Article Text

Abstract
Background: A study was undertaken to assess both oxidative stress and inflammation in the lungs of patients with chronic obstructive pulmonary disease (COPD) during severe and very severe exacerbations compared with those with stable COPD, healthy smokers, and non-smokers. Two sites within the lungs were compared: the large airways (in sputum) and the peripheral airways (by bronchoalveolar lavage (BAL)).
Methods: BAL fluid cell numbers and levels of tumour necrosis factor (TNFα) and interleukin (IL)-8 were measured as markers of airway inflammation and glutathione (GSH) levels as a marker of antioxidant status. Nuclear translocation of the pro-inflammatory transcription factors nuclear factor-κB (NF-κB) and activator protein 1 (AP-1) were also measured by electromobility shift assay in BAL fluid leucocytes and lung biopsy samples.
Results: Influx of inflammatory cells into the peripheral airways during exacerbations of COPD was confirmed. Increased IL-8 levels were detected in BAL fluid from patients with stable COPD compared with non-smokers and healthy smokers, with no further increase during exacerbations. In contrast, IL-8 levels in the large airways increased during exacerbations. GSH levels were increased in the BAL fluid of smokers (444%) and patients with stable COPD (235%) compared with non-smokers and were reduced during exacerbations (severe 89.2%; very severe 52.3% compared with stable COPD). NF-κB DNA binding in BAL leucocytes was decreased in healthy smokers compared with non-smokers (41.3%, n = 9, p<0.001) but did not differ in COPD patients, whereas AP-1 DNA binding was significantly decreased during exacerbations of COPD.
Conclusion: There is evidence of increased oxidative stress in the airways of patients with COPD that is increased further in severe and very severe exacerbations of the disease. This is associated with increased neutrophil influx and IL-8 levels during exacerbations.
- AP-1, activator protein 1
- COPD, chronic obstructive pulmonary disease
- EMSA, electrophoretic mobility shift assay, γ-GCS, gamma-glutamylcysteine synthetase
- GSH, glutathione
- IL, interleukin
- NF-κB, nuclear factor-κB
- TNFα, tumour necrosis factor α
- chronic obstructive pulmonary disease
- nuclear factor-κB
- interleukin 8
- oxidative stress
- glutathione
- inflammation
Statistics from Altmetric.com
- AP-1, activator protein 1
- COPD, chronic obstructive pulmonary disease
- EMSA, electrophoretic mobility shift assay, γ-GCS, gamma-glutamylcysteine synthetase
- GSH, glutathione
- IL, interleukin
- NF-κB, nuclear factor-κB
- TNFα, tumour necrosis factor α
- chronic obstructive pulmonary disease
- nuclear factor-κB
- interleukin 8
- oxidative stress
- glutathione
- inflammation
Inflammation is a prominent feature of chronic obstructive pulmonary disease (COPD) as shown by the presence in the airways of activated neutrophils and macrophages and increased numbers of inflammatory mediators.1,2 A current hypothesis in the pathogenesis of COPD is that the increased oxidant burden—both directly as a result of smoking or indirectly by the release of increased amounts of reactive oxygen species from airspace leucocytes—may not be adequately counterbalanced by the lung antioxidant systems, resulting in oxidative stress. An excess of oxidants may then lead to enhanced pro-inflammatory gene expression and protein release, inactivation of antiproteases, and oxidative tissue injury leading to COPD.
The presence of oxidative stress in the airways of smokers and patients with COPD has been shown by increased products of lipid peroxidation and altered antioxidant status.3–5 Transcription factors such as nuclear factor-κB (NF-κB) and activator protein 1 (AP-1) that are important for gene transcription of the inflammatory cytokines associated with airway inflammation in COPD—such as interleukin (IL)-8, tumour necrosis factor α (TNFα), and IL-6)1,2,6—are oxidant sensitive.7 We also recently found an increase in NF-κB nuclear translocation in airway leucocytes from COPD patients during an exacerbation compared with leucocytes from stable patients.8 Furthermore, we found the sequestration of neutrophils in the pulmonary micropreparative increased during active cigarette smoking and in COPD patients undergoing an exacerbation, an effect that may be oxidant mediated.9–11 These data suggest that lung oxidative stress and inflammation in COPD may be linked.
