10 female Hartley strain guinea pigs (Charles River, Montreal, Quebec) weighing approximately 350 g were exposed to the smoke or air (control) of seven University of Kentucky 2R1 cigarettes 5 days per week for up to 6 months. The smoking apparatus and details of the exposure methods have been described previously.7 Additional groups were treated with AZ11557272 by gavage 1 h before smoke at a dose of 100 mg/kg. AZ11557272 is an orally bioavailable low molecular weight non-hydroxamate MMP-9/MMP-12 inhibitor developed by AstraZeneca. It has minimal action against other MMPs: >3 orders of magnitude selectivity for MMP-9 and MMP-12 over TACE and MMP-1, 2–3 orders of magnitude selectivity over most other MMPs including MMP-14, and <1 order of magnitude selectivity over MMP-8. The dose of 100 mg/kg was selected on the basis of range finding experiments to determine a dose that completely suppressed smoke-induced increases in lavage neutrophils 24 h after a single smoke exposure (see results and figures in online supplement available at http://thorax.bmj.com/supplemental). Using the regimen described above, measured blood levels of AZ11557272 were 50 times the IC50 for MMP-12 and 60 times for MMP-9 at 24 h after dosing. The exact numbers of animals analysed at 1 month were 6 control, 10 smoke-exposed and 10 exposed to AZ11557272; at 3 months 8, 8, and 10, and at 6 months 6, 10, and 8, respectively. All procedures were approved by the University of British Columbia Animal Care Committee.
Collection of lavage fluid and lung tissue
At 1, 3 and 6 months of smoking, animals were killed by urethane anaesthesia overdose followed by exsanguination. Blood was collected for serum TNFα measurements. The left lower lobe was lavaged five times with 2.5 ml saline, the cells were collected by centrifugation and total cell counts and differential counts were performed. For the animals exposed for 6 months, the lavaged lobe was then inflated with agarose at a pressure of 25 cm H2O, fixed in formalin for 24 h, and embedded and sectioned for histological examination. The left upper lobe was snap frozen and used for whole lung Western blot analysis for metalloprotease levels. The right lower lobe was lavaged with cold distilled water and the lavageate was used for desmosine analysis.
Pulmonary and cardiac function tests
Pulmonary and cardiac function tests were performed at 6 months before animal sacrifice using methods described previously.7,8
Morphometric analysis: airspace size
Morphometric analysis followed the approach of Thurlbeck.9 The lung lobes were sectioned serially in a sagittal fashion. Using a random selection process, slices were selected and submitted for paraffin embedding and sectioning at 5 μm thickness followed by haematoxylin and eosin staining. Using a random selection process, 15 fields were photographed at 10× magnification. The ImagePro system with a grid of 130 lines and 250 points with a line length of 2730 μm was used to count the numbers of intercepts, and the mean airspace size (Lm) and surface to volume ratio (Sv) were calculated.
Morphometric analysis: small airway wall thickness
Five animals from each group were examined. All membranous bronchioles were analysed regardless of orientation. To measure wall thickness, a line was cast across the largest diameter perpendicular to the longitudinal axis of the airway. The external bronchiolar diameter was measured as the distance between adventitial borders, and the internal bronchiolar diameter was measured as the distance between basement membranes. Wall thickness was calculated as the difference between the external and internal bronchiolar diameters.
Lavage desmosine analysis
Desmosine, a marker of elastin breakdown, was measured by high performance liquid chromatography as described previously.10
Serum TNFα levels
Serum TNFα levels were measured using the L929 cell assay as previously described.5
Western blots for whole lung metalloprotease levels
These were performed using methods described previously.11 The catalytic domain of guinea pig MMP-12 was cloned at AstraZeneca (Lund) by PCR using first strand cDNA from guinea pig lung (Genelink Cat no 10-2107-05) and primers 5′cattcatatgcctcgatgtggagtgcccgat and 5′attgcggccgcctaaatttcgtaagcagcttgaat containing restriction sites for NdeI and NotI. A pT7-based expression vector was used and insoluble protein was expressed in E coli. Active protein was obtained by protein refolding and was used to generate a polyclonal rabbit antibody.
