Article Text

Abstract
Background: Apoptosis of alveolar septal cells has been linked to emphysema formation. Nitrogen dioxide, a component of cigarette smoke, has been shown to induce alveolar epithelial cell apoptosis in vitro. It is hypothesised that exposure of rats to nitrogen dioxide may result in increased alveolar septal cell apoptosis in vivo with ensuing emphysema—that is, airspace enlargement and loss of alveolar walls.
Methods: Fischer 344 rats were exposed to 10 ppm nitrogen dioxide for 3, 7, 21 days or 21 days followed by 28 days at room air. Age-matched control rats were exposed to room air for 3, 21 or 49 days. Lungs fixed at 20 cm fluid column, embedded in paraffin wax, glycol methacrylate and araldite, were analysed by design-based stereology. Alveolar septal cell apoptosis (transferase dUTP nick end labelling assay, active caspase 3) and proliferation (Ki-67), airspace enlargement, total alveolar surface area, and absolute alveolar septal volume as well as the ultrastructural composition of the alveolar wall were quantified.
Results: Nitrogen dioxide resulted in an eightfold increase in alveolar septal cell apoptosis at day 3 and a 14-fold increase in proliferation compared with age-matched controls. Airspace enlargement, indicated by a 20% increase in mean airspace chord length, was evident by day 7 but was not associated with loss of alveolar walls. By contrast, nitrogen dioxide resulted in an increase in the total surface area and absolute volume of alveolar walls comprising all compartments. The ratio of collagen to elastin, however, was reduced at day 21. Lungs exposed to nitrogen dioxide for 21 days exhibited quantitative structural characteristics as seen in control lungs on day 49.
Conclusions: Nitrogen dioxide exposure of rats results in increased alveolar septal cell turnover leading to accelerated lung growth, which is associated with an imbalance in the relative composition of the extracellular matrix, but fails to induce emphysema.
- COPD, chronic obstructive pulmonary disease
- TUNEL, transferase dUTP nick end labelling
Statistics from Altmetric.com
Emphysema is defined as the “abnormal permanent enlargement of the airspaces distal to the terminal bronchioles, accompanied by destruction of their walls”.1 The pathogenetic pathways leading to emphysema are still a matter of debate. At present there are two major concepts which, although not necessarily contradictory, have clearly different perspectives of the sequence of pathogenetic events. The classical concept focuses on the inflammation-associated imbalance of proteases and antiproteases. This is thought to be the primary cause of degradation of matrix components, which subsequently results in the loss of alveolar septal walls.2 On the basis of recent studies, an alternative concept has been developed, which focuses on the apoptosis of endothelial and/or alveolar epithelial cells as being the primary event in the pathogenesis of emphysema.3,4
Experimental induction of the apoptosis of pulmonary endothelial cells—for example, by blockade of vascular endothelial growth factor receptor 25 or lung-targeted inactivation of vascular endothelial growth factor,6 or of alveolar epithelial cells by transfer of active caspase 37 or cigarette smoke exposure8—were shown to result in airspace enlargement. Additional evidence for the importance of apoptosis in the pathogenesis of emphysema comes from several human studies, which showed increased levels of apoptotic alveolar septal cells in patients with emphysema.9–11 Notably, programmed cell death appeared to be predominant in alveolar epithelial cells of patients with emphysema, whereas apoptosis of endothelial cells was observed less frequently.10,11
The major risk factors for chronic obstructive pulmonary disease (COPD), which comprises chronic bronchitis and emphysema, are cigarette smoking and indoor air pollution from burning fuels.12 Until now, however, studies of experimental animal models based on induction of emphysema by inhalation of, for example, cigarette smoke have shown contradictory results regarding the involvement of apoptosis of alveolar septal cells in the development of emphysema.8,13
Nitrogen dioxide is an important component of cigarette smoke with reported emissions of up to 0.73 mg nitrogen dioxide per cigarette.14 Exposure of rats to nitrogen dioxide results in airway inflammation which, as in human COPD, is dominated by alveolar macrophages and neutrophilic granulocytes.15,16 Further, nitrogen dioxide induces apoptosis of alveolar epithelial cells in vitro with actively dividing cells and cells at the leading edge of a wound being particularly susceptible.17 As short-term exposure of rats to nitrogen dioxide in vivo results in an initial phase of alveolar epithelial injury followed by a phase of epithelial repair,18 we hypothesised that the exposure of rats to a nitrogen dioxide-containing atmosphere may result in an increase in apoptosis of alveolar epithelial cells in vivo with ensuing emphysema.
MATERIALS AND METHODS
Experimental protocol
Forty-two male Fischer 344 rats (Charles River, Sulzfeld, Germany), 181 (5) g body weight, 8–10 weeks of age, were divided into seven groups. Three groups (n = 6 each) were exposed for 3, 7 or 21 days (23 h/day, 7 days/week) to an atmosphere containing 10 ppm nitrogen dioxide (Messer Griesheim, Duisburg, Germany) as described elsewhere.19 An additional group (n = 6) was exposed to nitrogen dioxide for 21 days followed by exposure to room air for 28 days. The concentration of nitrogen dioxide was monitored by a nitrogen dioxide-sensitive electrochemical element (ECS 102-1, MP Sensor System, Munich, Germany). Age-matched control groups (n = 4–8) were exposed to room air for 3, 21 or 49 days. Mean food consumption per day was monitored. Body weights were measured at the end of each exposure period. The animal experiments were approved by the regional government (Regierungspräsidium Giessen, Dezernat V.54, Giessen, Germany).