Glutathione (GSH) is the principal small molecular weight thiol in the lungs which, together with its redox enzymes, provides an important protective antioxidant system. Studies show depletion of GSH in lung cells in vitro and in rat lungs in vivo following acute exposure to cigarette smoke, with a rebound increase in levels over time.12,13 These findings are in agreement with the high levels of GSH reported in the epithelial lining fluid of smokers.14 Changes in GSH levels were found to be due to inhibition (by acute smoking) and then enhanced activation of the rate limiting enzyme for GSH production, gamma-glutamylcysteine synthetase (γ-GCS).15 The regulation of this gene was linked with the redox sensitive transcription factor, AP-1.15
Exacerbations of COPD are considered to reflect worsening of the underlying chronic inflammation in the airways. However, there is little information on the inflammatory responses in the lungs in exacerbations of COPD, particularly during severe exacerbations. Patients with COPD are known to have increased numbers of activated neutrophils in their airways. These neutrophils are believed to be attracted to the airways by the cytokines IL-8 and TNFα which are present in increased levels in the lungs of patients with stable COPD.1 Further increases in the levels of cytokines and also increased cell numbers in the large airways have been reported during exacerbations of the disease.2 Moreover, the frequency of exacerbations is associated with disease severity16 and a further increase in airways inflammation.2 These patients have a reduced quality of life and significantly more hospital admissions and a longer time in hospital, thereby adding to the socioeconomic burden.
The aim of this study was to assess the oxidant/antioxidant imbalance and markers of inflammation in both the proximal and distal compartments of the lungs in patients with severe and very severe exacerbations of COPD. It is known that samples from the different compartments of the lungs yield differences in cell numbers, markers of inflammation and oxidative stress. We therefore set out to assess the extent and location of inflammation in these patients. Specifically, we studied the antioxidant GSH, cell numbers, and the inflammatory mediators TNFα and IL-8 in proximal and distal airways.
METHODS
Study subjects
Three groups of patients with COPD were studied. A group with moderate COPD (GOLD stage II), diagnosed in accordance with GOLD guidelines,17 were studied when clinically stable at least 6 weeks after their last exacerbation, and two groups of patients with severe COPD were studied during exacerbations with severity assessed by healthcare utilisation according to the European Respiratory Society COPD guidelines.18 Exacerbations were defined as an increase in symptoms beyond normal day-to-day variation requiring a change in medication and hospital admission. According to a recent consensus statement on exacerbations, these patients could be divided into those with severe exacerbations (GOLD stage III) requiring hospital admission, and those with very severe exacerbations (GOLD stage IV) who had respiratory failure and required intervention with mechanical ventilation.18 Patients with severe or very severe exacerbations of COPD were studied within 48 hours of admission to hospital or the intensive care unit. Exclusion criteria were age less than 40 years; pneumonia or other lung diseases; a history suggestive of asthma; and treatment with oral corticosteroids, non-steroidal anti-inflammatory agents, or theophylline preparations during the 2 weeks before admission. Healthy non-smokers and smokers formed control groups for comparison. Smokers had not smoked for 12 hours before bronchoscopy to exclude the acute effects of smoking.
All subjects were informed of the nature and purpose of the study and gave their consent or, in the case of ventilated patients, consent was obtained from their relatives. The studies were approved by the local hospital ethics committees in Barcelona and Palma de Mallorca for the studies in patients with exacerbations and by the Lothian ethics committee for studies in healthy subjects, smokers, and those with stable COPD studied in Edinburgh. All procedures were standardised across the three study centres.
Bronchoscopy and bronchoalveolar lavage (BAL)
All study subjects underwent bronchoscopy. Patients with very severe exacerbations were ventilated and bronchoscopic examination was undertaken via an endotracheal tube. The other groups had fibreoptic bronchoscopy under local anaesthetic. Topical lignocaine was applied to the nasopharynx (0.5 ml of 0.4% solution and 2 ml of 0.2% solution) and to the vocal cords and major airways (6 ml of 2% solution). Subjects were given oxygen at 2 l/min throughout the procedure. Two separate samples were obtained: (1) large airways secretions and (2) bronchoalveolar lavage (BAL) fluid from a segment of the middle lobe or lingula. Resident mucus was aspirated from the trachea and main bronchi to represent a large airways sample. The collection receptacle was then changed before BAL was performed using up to 240 ml warmed saline in 30 ml aliquots and aspirated immediately using a low suction rate. This technique samples the distal airways and alveoli.19
A biopsy sample was taken from a segmental bronchus where possible and frozen immediately at −80°C. Biopsy specimens were homogenised in cytoplasmic extraction buffer containing 10% Nonidet-40 (NP40) as described for electrophoretic mobility shift assay (EMSAs) and nuclear proteins extracted as detailed below.
Processing of proximal and distal airway samples
The sample of proximal large airway secretions was weighed and an equal volume of dithiotreitol (DTT) (stock 1% in distilled water diluted to 0.1% in phosphate buffered saline (PBS)) was added. The sample was mixed on ice for 15 minutes, following which an equal volume of PBS was added and the solution filtered through 48 μm nylon gauze. The supernatant was centrifuged at a relative centrifugal force of 1050g for 10 minutes at 4°C. The cell pellet was discarded and aliquots of the spun supernatant were stored at −70°C.