For detection of MMP-12, the membranes were incubated in 1:3000 anti-guinea pig MMP-12. For detection of MMP-1, -2, and -9, antibodies were purchased from Lab Vision Corporation (Fremont, California, USA) and used at 1:1000 (anti MMP-9), 1:300 (anti MMP-1) and 1:500 (anti MMP-2). The second antibody was horseradish peroxidase-conjugated goat anti-rabbit IgG from Santa Cruz. Detection was by chemiluminescence and densitometry was performed on the films.
Statistical analysis
Differences between treatment groups were analysed by analysis of variance (ANOVA). For the pressure-volume curves, repeated measures ANOVA was used. Using the entire set of animals as one group, Pearson correlations were performed between measures of airspace size or surface to volume ratio and lavage cell counts, desmosine and serum TNFα, and corrections for multiple comparisons were applied as appropriate.
For analysis of airway remodelling we constructed probability plots for internal bronchial diameter and wall thickness within each animal and found that the data was normally distributed, thus allowing us to use mean animal values. Comparisons of the internal bronchiolar diameters of the three treatment groups were not significantly different, indicating that we had sampled equivalently sized airways in all groups. Using analysis of variance, we then tested for differences in airway wall thickness between groups using individual animal values.
RESULTS
Graphs showing the dose range finding data at 24 h after a single smoke exposure are shown in figs 1–3 in the online supplement available at http://thorax.bmj.com/supplemental. AZ11557272 suppressed macrophage influx and serum TNFα at levels as low as 3 mg/kg, a dose that produces a blood level about 10 times the IC50 at 24 h after exposure, but suppression of neutrophil influx required a dose of 30 mg/kg. Because, in acute experiments, lavage neutrophil numbers correlate with lavage desmosine, a measure of elastin breakdown,12 we selected a dose of 100 mg/kg to ensure complete neutrophil suppression in the chronic exposure model (see later).

Lavage inflammatory cells. Smoke exposure increased both neutrophil (PMN) and macrophage (MAC) numbers at all time periods (3 months not shown), and these increases were abolished by treatment with AZ11557272. In this and all other figures, the numbers of animals at 1 month are 6 controls, 10 smoke-exposed and 10 exposed to AZ11557272; at 3 months 8, 8 and 10; and at 6 months 6, 10 and 8, respectively. Values are mean (SD). *p⩽0.001 compared with controls.
Figure 1 shows lavage neutrophil and macrophage counts at 1 and 6 months. Smoke exposure produced marked increases (4–10-fold) in lavage neutrophils and roughly doubled lavage macrophage numbers at all time periods (3 month time period not shown); AZ11557272 completely abolished these increases.
Figure 2 shows lavage desmosine and serum TNFα levels. Desmosine levels were increased 2–3 times in the smokers, and this increase was completely prevented by treatment with AZ11557272 at all time periods (3 month time period not shown). Smoke caused a 2–2.5-fold increase in serum TNFα and this increase was abolished by treatment with AZ11557272 (3 month time period not shown).

Lavage desmosine and serum tumour necrosis factor (TNF) α levels at 1 and 6 months. Desmosine, a marker of elastin breakdown, was increased by smoke exposure at all time points; AZ11557272 completely prevented this effect. TNFα levels were increased by cigarette smoke exposure at all time points and AZ11557272 completely prevented this effect. Values are mean (SD). *p⩽0.001 compared with controls.
Figure 3 shows whole lung Western blots against MMP-9 and MMP-12. Levels of MMP-9 were increased about 75% at all time periods; for MMP-12 there was a much more marked increase (more than twofold) at 1 month and smaller increases at 3 and 6 months. Treatment with AZ11557272 reduced the smoke-mediated increase in both MMPs. Western blots for MMP-1 and MMP-2 showed only small and mostly non-significant changes, but with a general trend toward increased levels in the smokers and decreased levels in animals treated with AZ11557272 (data not shown). Because these are whole lung samples, the increases in specific anatomical compartments such as macrophages may be considerably greater.