Fixation and tissue sampling
Immediately after exposure, rats were killed. Lungs were fixed by airway instillation with 4% phosphate-buffered paraformaldehyde at a pressure of 20 cm fluid column. After overnight immersion into fresh fixative, lung volume was determined by fluid displacement, and two to three fractions of lung slices were collected by systematic uniform random sampling as described previously.20
Embedding in glycol methacrylate and araldite
One fraction of lung slices and a systematic uniform random subsample of tissue blocks from the second fraction of slices were post-fixed in 1% glutardialdehyde, 1% paraformaldehyde in 0.1 M sodium cacodylate buffer followed by osmication and en bloc staining with aqueous uranyl acetate before dehydration. Complete lung slices were embedded in glycol methacrylate for stereological analysis of emphysema. Tissue blocks subsampled from the second fraction of slices of lungs exposed for 3–21 days to nitrogen dioxide or room air were embedded in araldite for quantification of the components of the alveolar wall by means of transmission electron microscopy.
Embedding in paraffin wax
The third fraction of lung slices was dehydrated and embedded in paraffin wax for stereological analysis of cell proliferation and apoptosis, and identification of apoptotic cell types by means of double labelling.
Demonstration of cell proliferation and apoptosis
Cell proliferation was assessed by immunohistochemistry using the proliferation marker Ki-67 as described elsewhere.21 Apoptosis was assayed using terminal transferase dUTP nick end labelling (TUNEL) and immunohistochemistry for active caspase 3.22 To identify the phenotype of apoptotic cells, double staining by TUNEL assay and immunohistochemistry for epithelial and endothelial cell markers were performed as described previously.22
Stereology at the light microscopical level
Richardson-stained glycol methacrylate sections were used for stereological quantification of airspace enlargement (independent measurements of mean airspace chord length, alveolar surface density and volume–weighted mean airspace volume), total alveolar surface area and total volume of alveolar septal wall tissue according to standard techniques by point and intersection counting.20 In addition, point counting was performed on glycol methacrylate sections to quantify the total volumes of inflammatory cells, alveolar macrophages and intra-alveolar polymorphonuclear granulocytes. Paraffin wax sections stained for Ki-67 and for DNA single strand breaks by TUNEL assay were used for stereological quantification of cell proliferation and apoptosis, respectively.21,22 Briefly, the fractions of alveolar wall occupied by proliferating or apoptotic alveolar septal wall cells were assessed by counting the number of test line intersections with alveolar wall associated with cells stained for Ki-67 or TUNEL, respectively, relative to the total number of intersections with alveolar wall surface. As we were mainly interested in the effect of exposure on the alveolar septal wall architecture, only those cells were included in the analysis that were part of the alveolar septal wall, whereas free apoptotic or proliferating cells present in the alveolar air space were not considered. Analyses were performed on a computer-based Olympus BX 51 light microscope equipped with a Cast-Grid 2.01 system (Olympus, Denmark).
Stereology at the electron microscopical level
Ultra thin sections of araldite-embedded tissue blocks were collected on 200 µm mesh grids and stained accordingly.23 For quantification of the volume fractions of the components of the alveolar wall—that is, alveolar epithelium, capillary endothelium, and interstitial tissue including elastin and collagen deposition—point counting was performed on each upper left corner of the grid. A transparent sheet with 140 equidistant test points was superimposed onto a television monitor, which was connected to a Zeiss EM 900 transmission electron microscope (Carl Zeiss, Oberkochen, Germany), and used for counting all points hitting the respective component relative to the total number of hits on the alveolar wall at a final magnification of 60.632. As described earlier,24 the total volume of each component was obtained by multiplying the volume fraction by the volume of the reference space, the alveolar wall.
Statistics
Mean (SD) values are given unless indicated otherwise. Differences between experimental groups were tested for significance with parametric one-way analysis of variance followed by post hoc multiple comparisons (Tukey’s test) provided that normality and equal variance were given at p>0.1. Otherwise, non-parametric one-way analysis of variance on ranks was used. Student’s t test was used to test for differences between pairs of groups. Significance of differences between groups was considered for p<0.05. Spearman rank order correlation analysis was performed to test for relationships between proliferation, apoptosis and inflammation. All statistical analyses were performed using the SigmaStat V.3.1 software program. p Values <0.05 were considered to be significant.
RESULTS
General effects of nitrogen dioxide exposure
The initial mean body weight of the animals before exposure was 181 (5) g. Rats exposed to nitrogen dioxide showed no significant gain in body weight during the first week of exposure (table 1).
Body weights and food consumption
During the following 2 weeks, they had a weight gain of about 20 g, whereas control rats exposed to room air for the same period of time had an increase in body weight of about 70 g. This was because of a significantly lower amount of mean food consumption in rats exposed to nitrogen dioxide, which was 36% of the mean food consumption of control animals during the first 3 days of exposure and about 70% during the last 2 weeks (table 1). Animals kept in room air for 28 days after 21 days of exposure to nitrogen dioxide consumed considerably more food than during exposure to nitrogen dioxide, and achieved a body weight of 280 (11) g at the end of the experiment, which was not significantly different from the mean body weight of 297 (13) g of the control rats exposed to room air for 49 days (table 1).
