Article Text

Abstract
Background The genetic basis for developing asthma has been extensively studied. However, association studies to date have mostly focused on mild to moderate disease and genetic risk factors for severe asthma remain unclear.
Objective To identify common genetic variants affecting susceptibility to severe asthma.
Methods A genome-wide association study was undertaken in 933 European ancestry individuals with severe asthma based on Global Initiative for Asthma (GINA) criteria 3 or above and 3346 clean controls. After standard quality control measures, the association of 480 889 genotyped single nucleotide polymorphisms (SNPs) was tested. To improve the resolution of the association signals identified, non-genotyped SNPs were imputed in these regions using a dense reference panel of SNP genotypes from the 1000 Genomes Project. Then replication of SNPs of interest was undertaken in a further 231 cases and 1345 controls and a meta-analysis was performed to combine the results across studies.
Results An association was confirmed in subjects with severe asthma of loci previously identified for association with mild to moderate asthma. The strongest evidence was seen for the ORMDL3/GSDMB locus on chromosome 17q12-21 (rs4794820, p=1.03×10(−8) following meta-analysis) meeting genome-wide significance. Strong evidence was also found for the IL1RL1/IL18R1 locus on 2q12 (rs9807989, p=5.59×10(−8) following meta-analysis) just below this threshold. No novel loci for susceptibility to severe asthma met strict criteria for genome-wide significance.
Conclusions The largest genome-wide association study of severe asthma to date was carried out and strong evidence found for the association of two previously identified asthma susceptibility loci in patients with severe disease. A number of novel regions with suggestive evidence were also identified warranting further study.
- Severe asthma
- GWAS
- AUGOSA
- GABRIEL
- asthma
- asthma mechanisms
- asthma epidemiology
- perception of asthma/breathlessness
- respiratory measurement
- respiratory muscles
- COPD mechanisms
- airway epithelium
- allergic lung disease
- COPD epidemiology
- asthma guidelines
- cystic fibrosis
- exhaled airway markers
- lung physiology
- paediatric asthma
- paediatric lung disaese
- asthma pharmacology
- COPD pharmacology
- cytokine biology
- macrophage biology
- clinical epidemiology
- tobacco and the lung
- COPD exacerbations
- asthma in primary care
- bacterial infection
- emphysema
- eosinophil biology
- pulmonary eosinophilia
- respiratory infection
- occupational lung disease
- COPD pathology
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
- Severe asthma
- GWAS
- AUGOSA
- GABRIEL
- asthma
- asthma mechanisms
- asthma epidemiology
- perception of asthma/breathlessness
- respiratory measurement
- respiratory muscles
- COPD mechanisms
- airway epithelium
- allergic lung disease
- COPD epidemiology
- asthma guidelines
- cystic fibrosis
- exhaled airway markers
- lung physiology
- paediatric asthma
- paediatric lung disaese
- asthma pharmacology
- COPD pharmacology
- cytokine biology
- macrophage biology
- clinical epidemiology
- tobacco and the lung
- COPD exacerbations
- asthma in primary care
- bacterial infection
- emphysema
- eosinophil biology
- pulmonary eosinophilia
- respiratory infection
- occupational lung disease
- COPD pathology
Key messages
What is the key question?
-
The aim of this study was to identify genetic determinants of severe asthma and to evaluate whether susceptibility to severe asthma differs from that of mild to moderate asthma.
What is the bottom line?
-
The first genome-wide association study of severe asthma was undertaken which identified the contribution of some but not all genetic loci previously associated with mild to moderate disease. Suggestive evidence for a number of novel loci associated with severe disease is also reported.
Why read on?
-
Novel loci, which may be specific to severe asthma, potentially provide further insight into disease mechanisms and warrant further study.