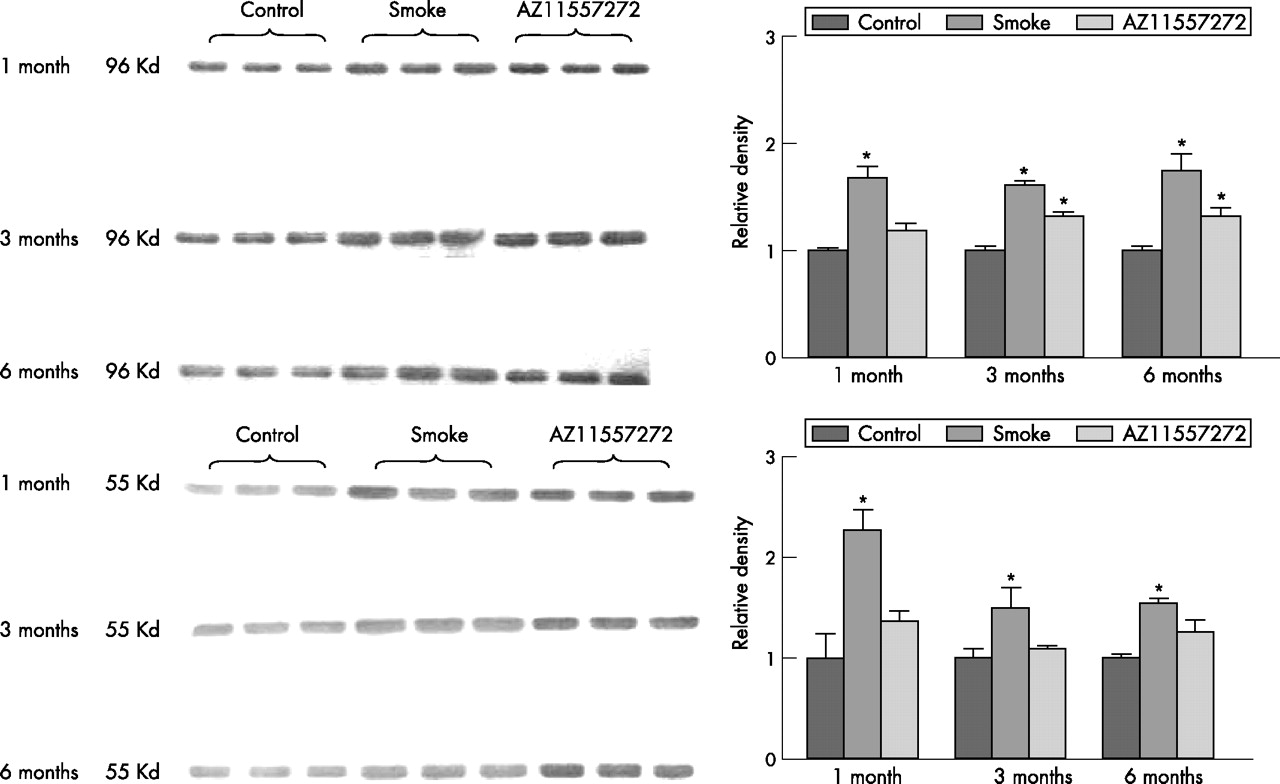
Top: Whole lung Western blot analysis of matrix metalloprotease (MMP)-9 levels on three animals from each treatment group. Smoke exposure significantly increased whole lung MMP-9 levels at all time points and AZ11557272 partially prevented this effect. Values are mean (SD). *p<0.01 control vs smoke-exposed animals; p<0.05 control vs AZ11557272-exposed animals. Bottom: Whole lung Western blot analysis of MMP-12 levels on three animals from each treatment group. Smoke markedly increased whole lung MMP-12 at 1 month and produced lesser but significant increases at 3 and 6 months. AZ11557272 completely prevented this effect. Values are mean (SD). *p<0.01 control vs smoke-exposed animals.