BAL fluid from the distal airspaces was filtered through sterile gauze swabs and centrifuged at 242g for 10 minutes at 4°C. The supernatant was decanted and spun at 1050g for 10 minutes at 4°C and aliquoted for storage at −70°C for later analysis. The cell pellet obtained from the BAL fluid was washed in PBS and counted with a haemocytometer. Viability was ascertained by exclusion of trypan blue. Cytospins were prepared in a Shandon cytospin 3 (Shandon, Pittsburgh, PA, USA) and stained with Diff-Quick stain (Merz Dade, Switzerland). Cell differential counts were determined by counting 200 cells per sample.
Assays
All reagents were obtained from Sigma (Poole, UK) unless stated otherwise.
Glutathione
Reduced glutathione (GSH) was measured in BALF fluid by the method of Tietze20 with modifications for a 96 well plate.21 Large airway secretions were not assessed for GSH as the presence of DTT interferes with the GSH assay (by interacting with GSH reductase). The reaction rate of DTNB, NADPH, and GSH reductase was measured over 6 minutes at an absorbance of 410 nm using a Dynex MRX plate reader. A standard curve of GSH in the range of 0.25–16 μg/ml was used. The results were expressed as ng GSH/mg total protein, as measured by the Pierce bicinchoninic acid (BCA) protein assay (Pierce and Warriner (UK) Ltd, Chester, UK).
IL-8 and TNFα ELISAs
IL-8 and TNFα concentrations in large airway and BAL fluid samples were assessed by a human enzyme-linked immunosorbent sandwich assay (ELISA) using specific monoclonal biotinylated antibodies (R&D Systems, Oxon, UK) and a streptavidin peroxidase (Dako Ltd, Ely, UK)/3,3′,5,5′-tetramethylbenzidine (Boehringer Mannheim, East Sussex, UK) detection system. Sulphuric acid was added to terminate the reaction and the optical density was determined at 450 nm. Cytokine levels were expressed as pg/ml or ng/ml. For BAL fluid, the data were expressed per mg protein (as measured by the Pierce BCA protein assay) to account for the different volumes of saline instilled during lavage. DTT and lignocaine did not interfere with the ELISAs (data not shown).
Electrophoretic mobility shift assay (EMSA)
Nuclear proteins were extracted from mixed leucocytes obtained from BAL fluid and lung biopsies by the method of Andrews and Faller.22 The cells were incubated in cytoplasmic extraction buffer (10 mM Hepes-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT and 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 10 μM 4-(2-aminomethylbenzensulfonyl fluoride)) for 15 minutes on ice and lysed with 10% Nonidet-40 (NP-40). After centrifugation the nuclei were resuspended in hypertonic nuclear extraction buffer (20 mM Hepes-KOH, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 25% glycerol containing 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 10 μM 4-2(aminomethylbenzenesulfonyl fluoride, 10 μM glycerophosphate, 1 μM Na3VO4) for 20 minutes on ice. The samples were centrifuged and the supernatant stored at −70°C. Biopsy specimens were homogenised in cytoplasmic extraction buffer and the procedure followed as for BAL fluid leucocytes.
EMSAs were performed as previously described.23 Seven μg of nuclear protein (as determined using the Bio-Rad protein assay) were incubated in binding buffer (50 mM Tris-HCl pH 7.0, 500 mM KCl, 5 mM DTT, 5 mM EDTA, 2.5 mM MgCl2, 40% glycerol with 1 μg poly(dI-dC).poly(dI-dC) added per reaction) with γ-32P-ATP (Amersham, Buckinghamshire, UK) end labelled consensus oligonucleotides for NF-κB and AP-1 (Promega, Southampton, UK). The sequences of oligonucleotides used for EMSAs were: NF-κB sense 5′-AGTTGAGGGGACTTTCCCAGG-3′, AP-1 sense 5′-CGCTTGATGAGTCAGCCGGAA-3′. Binding was allowed to proceed for 20 minutes at room temperature and electrophoresed on a 6% non-denatured polyacrylamide gel in 1X Tris-buffered-EDTA (TBE) buffer. Specificity was determined by 100 fold excess addition of a cold unlabelled corresponding oligonucleotide (cold competitor) and a different oligonucleotide (non-competitor). Gels were dried onto Whatman paper and exposed for autoradiography. The retarded oligonucleotides were visualised by a Storm 860 phospho-imager and the intensity of the NF-κB bands were quantified using ImageQuant Analysis (Molecular Dynamics Inc, Buckinghamshire, UK).
Statistical analysis
Spirometric data, differential cell counts, and leucocyte NF-κB DNA binding data were normally distributed and were expressed as mean (SE) values. Differences between the means were assessed by a one-way analysis of variance (ANOVA). Student-Newman-Keuls post tests were performed when a significant difference between the groups was obtained. Data for non-normally distributed variables (GSH, TNFα and IL-8 levels, and leucocyte and biopsy AP-1 and biopsy NF-κB) were expressed as medians with interquartile ranges (IQR), and statistical analysis was performed by Kruskal-Wallis ANOVA and Dunn’s multiple comparison post hoc pairwise comparison when significant differences were found. A p value of <0.05 was considered significant. To assess the relationship between variables, linear regressions were performed on all the data as the numbers were too low in the subgroups to confirm such correlations.