Apoptosis and proliferation of alveolar septal cells
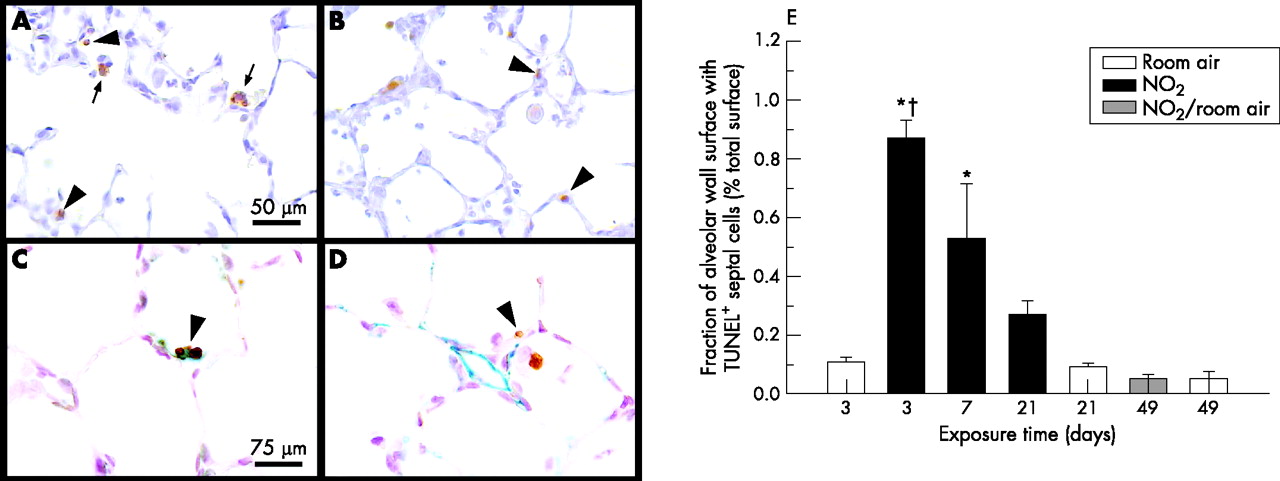
In rat lungs exposed to nitrogen dioxide for 3 days, numerous apoptotic cells were observed in alveolar septal walls and in the alveolar space by TUNEL assay (fig 1A) and immunohistochemistry for active caspase 3 (fig 1B). By contrast, apoptotic cells were extremely rare in lungs of animals exposed to room air (not shown). At day 3 of exposure to nitrogen dioxide, the fraction of alveolar surface area that was associated with apoptotic cells of the alveolar septal wall was significantly increased to more than eightfold in comparison with age-matched control animals (fig 1E). This increase exclusively reflects the increase in apoptosis of alveolar septal wall cells, as free apoptotic cells present in the alveolar air space were not considered in our analysis. With increasing exposure time, a continuous decrease of apoptotic events in alveolar septal wall cells was recorded. However, the level of alveolar septal wall apoptosis remained significantly higher than in control animals exposed to room air.

Apoptosis of alveolar septal cells. Apoptotic alveolar septal wall cells (arrowheads) are shown by (A) TUNEL assay and (B) indirect immunohistochemistry against active caspase 3 in lungs exposed to nitrogen dioxide for 3 days; arrows indicate alveolar macrophages with engulfed apoptotic bodies. (C) Apoptotic cells are identified as alveolar epithelial type II cells by double staining using TUNEL assay (brown staining) and indirect immunohistochemistry against surfactant protein D (greenish staining), whereas (D) co-staining against aquaporin 1 (greenish staining) shows that endothelial cells do not undergo apoptosis (brown staining); lungs were exposed to nitrogen dioxide for 3 days. (E) Quantification of TUNEL assay (as shown in A) shows a considerable increase in the apoptosis of structure forming alveolar septal wall cells at day 3 and day 7 of exposure to nitrogen dioxide. The magnifications used in (A) and (B), and in (C) and (D), were the same. Significant differences between groups (p<0.05, one way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test) are indicated as follows: *vs all groups exposed to room air and vs NO2/room air; †vs NO2 21 days.
Double stainings using the epithelial and microvascular endothelial markers surfactant protein D and aquaporin 1 showed that most of the apoptotic cells were of epithelial origin (fig 1C). Apoptosis of microvascular endothelial cells was not observed (fig 1D).
Immunohistochemistry for the proliferation marker Ki-67 (fig 2A–D) showed large numbers of proliferating cells at day 3 of exposure to nitrogen dioxide. The fraction of alveolar surface area associated with proliferating alveolar septal cells was significantly increased to approximately 14-fold above the level of the respective age-matched control group (fig 2E). At day 7, the number of proliferating cells had dramatically decreased and was no longer significantly different from control group levels.