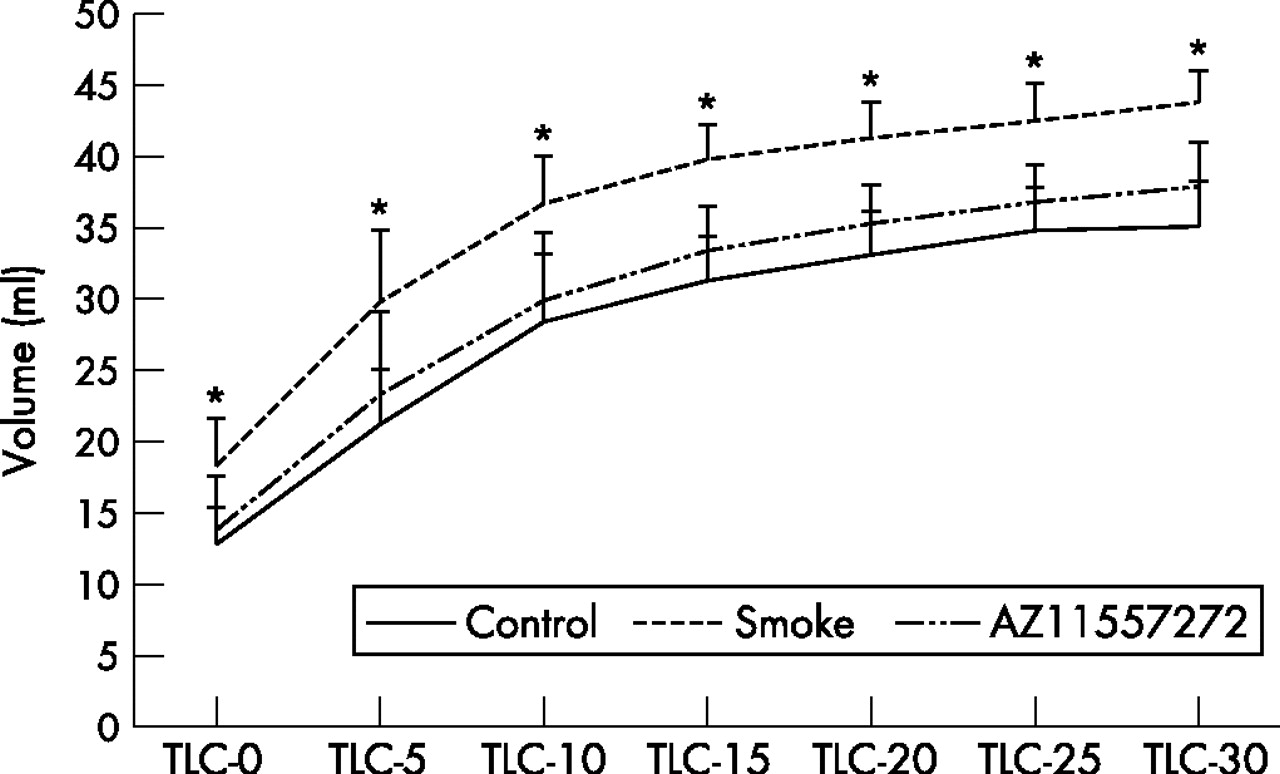
Figure 4 shows the pressure-volume curves for the animals exposed to smoke for 6 months. At all distending pressures, lung volumes were significantly greater in smoke-exposed animals than in controls. The pressure-volume curves from animals treated with AZ11557272 were not significantly different from the controls. Pulmonary function measurements are shown in fig 4 in the supplementary file available online at http://thorax.bmj.com/supplemental. Total lung capacity, vital capacity and residual volume were all increased by a small but significant amount in the smoke-exposed animals and AZ11557272 provided about 70% protection. Functional residual capacity was also increased in the smokers and decreased by AZ11557272, but the differences were not statistically significant. No differences were seen for expiratory reserve volume (ERV).

Pressure-volume curves after 6 months of smoke exposure. Mice exposed to smoke had significantly increased total lung capacity (TLC) at all inflating pressures. AZ11557272 completely prevented this effect. Values are mean (SD). *p⩽0.01 control vs smoke-exposed animals; p⩽0.01 AZ11557272-treated vs smoke-exposed animals (except p<0.03 at baseline and 5 cm H2O pressure). Analysis by repeated measure ANOVA.
Pulmonary artery pressures are shown in fig 5 in the online supplement available at http://thorax.bmj.com.supplemental. Mean pulmonary artery pressure was increased about 25% in the smokers and AZ11557272 had no protective effect.