RESULTS
Subjects
Twenty one patients with COPD were studied: seven with moderate COPD were studied when stable, seven with a severe exacerbation, and seven with a very severe exacerbation of COPD and respiratory failure. Ten non-smokers and 12 smokers with no respiratory symptoms and normal spirometric data were studied for comparison. The characteristics of the subjects at the time of enrolment into the study are shown in table 1. All healthy subjects had normal lung function. Non-smokers and healthy smokers were younger than those with stable COPD and those with severe exacerbations. Patients with very severe exacerbations of COPD had a significantly greater number of pack years of smoking than healthy smokers (p<0.05), although all smokers had at least a 22 pack year history. Significant differences in % predicted forced expiratory volume in 1 second (FEV1) and ratio of FEV1 to forced vital capacity (FVC) are evident between the groups (table 1).
Characteristics of subjects
BAL fluid cell counts
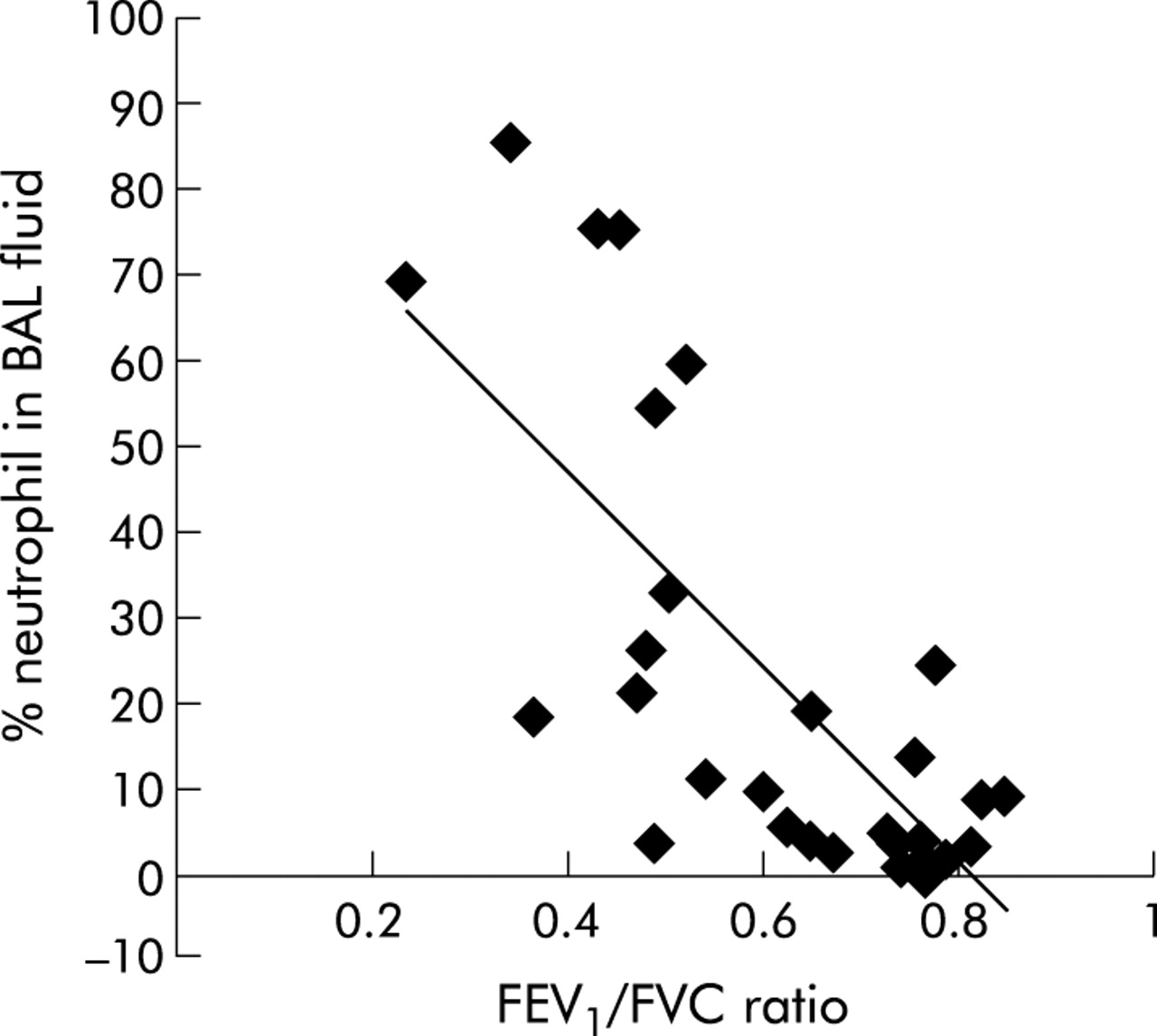
The percentage recovery of BAL fluid and differential cell counts for BAL fluid leucocytes are shown in table 2. Significantly less fluid was aspirated in patients with moderate and very severe COPD than in non-smokers. The total BAL fluid cell count was significantly reduced in patients with moderate COPD and was significantly higher in those with very severe COPD than in healthy smokers and non-smokers, respectively. Patients with an exacerbation of COPD had increased numbers of neutrophils in the BAL fluid compared with healthy smokers and non-smokers. A concomitant decrease in mononuclear cells was seen in patients with very severe COPD. Moreover, the percentage of neutrophils present in the BAL fluid had a linear relationship with the FEV1/FVC ratio (r = −0.751, n = 34, p<0.0001; fig 1).