Introduction
Asthma is a chronic inflammatory condition of the airways characterised by recurrent episodes of reversible airway obstruction and increased bronchial hyper-responsiveness.1 Approximately 10% of patients with asthma are prone to severe exacerbations and remain symptomatic despite treatment with high-dose inhaled corticosteroids (ICS) and long-acting β2-adrenergic receptor agonists.2 This subgroup of patients disproportionately consume healthcare resources related to asthma and contribute the largest proportion of morbidity and mortality.3
The genetic basis for developing asthma has been extensively investigated; numerous candidate genes have been studied for association with asthma due to the potential biological effects on airway function of the relevant gene products, although replication has been inconsistent.4–10 In addition, recent genome-wide association studies (GWAS) in asthma have identified a widely replicated locus on chromosome 17q12-21 containing genes ORMDL3, CCL11 and GSDML, and additional genes including CHI3L1, IL1RL1 and WDR36 on chromosomes 1q31, 2q12, and 5q22 respectively.11–13
Recently, the biggest collaborative effort thus far investigating the genetic determinants of asthma was published by the GABRIEL consortium. This study consisted of 10 365 case subjects and 16 110 controls and observed genome-wide significance between asthma and single nucleotide polymorphisms (SNPs) within previously reported loci and genes, including genes IL18R1, HLA-DQ, IL33, and chromosome 17q12-21, the latter specific to childhood-onset disease.14 Subsequently, a large Australian collaborative effort carried out a GWAS in 2669 cases and 4528 controls. A selected number of identified loci were then followed up in a replication analysis following a meta-analysis with the results of the GABRIEL study. This paper provided independent support for loci reported by GABRIEL: IL18R1, IL33, ORMDL3 and IL2RB, and identified a further two loci reaching genome-wide significance in the combined analysis of all studies (n=57 800): IL6R and chromosome 11q13.5.15
Furthermore, the EVE consortium conducted a meta-analysis of North-American GWAS (n=5416 in meta-analysis, n=12 649 in replication) inclusive of individuals of European American, African American or African Caribbean, and Latino ancestry. This study reported that previously identified loci on 17q21, near IL1RL1, TSLP and IL33, were robust to ethnic differences showing significant association in all three ethnic groups.16
In addition, a single small GWAS (473 cases, 1892 controls) was conducted in 2009 on a population of patients with severe or difficult to treat asthma from The Epidemiology and Natural History of Asthma: Outcomes and Treatment Regimes (TENOR) study and identified association with multiple SNPs in the RAD50-IL13 and HLA-DR/DQ regions, although no loci reached conventional GWAS significance criteria.17
These studies have generally involved subjects with mild asthma. The aims of the current study were first to identify genetic determinants of severe asthma, and second to evaluate whether susceptibility to severe asthma differs from that of mild to moderate asthma.
Methods
Participants
Discovery cohort
We genotyped 1026 individuals of European ancestry with severe asthma based on the Global Initiative for Asthma (GINA) criteria.18 Only subjects in classes 3–5 were included, recruited across UK-based centres. Subjects were selected from individuals participating in the Difficult/Severe Asthma (BTS) study (n=290), supplemented with subjects from other centres. A total of 3353 control subjects without history of asthma or wheeze (clean controls) were collected from the UK and Western Australia, all of whom were of European ancestry.
Replication cohort
A replication cohort of 231 cases with more severe asthma based on clinical examination by a respiratory physician and treatment steps (ie, receiving ICS ≥400 μg in combination with a long- and short-acting β2-adrenergic receptor agonists and short-acting beta-agonist) and 1345 controls without asthma were recruited by the Australian Asthma Genetics Consortium (AAGC) study. All individuals from Australia were of European ancestry.
Baseline characteristics and participant recruitment of all study populations per centre are described in the online repository.