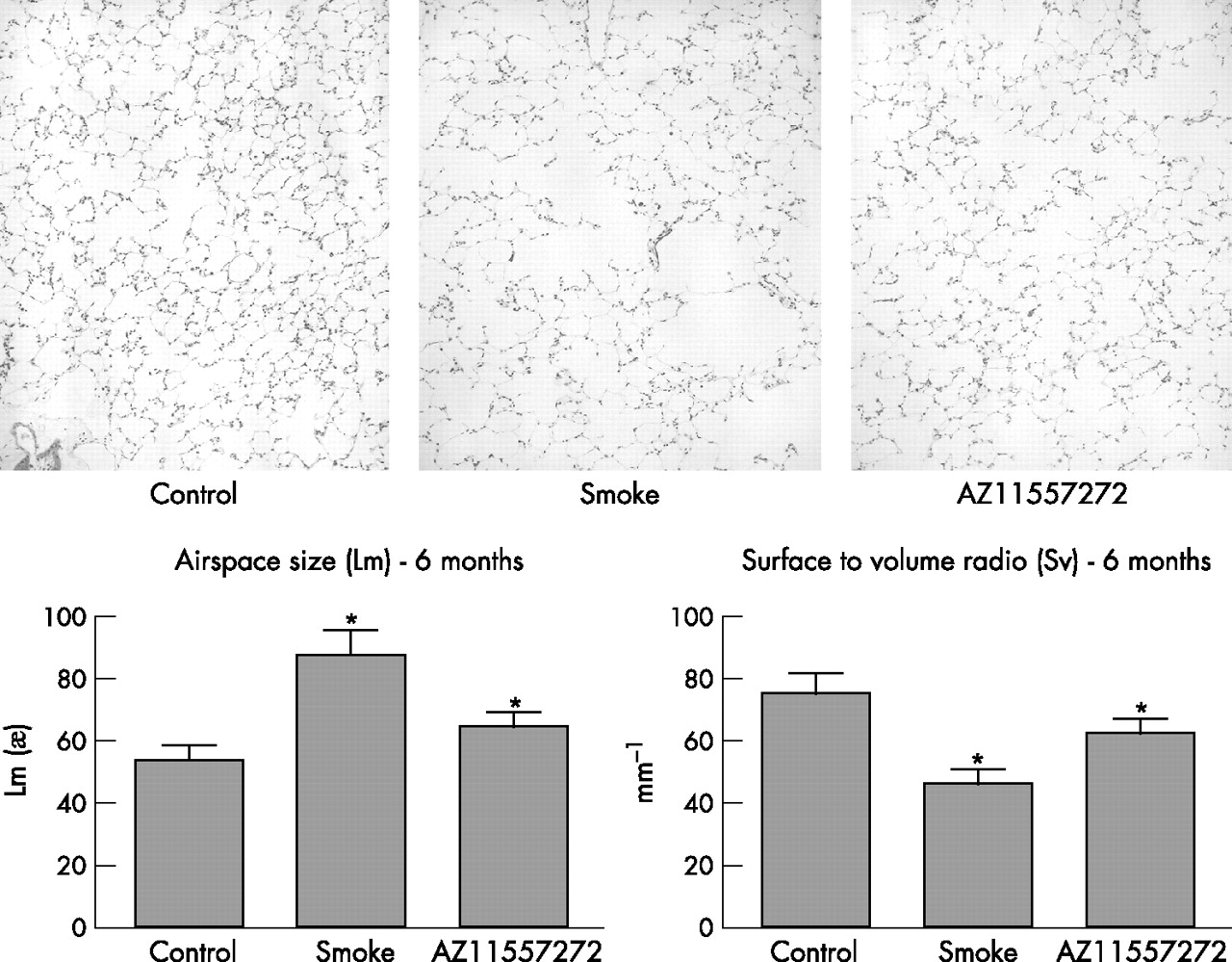
Top: Representative images of control, smoke-exposed and AZ11557272-treated animals at 6 months showing clear increases in airspace size with smoke and protection with AZ11557272. Lower left: airspace size measured as Lm. Smoke increased Lm and AZ11557272 provided 68% protection. Lower right: Surface to volume ratio (Sv): smoke decreased Sv and AZ11557272 provided 57% protection. Values are mean (SD). *p<0.0001 control vs smoke-exposed animals; p<0.001 control vs AZ11557272-treated group.
Figure 5 shows representative illustrations of airspace size in the control, smoke-exposed and AZ11557272-treated animals at 6 months. The protective effect of AZ11557272 is evident on visual inspection. The graphs in fig 5 show that mean airspace size was increased by 62% in the smokers and only 20% in the AZ11557272-treated animals; that is, there was 68% protection against emphysema. Similarly, the surface to volume ratio was decreased by 40% in the smokers and only by 17% in the AZ11557272-treated animals, indicating 57% protection.