Total and differential cell counts in bronchoalveolar lavage (BAL) fluid

Relation between percentage neutrophils in BAL fluid and FEV1/FVC ratio. A significant linear relationship was seen between the percentage of neutrophils in the BAL fluid and the severity of airways obstruction as assessed by FEV1/FVC ratio for all subjects (r = −0.751, n = 34, p<0.0001).
Measurements of oxidative stress
Healthy smokers had considerably higher GSH levels than non-smokers or the COPD patient groups (fig 2). GSH levels in patients with COPD were significantly lower than in healthy smokers but were still higher than in healthy non-smokers, with an even greater reduction observed during acute exacerbations.

Assessment of oxidative stress locally in the lungs. Increased levels of glutathione (GSH) were seen in BAL fluid from asymptomatic smokers. Patients with moderate COPD were studied in a stable condition and patients with moderate or severe COPD were studied during an exacerbation. Data are expressed as medians with interquartile range (box) and range (whiskers); numbers of subjects shown in brackets. ANOVA p<0.0001; **p<0.01 and ***p<0.001 v non-smokers; †p<0.05, †††p<0.001 v smokers; ‡p<0.01 v subjects with severe COPD exacerbations (multiple comparison post testing).
Inflammatory mediators
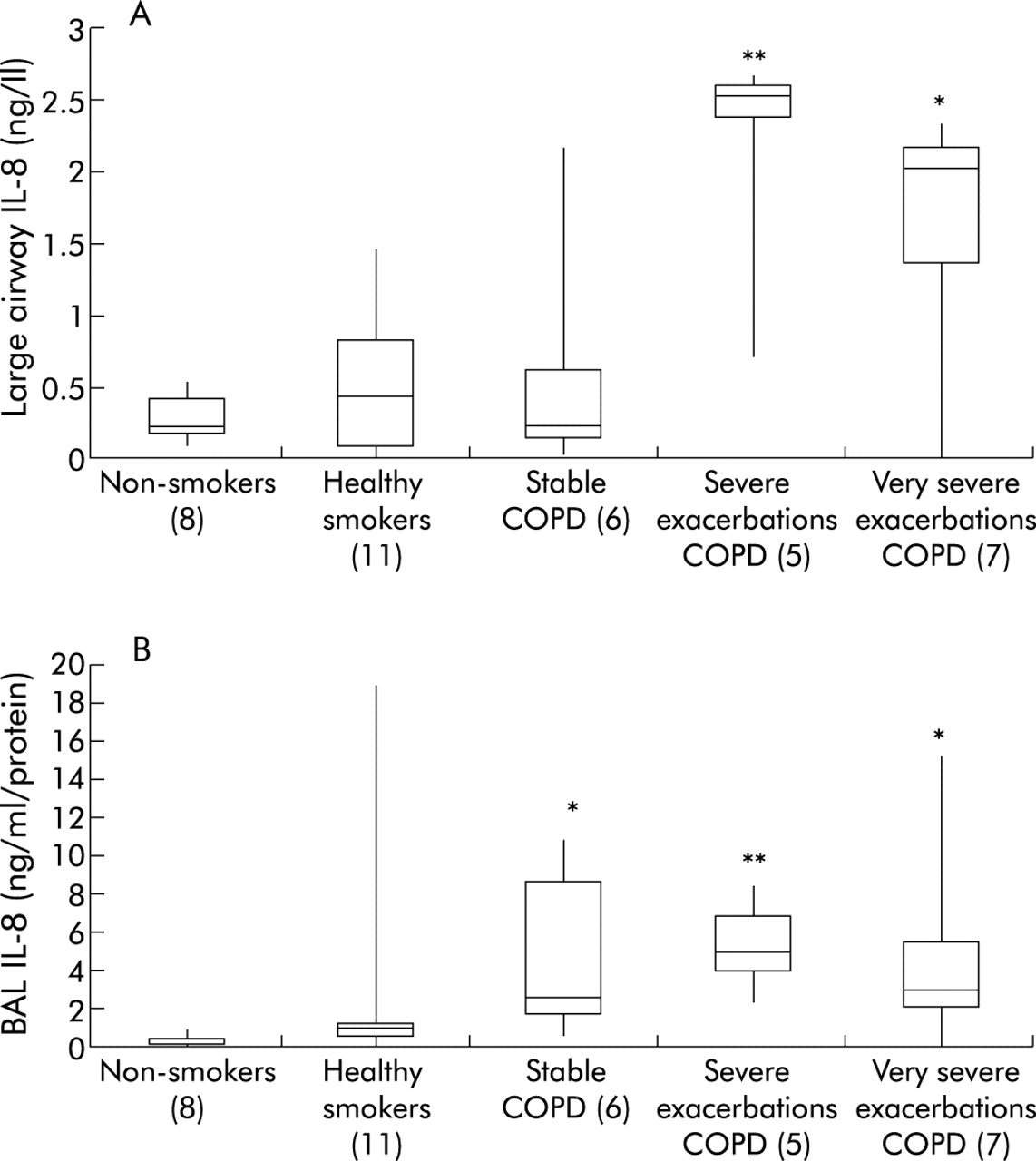
TNFα and IL-8 levels were measured in large airway secretions and BAL fluid. TNFα levels detected in airway secretions and BAL fluid were generally low and were not significantly different between the groups for either large airway secretions or BAL fluid (data not shown). Significantly higher levels of IL-8 were detected in large airway secretions during both severe and very severe exacerbations of COPD (fig 3A). Significantly higher levels of IL-8 were observed in the BAL fluid in all COPD patients, although the levels during exacerbations did not differ from those in stable patients (fig 3B). IL-8 levels in large airway samples had a linear relationship with the FEV1/FVC ratio (r = −0.592, p<0.01, n = 29; fig 4A); a similar relationship was seen in BAL fluid (r = −0.33, n = 35, p = 0.05; fig 4B).

Assessment of inflammation in the airways. IL-8 levels were determined in (A) large airway secretions and (B) BAL fluid for the different subject groups. Data are expressed as medians with interquartile range (box) and range (whiskers); numbers of subjects are shown in brackets. ANOVA values were p<0.01 for large airway secretions and p<0.001 for BAL fluid; *p<0.05, **p<0.01 v non-smokers (multiple comparison post testing).