Proliferation of alveolar septal cells. Proliferating alveolar septal wall cells (arrowheads) are shown by indirect immunohistochemistry against proliferation marker Ki-67 in lungs exposed to (A) nitrogen dioxide for 3 days, (B) room air for 3 days, (C) nitrogen dioxide for 7 days and (D) nitrogen dioxide for 21 days. (E) Quantification of Ki-67 staining (as shown in A–D) shows a significant increase in proliferation of structure-forming alveolar septal wall cells at day 3 of exposure to nitrogen dioxide. Insert in (A) shows that most Ki-67 positive cells are alveolar epithelial cells (A,E); asterisk denotes inflammatory cells in the alveolar airspace. The magnifications used in (A) and (B), and in (C) and (D), were the same. Significant differences between groups (p<0.05, one-way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test) are indicated as follows: * vs all groups.
Correlation analysis (performed in nitrogen dioxide-exposed lungs only) showed a significant positive correlation (r = 0.604, p = 0.008) between apoptosis and proliferation of structure forming alveolar septal wall cells, whereas no relationship was seen with any parameter in the inflammatory cells.
Inflammation
Rat lungs exposed to nitrogen dioxide for 3 days had a threefold increase in the total volume of alveolar macrophages compared with lungs exposed to room air (fig 3A), whereas polymorphonuclear granulocytes were only slightly but insignificantly increased (fig 3B). After 21 days of exposure to nitrogen dioxide, the total volumes of alveolar macrophages and of other inflammatory cells were increased by about twofold (fig 3A,C).

Quantification of total volumes of inflammatory cells (performed on 1 µm thick glycol methacrylate sections) shows (A) an increase in alveolar macrophages in lungs exposed to nitrogen dioxide for 3 and 21 days, (B) a slight but insignificant increase in polymorphonuclear granulocytes after 21 days of exposure to nitrogen dioxide, and (C) a significant increase in other inflammatory cells at day 21 of exposure to nitrogen dioxide. Significant differences between groups (p<0.05, one-way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test) are indicated as follows: * vs all groups exposed to room air and vs NO2/room air; † vs NO2/room air.
Airspace enlargement
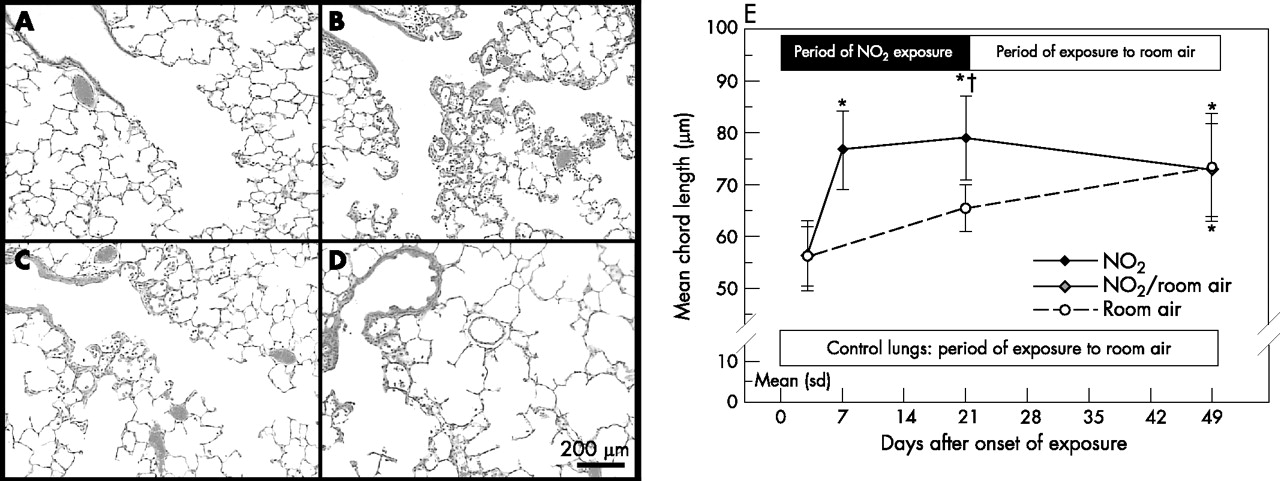
Control lungs exposed to room air showed a progressive increase in airspace size with age (fig 4A–E). In comparison with age-matched control lungs, animals exposed to nitrogen dioxide for 3 days showed normal indices of airspace size—that is, mean airspace chord length, alveolar surface density and volume-weighted mean airspace volume, respectively (table 2). However, airspace enlargement was evident at day 7 of exposure to nitrogen dioxide as indicated by significant increases in mean airspace chord length, surface density and volume-weighted mean airspace volume. Airspace enlargement further progressed until day 21: lungs exposed to nitrogen dioxide had an increase in mean airspace chord length of about 20% and in volume-weighted mean airspace volume of about 40% in comparison with age-matched control lungs. Although the mean chord length was considerably decreased in lungs exposed to nitrogen dioxide for 21 days followed by 28 days exposure to room air in comparison with lungs exposed to nitrogen dioxide for 21 days, volume-weighted mean airspace volume and surface density were not markedly different between these two groups (table 2). Hence, airspace enlargement appeared to persist after termination of exposure to nitrogen dioxide. Equivalent indices of airspace size were noted for age-matched animals exposed to room air for 49 days. The major difference between lungs exposed to nitrogen dioxide versus room air was that the airspace size of lungs exposed to nitrogen dioxide had already reached values that were equivalent to 49 days control lungs by day 7.