Figure 6 shows representative images of small airways (membranous bronchioles) from the three treatment groups and a graphical representation of mean airway wall thickness. Mean wall thickness was increased by 42% in the smokers (p = 0.02 vs controls), and this effect was completely prevented by AZ11557272.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Top: Representative images of control, smoke-exposed and AZ11557272-treated animals at 6 months showing a representative membranous bronchiole. Smoke increased bronchiolar wall thickness (that is, produced small airway remodelling) and this was prevented by AZ11557272. Graphical representation of data from all the animals is shown below. Values are mean (SD). *p<0.02 vs controls.
Table 1 shows correlations between surface to volume ratio and other measures at 6 months, and also correlations between serum TNFα levels and other measures at 1 and 3 months. These calculations were performed over the entire set of animals. In univariate analyses, at 6 months there was a strong inverse correlation between surface to volume ratio and lavage desmosine levels (r = −0.82, p<0.0001), serum TNFα (r = −0.87, p<0.0001), and less strong but still highly significant correlations with inflammatory cell numbers in lavage fluid. Almost identical positive correlations were seen with airspace size and desmosine, serum TNFα levels and inflammatory cell numbers (data not shown). At all time periods, serum TNFα levels showed strong correlations with desmosine levels and with numbers of lavage inflammatory cells. Desmosine levels also showed somewhat less strong but still quite significant correlations with numbers of inflammatory cells.
Correlations between lavage desmosine levels, inflammatory cell numbers, serum TNFα levels and airspace enlargement
DISCUSSION
We have shown that an MMP-9/MMP-12 inhibitor with good selectivity over most other MMPs ameliorates emphysema and prevents smoke-induced increases in small airway wall thickness in a guinea pig model. One possible criticism of this study is the relatively high dose of compound used. In our acute range-finding model which uses a single smoke exposure with sacrifice at 24 h, AZ11557272 suppressed macrophage numbers and increased serum TNFα levels at a relatively low dose. However, because neutrophil numbers but not macrophage numbers in lavage correlate with increased levels of lavage desmosine in the acute smoke exposure model,12 we picked a considerably higher dose in order to suppress neutrophil influx (this phenomenon changes with chronic exposure models where, as here, there is a correlation between lavage desmosine levels and lavage macrophages as well). One could argue that the dose used in our chronic study was suppressing many MMPs but, given the relative selectivity data described in the Methods section, the blood levels of AZ11557272 achieved should not have suppressed most MMPs other than MMP-9 and MMP-12. In particular, it will not suppress MMP-1 which has been implicated in human emphysema (see below), but probably suppressed MMP-8 (see below). These observations also suggest that the exact roles played by MMP-9 and MMP-12 (and other MMPs) in the inflammatory response to smoke as well as the genesis of emphysema and small airway remodelling are complex.
Although there has been evidence for a number of years that proteases other than serine proteases are present in increased amounts in the lungs of smokers (indeed, even in 1983, Janoff et al13 noted that 50% of the elastase activity in lavage fluid from cigarette smokers was metalloprotease-derived), the central role of metalloproteases was really brought to the fore by the observation that mice lacking MMP-12 were completely protected against smoke-induced emphysema.4 Subsequently it has been shown that different synthetic broad spectrum metalloprotease inhibitors provide 75–100% protection in mice14,15 (see below and table 2).
Protection against smoke-mediated increases in airspace size
These observations have led to the assumption that metalloproteases are also crucial effectors of emphysema in humans. MMP-1, MMP-2, MMP-9, MMP-1416–19 and, in some but not all reports, MMP-1218,20–23 can be found in greater amounts in sputum or lavage fluid or cultured alveolar macrophage supernatant from smokers with emphysema than in those without emphysema. Based on data from cultured macrophages, it has been proposed that it is specifically MMP-9 that is the crucial MMP in humans.24,25 Data on metalloprotease gene promoter region polymorphisms have also suggested a role for MMP-1, MMP-12 and possibly MMP-9 in COPD.26,27 However, the actual role of metalloproteases in general and of specific types of metalloproteases in humans is unknown.
A major complication in extrapolating from mouse models is the finding that different species produce different types of metalloproteases or relatively greater or lesser amounts of a given metalloprotease, and that these metalloproteases sometimes have different functions. For example, mice do not have a true MMP-1 (a collagenase), and mice and humans differ in some models in the metalloproteases that break down elastin, an important part of the pathogenesis of emphysema. Thus, in explanted aortic fragments, both murine and human macrophages use basal levels of MMP-12 to degrade elastin, but human macrophages stimulated with plasminogen switch on production of MMP-7 which becomes the major elastolytic enzyme in this model, whereas this phenomenon does not occur with murine macrophages.28 In addition, despite evidence that smoke-stimulated human alveolar macrophages produce large amounts of MMP-9,24 in the mouse knockout model MMP-9 does not prevent the development of emphysema (S D Shapiro, personal communication).