Relation between IL-8 levels and the FEV1/FVC ratio in (A) the large airways and (B) BAL fluid. A significant linear relationship was evident in the large airways (r = −0.592, n = 29, p<0.001) and BAL fluid (r = −0.33, n = 35, p<0.0001) for all subjects.
Transcription factor nuclear translocation of NF-κB in lung biopsies and BAL leucocytes
Transcription factor DNA binding was assessed for nuclear proteins isolated from both bronchial biopsy specimens and BAL fluid leucocytes. From the samples obtained, this was only possible for non-smokers, healthy smokers, and those with severe exacerbations. NF-κB nuclear binding in BAL leucocytes was lower in healthy smokers than in non-smokers (fig 5A), but this was not observed for leucocytes from patients with COPD. Patients with severe exacerbations of COPD had lower AP-1 nuclear binding in BAL fluid leucocytes than smokers or healthy subjects (fig 5B). There was insufficient sample to determine NF-κB or AP-1 in BAL fluid leucocytes from patients with stable COPD. There was no significant difference in NF-κB and AP-1 DNA binding activity in lung biopsy specimens (table 3), partly due to the small numbers available for analysis.
DNA binding of transcription factors NF-κB and AP-1 in lung biopsy specimens

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Electrophoretic mobility shift assays (EMSAs) for (A) NF-κB and (B) AP-1 DNA binding in BAL fluid leucocytes. Data are expressed as mean (SE) values or as medians with interquartile range (box) and range (whiskers) for the different subject groups; numbers of subjects are shown in brackets. Measurements for patients with stable COPD are not shown as DNA binding was only assessed in two samples for this group. ANOVA values were p<0.001 for NF-κB and p<0.05 for AP-1; ***p<0.001 v controls; †p<0.05 v healthy smokers (multiple comparison testing).
DISCUSSION
Increased neutrophils were found in the distal airspaces of patients with COPD, which increased further during exacerbations of the disease. The percentage of neutrophils in the distal airspace had a negative linear relationship with the severity of airways obstruction as assessed by the FEV1/FVC ratio. This increase in neutrophil numbers is concomitant with an increase in IL-8 levels in the proximal airway secretions in patients with stable and exacerbated COPD compared with smokers and non-smokers, and an increase in IL-8 levels in the distal airspaces in exacerbations of COPD. A new finding in this study is the depletion of GSH in the airspaces in exacerbations of COPD, indicating increased oxidative stress.