Genotyping and procedures
Participants were genotyped using the Illumina HumanHap550K, 610K, 660K, and 1.2M SNP chip platforms (Illumina, San Diego, California, USA). To minimise bias due to the use of different genotyping platforms in case and control cohorts, only the 490 303 SNPs in common across all six platforms were used. Furthermore, the use of principal components analysis (PCA) covariates provides some correction for assay effects.19 We also excluded any SNPs that showed a significant difference in allele frequencies between the three control groups (p<10(−6)). However, it is not possible to completely eliminate bias due to genotyping platform and centre and hence we sought replication. A table describing the number of samples genotyped on each platform has been included (online table E1). Within each study, individuals with <90% of SNPs called were excluded and SNPs were excluded if they had low call rates (proportion of genotypes called <90%), were not in Hardy-Weinberg equilibrium (HWE, p<10(−4)), had a low minor allele frequency (MAF<1%) or with differential missingness between cases and controls (p<10(−4)). PCA was carried out to detect outlying samples and to correct for residual population structure using EIGENSOFT V.3.0 (online figure E1).19
Statistical analyses
Association tests of genotyped SNPs were carried out using PLINK V.1.07 with an additive genetic model with the first 10 principal components as covariates.20 We tested association with 480 889 SNPs present across all cohorts; 21 SNPs showing a significant difference in allele frequencies (p<10(−6)) between the three control groups were removed. We identified regions of interest as those with a sentinel SNP showing association with asthma (at a threshold of p<5×10(−5)) with at least one additional SNP within 500 Kb also reaching a threshold of p<5×10(−5).
1000 Genomes imputation
Imputation was used to improve the resolution of regions identified for association from genotyped data. Genotyped SNPs were used for imputation to 6.9 million SNPs using the June 2010 release of the 1000 Genomes CEU reference panel comprising 120 individuals genotyped at 6 858 242 SNPs.21 Haplotypes were phased by comparing genotypes across our 4279 cases and controls with all alleles defined on the positive strand using MaCH.22 Imputation of genotypes was carried out by comparing haplotype blocks in our phased samples with those in the 1000 Genomes reference panel using minimac.23 A measure of confidence in the imputation is given by the metric r2 imp which is an estimate of the correlation between imputed and true genotypes ranging from 0 to 1 (1 for genotyped SNPs). Quality control was carried out to exclude SNPs with MAF<1% or imputation quality r2 imp <0.3 (recommended r2 imp filter to exclude 70% of poorly imputed SNPs).22 Association tests were performed with ProbABEL using a logistic model with the dose of the effect allele (on a continuous scale between 0 and 2 reflecting imputation uncertainties) as the independent variable and 10 ancestry principal components derived from our genotyped SNPs as covariates.24 Post-association filters were applied to remove SNPs showing significant association in control–control comparisons (p<10−6) leaving 6 103 628 SNPs.
Replication and meta-analysis
Subjects from the AAGC study were used to test replication of 24 SNPs identified in the discovery GWAS and subsequent imputation analyses. This SNP list consisted of six genotyped SNPs from the regions identified in genotype analyses, four imputed SNPs in the same regions with a lower p value than the original genotyped SNPs, two SNPs with p>10(−5) responsible for secondary peaks in regions with known asthma genes, and 12 SNPs from new regions identified through imputation. In order to select the best candidate imputed SNPs for replication we used a lower p value of <10(−5) and a stricter imputation quality (r2 imp≥0.7) than used for fine-mapping around genotyped SNPs. Statistical significance for replication was assessed using a 5% significance threshold and results of inverse-variance weighted meta-analysis assessed using conventional criteria for genome-wide significance (p=5×10(−8)).25
Evaluation of GABRIEL loci
We also investigated the contribution of polymorphisms identified for mild to moderate asthma by the GABRIEL consortium in our severe asthma population. We examined regions within 500 Kb of SNPs reported to be associated with asthma in the GABRIEL study, including both regions reported to have reached genome-wide significance (p≤7.2×10(−8)) and those providing suggestive evidence of association (p≤5×10(−7)) in GABRIEL.
Comparison of severe versus mild to moderate asthma
A total of 1028 individuals of European ancestry with a history of doctor diagnosed asthma at GINA steps 1 or 2 were collected from the WTCCC2, T1DGC and Busselton populations. A GWAS was then carried out comparing the 1026 patients with severe asthma against the 1028 patients with mild to moderate asthma. All genotyping and quality control procedures were conducted as above for the severe asthma versus clean control analyses. We tested association with 488 889 SNPs present across all cohorts. Imputation was used to improve resolution of identified regions and replication was assessed in the AAGC study using a comparison of the 231 severe cases versus 1085 patients with mild to moderate asthma identified as never having received steroid medication in their lifetime.