Quantitative morphological data obtained by light microscopy: absolute volumes (mm3)

Histopathology of terminal bronchioles and gas exchange area. H&E stainings of paraffin sections show (A) normal histology in lungs exposed to room air for 21 days, whereas lungs exposed to nitrogen dioxide for (B) 3 days, (C) 7 days and (D) 21 days show inflammation and airspace enlargement. (E) Quantification of mean chord length as an indicator of changes in airspace size (performed on 1 µm thick glycol methacrylate sections) shows a continuous increase in mean chord length in control lungs and an accelerated increase in lungs exposed to nitrogen dioxide. All micrographs were taken at identical magnification. Significant differences between groups (p<0.05, one-way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test) are indicated as follows: * vs room air 3 days and NO2 3 days; †vs room air 21 days.
Loss of alveolar walls
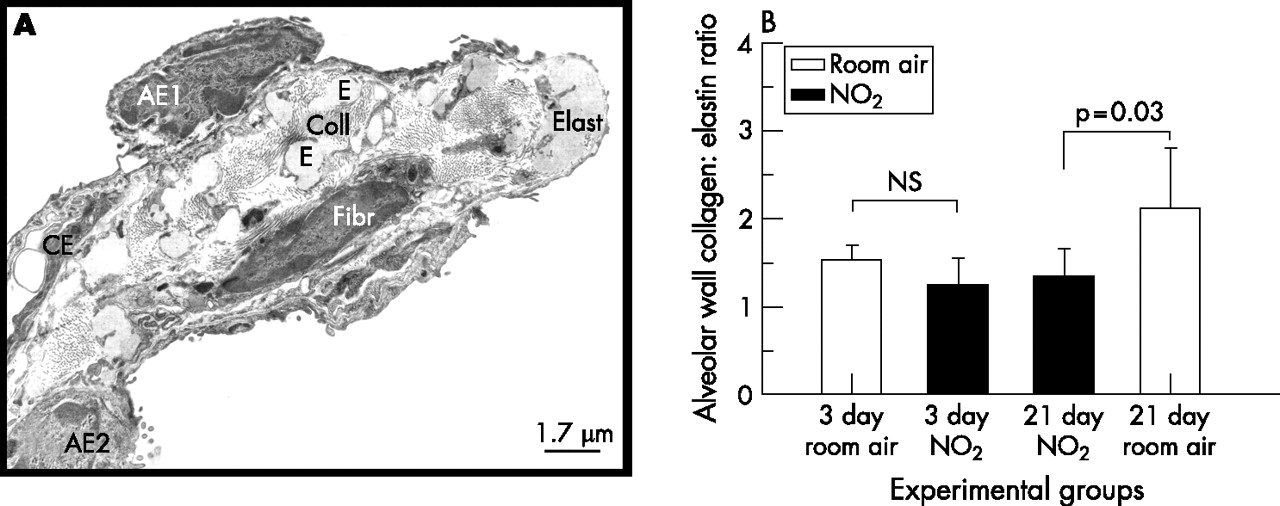
Despite the fact that airspace enlargement was prominent in lungs exposed to nitrogen dioxide for 21 days compared with age-matched control lungs, we did not find any sign of a loss of alveolar walls. By contrast, absolute volume and total surface area of alveolar septal walls were increased in nitrogen dioxide-exposed rats (day 21) by 53% and 29%, respectively, compared with age-matched controls (table 2, figs 5 and 6). Alveolar septal wall volume continued to increase after proliferation and apoptosis had returned to normal. There were no significant differences in the absolute volume of the non-parenchymal compartment. The increases in volume and surface area of alveolar septal walls were even more prominent when mass-specific parameters are considered (data not shown). Quantitative ultrastructural analysis showed that the increased alveolar septal wall volume at day 3 of nitrogen dioxide exposure was exclusively due to an increase in the volume of the alveolar epithelium (fig 7). Whereas no further increase was noted in the alveolar epithelial volume of nitrogen dioxide-exposed lungs, the volumes of capillary endothelium and interstitial tissue as well as of collagen and elastin were increased at day 21 compared with age-matched control lungs. Notably, the collagen-to-elastin ratio was significantly lower in nitrogen dioxide-exposed lungs at day 21 compared with control lungs exposed to room air for 21 days (fig 8).

Quantification of total alveolar surface area as an indicator of loss of alveolar walls (performed on 1 µm thick glycol methacrylate sections) shows a continuous increase in total alveolar wall surface in control lungs and an accelerated increase in lungs exposed to nitrogen dioxide. Significant differences between groups (p<0.05, one-way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test) are indicated as follows: *vs room air 3 days, 21 days and vs NO2 3 days, 7 days.

Quantification of total alveolar septal volume as an indicator of a potential loss of alveolar walls (performed on 1 µm thick glycol methacrylate sections) shows an increase in alveolar wall volume in control lungs and an accelerated increase in lungs exposed to nitrogen dioxide. Significant differences between groups (p<0.05, one-way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test) are indicated as follows: *vs room air 3 days, 21 days.