Although the pathological events that lead to the development of airways obstruction and exacerbations in COPD are still not completely understood, inflammation of the airways appears to be a critical factor. Indeed, by examining lung tissue sections, Hogg and colleagues found an increase in airways inflammation—evidenced by the enhanced presence of mucous exudates and inflammatory leucocytes—to be associated with COPD disease severity.24 Oxidative stress is thought to be an important component of inflammation through the activation of oxidant sensitive transcription factors leading to increased transcription of pro-inflammatory genes. Critical to the effects of oxidative stress is the protective counterbalance of antioxidant systems. A shift in this oxidant/antioxidant balance could result in an increase in oxidative stress which may cause cellular damage. In this regard, GSH appears to be an important antioxidant in the lungs and is present in high concentrations in epithelial lining fluid.14
A major limitation of our study is the fact that exacerbations were studied in patients with severe and very severe underlying COPD and compared with stable patients with moderate disease. Our interpretation of differences between exacerbations and the stable state may therefore actually be a reflection of differences in disease severity. However, increased numbers of neutrophils and their markers are seen in the airways during both severe and very severe exacerbations compared with stable disease and increased disease severity.25,26 Also, exacerbations are more common with increasing disease severity. Thus, although we cannot directly attribute our findings to the exacerbated condition in our patients, exacerbations and disease severity are very much associated with regard to the inflammatory response. We have confirmed that patients with COPD have increased numbers of neutrophils in the distal airways compared with healthy smokers and non-smokers. We also found substantial increases in neutrophil numbers during severe exacerbations of the disease, particularly during very severe exacerbations resulting in respiratory failure and the need for mechanical ventilation. In contrast, we found a significantly lower total cell count in patients with stable moderate COPD than in healthy smokers and patients with very severe COPD. Linden and colleagues27 also reported a lower total cell count in patients with airways obstruction. The fact that IL-8 protein levels in BAL fluid in patients with stable COPD is equivalent to levels detected during exacerbations suggests that the lower cell numbers in stable COPD is not due to difficulty in retrieving the lavage fluid. Furthermore, the highest cell counts recovered were for patients with very severe COPD where lavage fluid recovery is also low.
Although increased total cell counts and increased percentage neutrophils in sputum and BAL fluid have been reported previously,28–31 few studies have assessed inflammation in the airways during acute exacerbations of COPD. Qiu and colleagues32 reported increased neutrophil recruitment and gene expression of neutrophil chemoattractant (CXC) receptors in bronchial biopsies from patients with acute severe exacerbations of COPD compared with those with stable COPD and non-smoking healthy subjects. Aaron and colleagues26 measured sputum TNFα and IL-8 levels during exacerbations and found them to be significantly increased. Bhowmik et al2 examined sputum inflammatory markers in stable subjects who had frequent exacerbations and reported increased levels of IL-8 and IL-6 and total cell counts. In addition, more sputum and greater purulence of sputum was found over time during exacerbations,16 suggesting the presence of more inflammation over time. Previous reports in patients with mild exacerbations have shown an increase in eosinophil numbers33 that was not seen in our study of severe exacerbations of COPD. It should be noted that the patients in our study groups were not age matched, and those with moderate COPD and very severe COPD were significantly older than non-smokers and healthy smokers. This was unavoidable as the COPD patients attending the Royal Infirmary in Edinburgh constitute an older age group and the healthy individuals willing to participate in the study are not generally so elderly.
A role for IL-8 in the recruitment to and activation of neutrophils in the airways of patients with COPD has been proposed as it is detected in sputum and BAL fluid from patients with COPD.30,34–36 Although Yamamoto et al36 found a negative correlation between IL-8 levels in sputum and FEV1/FVC in patients with stable COPD, we were unable to confirm that IL-8 levels are increased in large airway secretions from patients with moderate stable COPD but we did find increased IL-8 levels in the distal airspaces. Increased IL-8 levels and neutrophil numbers in BAL fluid have previously been reported in patients with stable COPD.31 No significant difference was found in TNFα levels in either large airways secretions or BAL fluid (data not shown). However, it should be noted that the DTT used to break up the sputum mucin can interfere with the measurement of TNFα by ELISA37 which might explain our lack of a result. Interestingly, IL-8 levels in both large airways secretions and BAL fluid had a negative linear relationship with the severity of airways obstruction as measured by the FEV1/FVC ratio. It should be noted that these correlations were performed on all subjects—not just patients with COPD—because of the small numbers in individual groups. We found no evidence of a linear relationship between the percentage neutrophils and IL-8 levels in BAL fluid; however, such a relationship has been reported by other workers.6,38 These data therefore suggest that the IL-8 level may be useful as an indicator of disease severity and that, in addition to IL-8, other factors such as leukotriene B431 may be responsible for the neutrophil influx in exacerbations and stable COPD.