Results
Genotype data for 933 cases and 3346 controls were available for the primary discovery analysis after quality control. Replication of identified SNPs was assessed in 231 cases and 1345 controls. Characteristics of the study cohorts are summarised in the online appendix. The test statistic inflation factor λ for the discovery GWAS was modest (λ=1.04). Results shown by the quantile–quantile plot suggest the presence of multiple loci with modest effects (figure 1A).

Quantile–quantile plot and Manhattan plot of study results. (A) Quantile–quantile plot of observed versus expected log10 p values for all tested genotyped single nucleotide polymorphisms (SNPs). (B) Manhattan plot of – log10 p values for all tested SNPs against genomic position. *Note: the signal on chromosome 13q31.1 was intergenic.
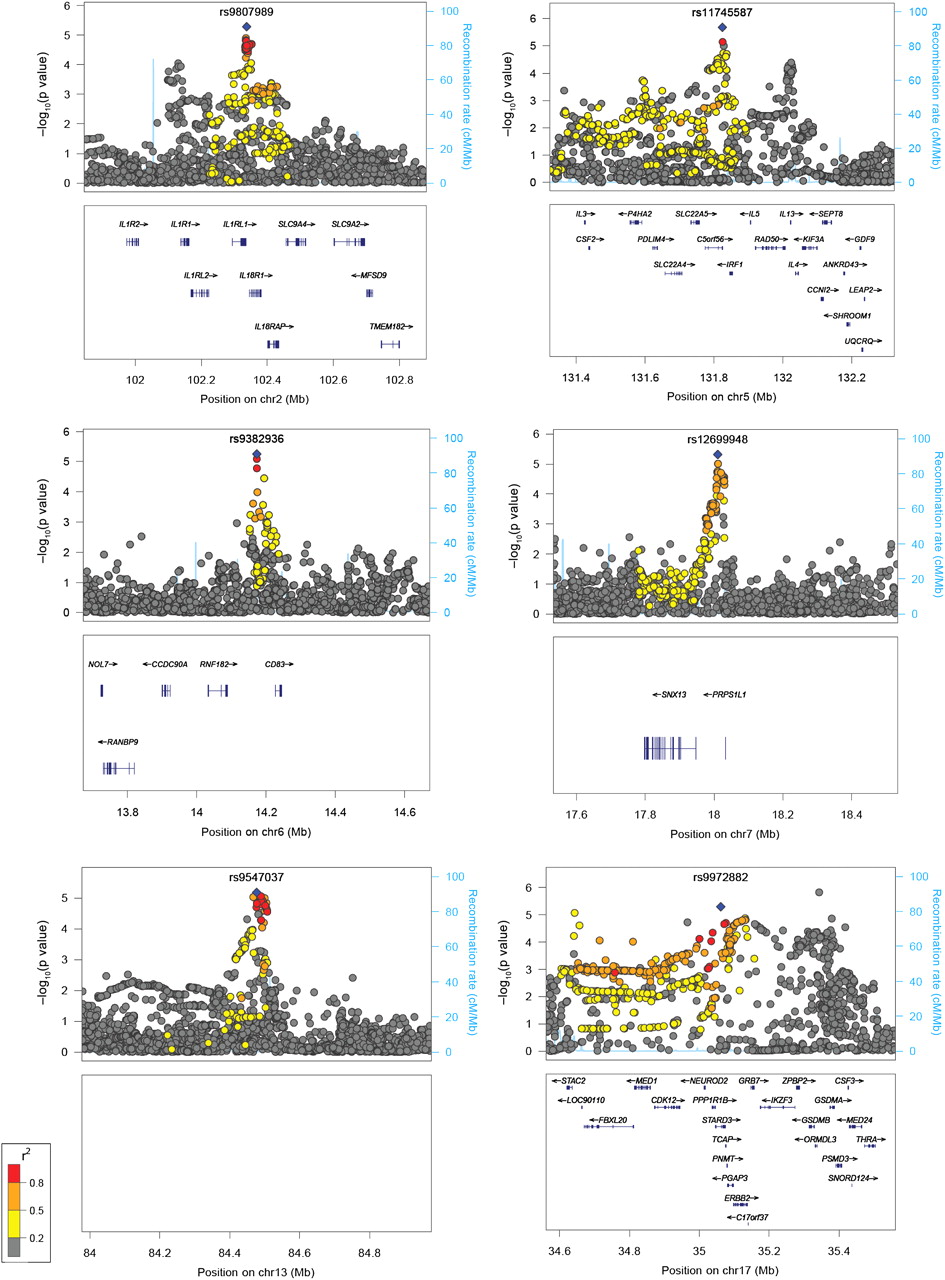
In the initial analysis of genotyped data in the discovery cohort, no SNPs met genome-wide significance for association with severe asthma using a conservative cut-off defined by the Bonferroni correction: p=1.04×10(−7) (figure 1B). We therefore went on to evaluate other potential loci with statistical significance below this threshold. A total of eight SNPs were identified with p<5×10(−5), with at least one other SNP within 500 kb with p<5×10(−5). Assessment for supporting evidence within the region for these SNPs suggests that six of these loci may contain susceptibility genes for severe asthma: rs3771166 within IL18RL1 on 2q12.1 (p=1.93×10(−5)), rs11745587 in the 3′ untranslated region (UTR) of C5orf56 on 5q31.1 (p=2.09×10(−6)), rs9382936 on 6p23 (p=5.61×10(−6)) tagging CD83, rs12699949 on 7p21.1 (p=1.19×10(−5)) tagging PRPS1L1, rs2496764 within an intergenic region on 13q31.1 (p=7.86×10(−6)), and rs1810132 within ERBB2 on 17q12-21 (p=1.73×10(−5)) (table 1, figure 2). We did not follow up those loci characterised by a single SNP within 500 kb showing an association (p<5×10(−5)); these loci are listed in online table E4.
Single nucleotide polymorphisms (SNPs) showing highest association signals for severe asthma

{kind=link}
{kind=link}
Region plots for suggestive loci. The region plots following imputation show the statistical significance of each single nucleotide polymorphism (SNP) on the –log10 scale as a function of chromosome position (National Center for Biotechnology Information (NCBI) build 36). The sentinel SNP is shown in blue and the correlation (r2) of each of the surrounding SNPs to the sentinel SNP is shown by their colour (see key). Fine scale recombination rate is plotted in blue.
Imputation was used to refine association signals within all six loci. This analysis identified a further four SNPs with a lower p value than the original genotyped SNP in five regions: rs9807989 on 2q12.1 (p=5.20×10(−6)) tagging IL18RL1, rs12699948 on 7p21.1 (p=4.84×10(−6)) tagging PRPS1L1, rs9547037 intergenic on 13q31.1 (p=6.60×10(−6)), and rs9972882 within STARD3 on 17q12-21 (p=5.17×10(−6)). Two imputed SNPs produced secondary peaks in regions with known asthma-associated genes: rs13035227 on 2q12.1 (p=8.91×10(−5)) tagging IL1RL1, and rs847 in the 3′ UTR of IL13 on 5q31 (p=4.05×10(−5)). An additional 12 SNPs with p<10(−5) and r2 imp=0.7 produced signals in new regions identified through imputation (1.19×10(−5)≤p≤2.82×10(−7)) (table 1, online figure E2).