Our observations help bridge the gap between mouse models and humans by showing that metalloproteases are involved in the pathogenesis of emphysema in a non-murine species and, further, that MMP-9 and/or MMP-12 are the most important types of metalloproteases involved. To our knowledge, the only previous study of a metalloprotease inhibitor in smoke-exposed guinea pigs was that of Selman et al29 who used CP-471 474, a broad spectrum inhibitor. They found 100% protection against increases in airspace size at 2 months but only 30% at 4 months, a result that implies that metalloprotease inhibition does not provide significant protection against emphysema in this species, contrary to what we have observed.
There is clear evidence in our model that smoke exposure produces increased levels of whole lung MMP-12 and MMP-9. The latter change must be interpreted with caution since much of the MMP-9 may be derived from smoke-evoked neutrophils, but the data on MMP-12 imply that this metalloprotease may be increased by smoke exposure in multiple species. Presumably the major source of MMP-12 in the guinea pigs is alveolar macrophages, but no direct data exist on this issue. By the same token, however, our data imply that MMP-9 and/or MMP-12 account for only about 70% of airspace enlargement in guinea pigs. The source of the remaining 30% of airspace enlargement is unclear. Metalloproteases other than MMP-9 and MMP-12 (particularly MMP-1 which has been shown to produce emphysema when expressed in transgenic mice30 and which is not inhibited by AZ11557272), or completely different types of proteases, or a protease-independent mechanism are all possibilities. As noted above, our protocol probably suppressed MMP-8 activity. Betsuyaku et al31 have reported parallel increases in MMP-8 and MMP-9 as a consequence of neutrophil influx in the lungs of patients with emphysema, but the actual role (if any) of MMP-8 in human emphysema is not known. It is possible, but appears unlikely, that suppression of MMP-8, a collagenase, is affecting our results in a major fashion.
One of the processes that has been proposed as a cause of emphysema is smoke-induced apoptosis of endothelial and/or epithelial cells. Chemical inhibition of vascular endothelial growth factor receptor 232 or intratracheal instillation of active caspase-3 or nodularin33 produces extremely rapid airspace enlargement in the absence of an inflammatory cell influx. In humans, some studies have found increased numbers of apoptotic cells in emphysematous lungs compared with non-emphysematous lungs.34–36 However, at least with caspase-3 or nodularin, the airspace enlargement is rapidly reversible, but this is not the case in our cigarette smoke models.37 Furthermore, although there was increased lavage elastolytic activity in the caspase-3 instillation model,33 the activity was maximal at pH 5.5 and the authors proposed that this represented activation of one or more cathepsins.
While our current observations do not address the issue of apoptosis directly, they do suggest that it is not the most important cause of airspace enlargement. Rather, we found a very strong correlation between measures of surface to volume ratio or airspace size and levels of lavage desmosine, a marker of elastin breakdown. Although there is a general assumption that markers of matrix breakdown should correlate with the severity of emphysema, this is the first demonstration that such a correlation exists, and the fact that AZ11557272 prevented increases in desmosine levels indicates that metalloproteases are—either directly or indirectly—driving matrix breakdown. These findings complement the recent report of Houghton et al38 that elastin fragments generated by proteolytic activity in the smoke-exposed lung drive monocyte recruitment and disease progression, and that MMP-12 is crucial to this process. These observations would fit with the comment made by Imai et al36 that a correlation between severity of emphysema and numbers of apoptotic cells does not necessarily imply that apoptosis causes emphysema, but could simply mean that damage to or loss of matrix leads to apoptosis of the overlying epithelial cells, a well known phenomenon.
In addition, in our model there were strong correlations of surface to volume ratio and serum TNFα levels, and of desmosine levels, serum TNFα levels and numbers of lavage neutrophils and macrophages. The conclusion from this whole set of observations is that the classic inflammation-driven model of matrix breakdown is the major driving force behind matrix destruction in emphysema.