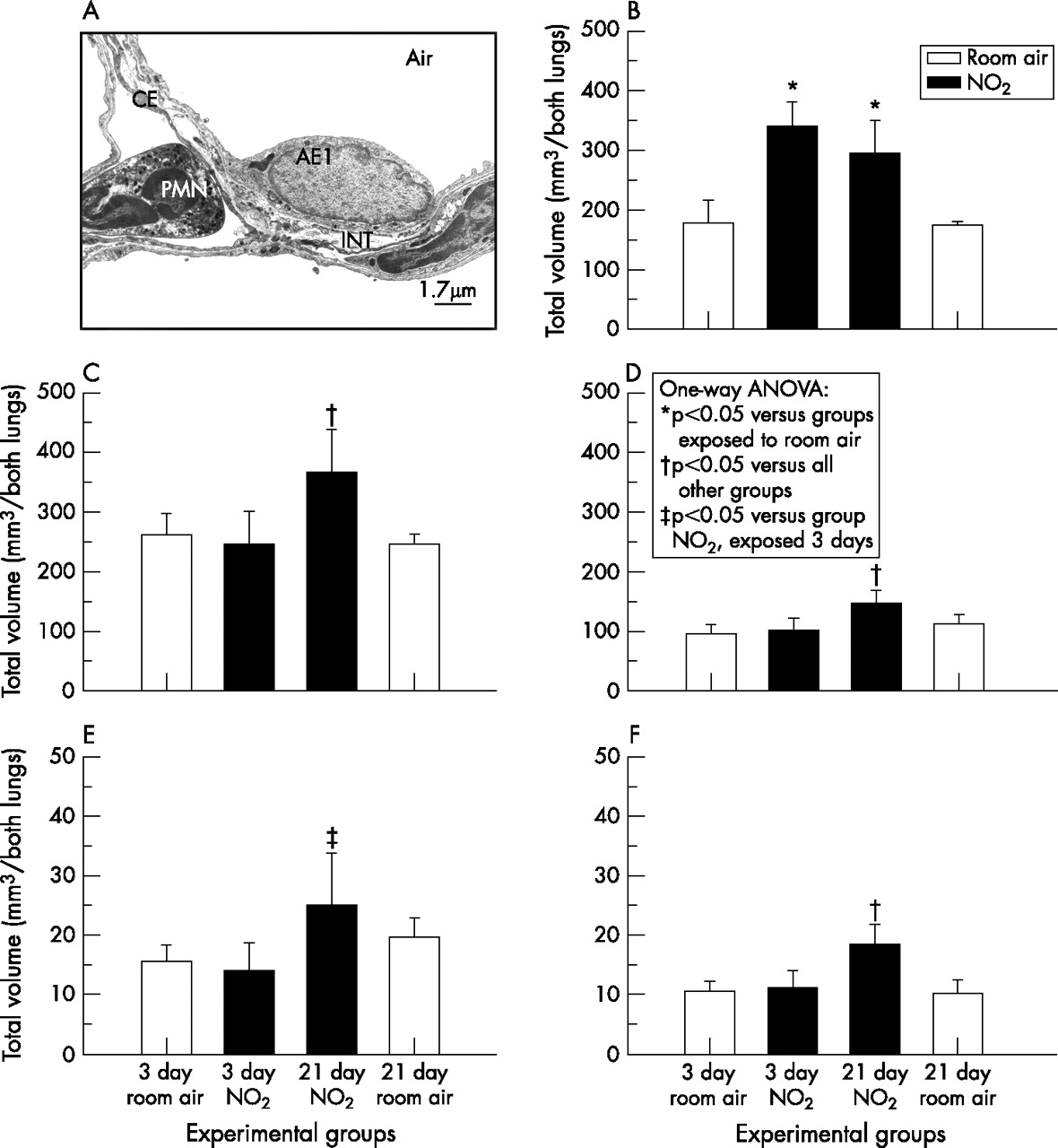
(A) Ultrastructure of alveolar septal wall of lung exposed to room air for 3 days. Electron microscopic quantification of different components of the alveolar septal wall showed an increase in the total volume of (B) the alveolar epithelium already on day 3 of exposure to nitrogen dioxide whereas the other components of the alveolar septal wall were not affected. At day 21 of exposure to nitrogen dioxide, an equivalent increase in volume was seen in all components of the alveolar wall; (B) alveolar epithelium, (C) interstitial tissue, (D) capillary endothelium, (E) collagen fibres and (F) elastin. Significant differences between groups (p<0.05, one-way analysis of variance followed by post hoc multiple comparisons according to Tukey’s test) are indicated. AE1, alveolar epithelial type 1 cell; Air, alveolar airspace; CE, capillary endothelium; INT, interstitial tissue; PMN, polymorphonuclear granulocyte.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Ultrastructure of extracellular matrix composition of alveolar septal wall entrance ring of lung exposed to room air for 3 days. (B) Electron microscopic quantification showed a significantly decreased collagen-to-elastin ratio in lungs exposed to nitrogen dioxide for 21 days compared with control lungs exposed to room air for 21 days. At day 3, no differences were seen between lungs exposed to nitrogen dioxide or room air. Significant differences between groups (p<0.05, Student’s t test) are indicated. AE1, alveolar epithelial type 1 cell; AE2, alveolar epithelial type 2 cell; CE, capillary endothelium; Coll, collagen fibrils; E, Elast, elastin fibres; Fibr, fibrocytes.