It has been reported that the IL-8 gene has both NF-κB and AP-1 binding sites in the promoter region that regulates its transcriptional activation.39 Both NF-κB and AP-1 are oxidant sensitive transcription factors that have been implicated in the regulation of numerous pro-inflammatory mediators pertinent to lung inflammation.7 We therefore assessed the nuclear translocation of NF-κB in bronchial biopsies and also in BAL fluid leucocyte populations. No significant difference in nuclear localisation of NF-κB was found in bronchial biopsies, although a trend was evident for increased NF-κB DNA binding in healthy smokers and patients with COPD. In contrast, BAL fluid leucocytes from healthy smokers showed a significant reduction in NF-κB DNA binding compared with non-smokers, while nuclear translocation of NF-κB during exacerbations of COPD did not differ from that in non-smokers. Since we obtained enough BAL fluid leucocytes from only two patients with stable COPD to determine NF-κB nuclear translocation (data not shown), we were unable to make a comparison with samples obtained during exacerbations. However, we have previously reported a comparison of NF-κB DNA binding in sputum leucocytes from patients with stable and acute exacerbations of COPD and found a significant increase during exacerbations.8 There are reports in the literature of both increased and decreased NF-κB activity for cigarette smoke exposure in vitro. Cigarette smoke exposure in vitro reduced peripheral blood monocyte NF-κB DNA binding activity.40,41 In contrast, acute cigarette smoke inhalation by guinea pigs induced NF-κB activation in alveolar macrophages.42 It appears that the variability in NF-κB activation/deactivation in response to cigarette smoke may be due to the site and cell type exposed and whether such exposure is acute or chronic.
A possible explanation for the reduced NF-κB DNA binding in BAL fluid leucocytes may be the excessive production of oxidants such as hydrogen peroxide and nitric oxide during exacerbations which inactivate inhibitory κB kinase (Iκ kinase; IKK),43,44 the enzyme responsible for the phosphorylation of IκBα, resulting in its degradation and activation of NF-κB. Alternatively, the lower levels of NF-κB DNA binding observed in BAL fluid leucocytes from healthy smokers may relate to the higher levels of antioxidants offering protection against oxidant stress by inhibiting NF-κB activation and translocation to the nucleus,45 leading to less gene activation.
In accordance with the latter, and as previously reported,4,14 we found higher levels of the antioxidant GSH in BAL fluid in smokers. We now report, for the first time, the levels of GSH in the distal airspaces of patients with COPD. Patients with stable COPD had higher than normal GSH levels in the BAL fluid which became depleted during exacerbations of the disease. The lower levels of GSH found in smokers with COPD compared with healthy smokers could facilitate NF-κB activation leading to inflammation, which is evident in the increased neutrophil numbers and IL-8 levels in the BAL fluid.
In healthy smokers, and possibly in patients with stable COPD, the increased GSH levels in response to oxidative stress are associated with an enhanced expression of γ-glutamylcysteine synthetase (γ-GCS), the main enzyme in the synthesis of GSH.15 In contrast, the reduced GSH levels in acute exacerbations may reflect an insufficient response to oxidative stress in the form of reduced γ-GCS activity resulting in lower GSH levels or direct depletion by an overwhelming oxidative burden from reactive oxygen species released by activated neutrophils. Support for the former mechanism comes from the observed decrease in DNA binding of the transcription factor AP-1 which regulates γ-GCS in exacerbations of COPD.15 γ-GCS, the rate limiting enzyme for GSH synthesis, was previously found to be downregulated in lung epithelial cells following acute exposure to cigarette smoke with a concomitant depletion of GSH.15 With inadequate protection, the lung tissue in these patients could be predisposed to injury. Indeed, in a study by Koul and colleagues,46 a decrease in lung GSH levels in mice exposed to cigarette smoke was associated with an increase in lipid peroxidation products. Assessment of lipid peroxidation markers in exhaled breath condensate also revealed an increase in patients with COPD compared with healthy smokers and non-smokers.47 Taken together, these data suggest an important role for GSH in the prevention of peroxidative lung damage in patients with COPD.
In conclusion, our data suggest that there is impaired antioxidant status in the peripheral airways of patients with stable COPD which is more pronounced in exacerbations. This is reflected in the presence of an inflammatory response, as shown by an increase in cell numbers and cytokine levels. A comparison of the parameters measured for very severe exacerbations compared with severe exacerbations of COPD revealed an increase only in the number of leucocytes in BAL fluid.
Acknowledgments
The authors thank all the subjects who participated in the study.
REFERENCES
Footnotes
-
The study was funded by Novartis Pharmaceutical Company, Horsham, UK, and part funded by FIS 00/0281 and Red Respira (RTIC C03/011 Fondo de Investigacion Sanitaria).
Linked Articles
- Airwaves