In this context, it is useful to compare the levels of protection we achieved with AZ11557272 and other antiproteases and anti-inflammatory agents reported in the literature. Since the published data are usually expressed in terms of airspace size (Lm), we have used that measure in compiling table 2. In general, MMP inhibitors or MMP knockout (in the case of MMP-12) appear to provide somewhat more protection than serine elastase inhibitors or knockout of neutrophil elastase, but there is still considerable variation from study to study and overlap among the groups. Comparable levels of protection are achieved using anti-inflammatory agents (roflumilast, simva-statin) or TNFα receptor knockout mice. While these data suggest that enzyme inhibitors or anti-inflammatory agents could produce quite significant protection, it should be noted that, in almost all the reported protocols, the drug treatment or target gene deletion is present from the first day of exposure to smoke. This is quite different from the situation in humans where drug treatment will probably be a relatively late intervention with the intent to prevent progression of established emphysema or airway disease.
Although it is now accepted that small airway remodelling is an important contributor to airflow obstruction,45 little is known about the pathogenesis of this lesion. The usual assumption is that small airway remodelling, like emphysema, is an effect of smoke-induced inflammation, but this assumption is more of an afterthought than an idea with evidence behind it.6 We have suggested elsewhere that, in fact, small airway remodelling may be caused by direct smoke effects that lead to the induction of profibrotic growth factors, particularly transforming growth factor β (TGFβ), in the airway walls.46 The fact that AZ11557272 protects against small airway remodelling is surprising and suggests that either smoke-induced inflammation is actually important in remodelling or, alternatively, that induction of MMPs in the airway wall itself plays a major role. This latter idea is attractive since MMPs not only reorganise matrix but are now known to release growth factors including insulin-like growth factors and TGFβ from matrix, effectively converting them to active forms.47 MMP-9 in particular has been shown to activate TGFβ in some model systems,48 and intratracheal instillation of TGFβ to mice has been shown to produce increased collagen in small airway walls without evoking an inflammatory response.49
Another unexpected finding in our model is the total lack of effect of AZ11557272 on smoke-induced pulmonary hypertension. In hypoxia50 or monocrotaline51 model pulmonary hypertension systems, administration of a broad-spectrum MMP inhibitor in vivo reduces vascular remodelling. Batimastat, a non-selective MMP inhibitor which functions mainly against collagenases, markedly reduced collagen breakdown and collagenolytic activity in the pulmonary arteries and reduced hypoxia-induced pulmonary hypertension.50 Inhibition of MMPs by GM6001, a very broad-spectrum metalloprotease inhibitor, in a cultured pulmonary artery system produced suppression of monocrotaline-induced cell proliferation and matrix production, followed by regression of vascular wall thickening.51 The inhibitor appeared to have its greatest effect on MMP-2, although activity of other MMPs was also decreased.
Given the species to species differences in the role of specific metalloproteases noted above, one needs to be cautious in drawing conclusions but it is possible that, in the guinea pig, MMP-9 and MMP-12 are not crucial to the development of smoke-induced vascular remodelling and pulmonary hypertension.
In summary, we have shown for the first time that MMPs have a role in the development of emphysema in a species other than the mouse and, more specifically, that MMP-9 and/or MMP-12 are the major players in this process. We have also shown that MMPs are important in the pathogenesis of small airway remodelling. Previously published data in the mouse using broad-spectrum MMP inhibitors14,15 have indicated a potential role for such compounds as a therapeutic approach to emphysema. Our findings extend these results and are of particular interest because of the known role of MMP-12 in driving emphysema in the mouse and the belief that MMP-9, and possibly MMP-12, are important in emphysema in humans. These observations thus provide support for the idea that MMPs also are likely to be important in the pathogenesis of emphysema and small airway remodelling in humans, and suggest some specific targets of antiprotease therapy.
REFERENCES
Supplementary materials
Files in this Data Supplement:
Footnotes
-
Published Online First 20 February 2007
-
P-OO and KT are employees of AstraZeneca and were involved in the design of the study, along with AC and JLW. AZ1557272 was developed at AstraZeneca R&D, Lund, Sweden and analyses of blood levels of AZ11557272 were performed in Lund. Guinea pig MMP-12 was cloned and an antibody developed in Lund. All other parts of the study and all other analyses were done in Vancouver. These four authors participated in the writing of this manuscript.
-
Supported by grant MOP 42539 from the Canadian Institutes of Health Research and a grant from AstraZeneca R&D, Lund, Sweden
Linked Articles
- Airwaves