A progressive age-dependent increase was noted in control lungs in all structural parameters related to the gas exchange region. Notably, total alveolar surface area and absolute alveolar septal volume of nitrogen dioxide-exposed rats (day 21) were largely equivalent to values characteristic of control animals at day 49. There was no noticeable change in alveolar septal wall volume and total alveolar surface area after exposure to nitrogen dioxide had been terminated, and no changes were noted after an additional exposure to room air for 28 days.
DISCUSSION
The effects of exposure of experimental animals to nitrogen dioxide vary widely—for example, with the species and strain exposed, the concentration and duration applied, as well as age and sex of the animals—as comprehensively reviewed by others.25 In our study of juvenile male Fischer 344 rats, which were 8–10 weeks of age at the beginning of the experiments, lungs exposed to an atmosphere containing 10 ppm nitrogen dioxide for 23 h per day for 3 days showed an increased turnover of alveolar septal cells indicated by an eightfold increase in apoptotic events and a 14-fold increase in proliferation in comparison with age-matched control lungs. Lungs exposed to nitrogen dioxide for 21 days exhibited quantitative structural characteristics equivalent to control lungs, which were 28 days older. As transfer of lungs to room air after 21 days of exposure to nitrogen dioxide did not result in a reversal of this process, we suggest that nitrogen dioxide induced an accelerated growth of the lung, which, however, was associated with an imbalance in the relative composition of the extracellular matrix indicated by a decreased collagen-to-elastin ratio.
In agreement with results from in vitro studies,17 we observed a significant increase in alveolar septal wall cell death in lungs exposed to nitrogen dioxide in vivo. Double stainings for apoptosis and markers of alveolar cell types showed that alveolar epithelial type II cells were the main target of nitrogen dioxide-induced apoptotic cell death, whereas vascular and capillary endothelial cells were not affected. Although experimental induction of epithelial and/or endothelial cell apoptosis has been linked to the development of airspace enlargement in various murine models5–7 including cigarette smoke exposure,8 apoptosis of alveolar epithelial cells appeared to predominate in human emphysema.10,11 Recent studies have shown that in the lungs of patients with emphysema, however, proliferation of alveolar septal cell was also increased.9,10 These findings indicate that emphysema may not simply be the consequence of the induction of apoptosis, but is more likely to be the result of an imbalance between apoptosis and proliferation in humans.9,10
Exposure of rats to nitrogen dioxide was shown to induce airway inflammation which, as in human COPD, was dominated by alveolar macrophages and neutrophilic granulocytes.15,16 Only a minor contribution of polymorphonuclear granulocytes was seen in our experiments. Whereas some studies have reported that exposure to nitrogen dioxide may result in emphysema,16 others have not found evidence for this.26 Unfortunately, all these studies relied only on the measurement of airspace enlargement to assess the presence of emphysema. An increase in airspace size alone, however, is insufficient to conclude that emphysema has developed.27 Stereological approaches to quantify emphysema has shown that, in human emphysema28 as well as in elastase-induced emphysema in the mouse,29 airspace enlargement is accompanied by a loss of alveolar walls as evidenced by a decrease in total alveolar surface area and total alveolar wall volume. Using a stereological approach to quantify airspace enlargement by independent measurements of mean airspace chord length, alveolar surface density (S/V ratio), and volume-weighted mean airspace volume, respectively, as well as quantification of total alveolar surface area and absolute alveolar septal wall volume, we showed that increased airspace enlargement during exposure to nitrogen dioxide was not associated with a loss of alveolar walls. On the contrary, lungs exposed to nitrogen dioxide for 21 days were characterised by a 29% increase in total alveolar surface area and a 53% increase in absolute septal volume in comparison with age-matched control lungs. Hence, relying on indicators of airspace enlargement alone would have led to the false conclusion that nitrogen dioxide induced apoptosis with subsequent emphysema. By contrast, inclusion of indicators of loss of alveolar walls—for example, total alveolar wall volume and total alveolar surface area—showed that the increase in apoptotic events observed in lungs exposed to nitrogen dioxide was over-compensated by the parallel increase in proliferation.
In early studies, Evans and coworkers showed that nitrogen dioxide results in hyperplasia of alveolar epithelial type II cells as a response to the initial damage of type I cells.30 In line with this, our quantitative ultrastructural analysis showed that, at day 3 of exposure to nitrogen dioxide only the alveolar epithelium exhibited an increase in the total volume, while all other compartments of the alveolar septal wall were unaffected. Hyperplasia of alveolar epithelial type II was followed by transformation of type II into type I cells.18 Although transformation is one mechanism for resolving hyperplasia, apoptosis of type II cells, as shown in this study, is another mechanism which is probably more important in quantitative terms than transformation.31 The significant correlation between the indices of proliferating and apoptotic cells in our study highlights the close link between apoptosis and proliferation of alveolar septal wall cells in this model. The decrease in both proliferation and apoptosis with exposure time may reflect some adaptive mechanisms induced—for example, in alveolar epithelial type II cells, which were previously shown to exhibit restored antioxidant superoxide dismutase acitivity at day 20,32 and/or in bronchoalveolar lavage cells, which showed an increase in glutathione peroxidase and superoxide dismutase enzyme activities in this model.33 However, we cannot discriminate whether the induction of these protective mechanisms were direct effects due to exposure or an indirect effect due to the reduced food intake (see below).
After 21 days of exposure to nitrogen dioxide, all compartments of the alveolar septum exhibited an increase in their total volume in comparison with room air exposure. However, no equivalent increase was seen in the collagen-to-elastin ratio, indicating that accelerated lung growth was associated with an imbalance in extracellular matrix deposition.
In this study, as in most studies performed in the field of emphysema research,6–8 animals were 8–10 weeks of age at the beginning of exposure—that is, they were juveniles. Considerable normal lung growth is observed in rats and mice up to an age of at least 5 months, and comprises an increase in total alveolar surface area and absolute alveolar wall volume as well as an age-dependent increase in airspace size.34,35 The quantitative morphological data obtained in our study of control animals compare well with data published on ageing male Fischer 344 rats summarised recently.35 In this review, the authors pointed out that only small changes occur in tissue volumes from 5 to 26 months of age with the exception of a reduction in the extracellular matrix component of the interstitium, whereas total airspace volume and airspace size increased with age.35 These structural changes are accompanied by age-related changes in the biochemical composition of the extracellular matrix as well as in lung function.36 Age-related changes in airspace size are also well documented in humans,37 as are age-related changes in lung function.38 Both an age-dependent decline in lung function and an increase in airspace size are significantly accelerated in smokers.39,40 Notably, data from a recent prospective study of California schoolchildren suggest that ambient nitrogen dioxide is a major contributor to the adverse effects of air pollution on lung development from the age of 10 to 18 years, which lead to clinically significant deficits—for example, in forced expiratory volume in 1 s as children reached adulthood.41 Although in the present study lung function parameters were not analysed, we have recently shown that exposure to nitrogen dioxide resulted in a progressive decline in expiratory airflow associated with an increase in airspace size as well as in volume-weighted mean alveolar volume in juvenile mice.42
From our study we cannot discriminate if the changes observed were a direct effect of the exposure to nitrogen dioxide or an indirect effect due to the significantly reduced food consumption (day 3: 36%, day 7: 44% of control animals at day 3; day 21: 69% of control animals at day 21). Calorie restriction (two thirds reduction for 2 weeks) in mice has recently been shown to result in a reduction in body weight of about 40% with a concomitant increase in apoptosis, a 25% reduction in total alveolar surface area and a reduction in total alveolar wall volume of about one third.43 Unlike these findings, exposure of rat lungs to nitrogen dioxide did not result in loss of body weight but only in a reduction in weight gain which may be due to the reduced activity, and hence metabolism, of nitrogen dioxide-exposed animals. We did not observe a loss of alveolar walls as a direct effect of exposure or as an indirect effect due to reduced food intake. Instead, a 29% increase in total alveolar surface area and a 53% increase in total alveolar wall volume were observed after 21 days of exposure to nitrogen dioxide versus room air. Wright and Churg44 pointed out that all of the animal models of emphysema using cigarette smoke exposure that have evaluated changes in body weight have shown a failure of smoke-exposed animals to gain weight. In a recent pair-fed study in mice, cigarette smoking for 4 weeks was shown to result in reduced weight gain equivalent to a food restriction group. In contrast with food restriction, smoke exposure caused a reduction in hypothalamic neuropeptide Y and fat mass, and regulated adipose cytokines, finally resulting in the reduced weight gain.45 Unfortunately, data regarding inflammation or histopathology were not recorded in this study. From human studies it is well documented that, in contrast with chronic starvation well-nourished adults who, for experimental or other reasons, have lowered their food intakes an apparently enhanced metabolic efficiency is observed resulting from changes in metabolic rates, which are disproportionate to the changes in body weight.46
In conclusion, nitrogen dioxide exposure of juvenile rats resulted in increased alveolar septal cell turnover as evidenced by an increase in apoptosis and proliferation of alveolar septal cells. Comparison with age-matched control lungs indicates that increased cell turnover was associated with an acceleration of normal lung growth as evidenced by an increase in airspace size, total alveolar surface area as well as total alveolar wall volume. Despite the increase in both collagen and elastin, the reduced collagen-to-elastin ratio in nitrogen dioxide-exposed animals indicates that extracellular matrix deposition was impaired and may result in altered lung mechanics in this model. We suggest that the changes observed are compatible with an accelerated lung growth of rat lungs exposed to nitrogen dioxide which, however, does not comprise a proportional growth of collagen and elastin. Exposure of juvenile rats to nitrogen dioxide may be a useful model to study mechanisms leading to the adverse effects of air pollution on postnatal lung development observed in children and young adults.41
Acknowledgments
We acknowledge the help of Bernd Müller and Petra Staats with the nitrogen dioxide exposure model, and the expert technical assistance of Roswitha Naumann and Tanja Rausch in all steps of organ preparation, tissue processing, sectioning and staining procedures.
REFERENCES
Footnotes
-
Funding: The study was supported by a grant from the German Ministerium für Bildung und Forschung (FKZ 01GC0103).
-
Competing interests: None.