Two loci were replicated in the AAGC study cohort. The first of these was on 17q12-21 by rs4794820 tagging the ORMDL3 locus (p=0.002). The second was on 2q12.1 by a cluster of three SNPs: rs3771166 within IL18R1 (p=0.001), rs9807989 tagging IL18R1 (p=0.003), and rs13035227 tagging IL1R1 (p=0.002). In non-replicated regions, a consistent direction of effect for the minor allele was seen across studies for 18 out of the 22 remaining SNPs. Following meta-analysis, the signal on ORMDL3 met conventional genome-wide significance (p=1.03×10(−8)) and the signal on IL18RL1 approached this threshold (p=5.59×10(−8)) (table 1, figure 2).
Next, we proceeded to test all SNPs reported in the GABRIEL study for both genome-wide significance and suggestive evidence for association with mild to moderate asthma to assess the degree of association with severe asthma in Asthma UK Genetics of Severe Asthma (AUGOSA) (online table E2 and E3 and figure E2). In general, as might be expected, we also found an association with these loci apart from rs2284033 on chromosome 22q12 (p=0.105) and rs11071559 on chromosome 15q22 (p=0.159).
A comparison of patients with severe asthma versus those with mild to moderate asthma was carried out (online figure E3). The test statistic inflation factor λ for this GWAS was again modest (λ=1.04). A single SNP met genome-wide significance in this analysis: rs981516 intergenic on 4p32.1 (p=3.34×10(−8), OR 1.50 95% CI 1.30 to 1.73). However, this was not replicated in the AAGC study (p=0.451) although the same direction of effect was seen for the minor allele.
Discussion
We conducted the largest severe asthma GWAS to date in a cohort of 933 cases defined by GINA steps three or above for severity and 3346 clean controls to determine if there are common genetic polymorphisms contributing to susceptibility to severe asthma.
Overall, we did not identify any novel SNPs meeting genome-wide significance. We carried out further analysis of results for polymorphisms just below this threshold to look for regions which did not meet standard genome-wide significance but had supporting evidence with at least one additional SNP with p<5×10(−5) within 500 Kb. Using this criterion, we identified six loci with suggestive evidence for association. Two of these loci, chromosomes 2q12 (p=5.20×10(−6)) and 17q12-21 (p=5.17×10(−5)) implicating the IL1RL1/IL18R1 and ORMDL3/GSDMB loci respectively have been previously reported by GWAS for association with mild to moderate asthma.11 ,13–16 Both of these loci replicated in a second cohort of 231 severe asthma cases and 1345 controls (2q12, p=0.001; 17q12-21, p=0.002). Evidence for the 17q12-21 locus became genome-wide significant following meta-analysis and was just below this threshold for the 2q12 locus, highlighting a potentially important role for these loci in asthma irrespective of severity.
A previous GWAS in which the main phenotype was blood eosinophil counts identified evidence for an association with asthma for SNPs in IL1RL1 and its ligand, IL33.13 Subsequently, asthma association of both loci have been shown to be robust to differences in ancestry.16 We report association with the same SNP, rs3771166 within IL18R1on chromosome 2q12. Rs3771166 was also reported by the GABRIEL consortium showing the same direction of effect for association with the minor allele. A stronger protective effect size is observed for this polymorphism within our severe asthma cohort (OR=0.79) compared with that shown in the GABRIEL study (OR=0.87).14 With current data, we are unable to determine if association is driven by the IL1RL1 or the IL18R1 gene. However, both genes are plausible biological candidates in the inflammatory cascade in the pathway to asthma pathogenesis.26 ,27
Associations with asthma and SNPs located on chromosome 17q12-21 have been reported and replicated across multiple study populations.11 ,14 ,28 ,29 Despite this, the region on 17q12-21 harbours a number of genes of poorly understood function. In combination with the complex linkage disequilibrium structure in this locus, it is difficult to be sure which genetic variants are causal. Furthermore, 17q12-21 has previously been suggested to be exclusive to childhood-onset disease.11 ,14 Although, we have not assessed age of onset in the current study, we found significant association with this locus despite studying predominantly adult subjects (mean age of disease onset: 21 years), thus challenging this hypothesis.
We report an additional four loci as showing suggestive evidence of association with severe asthma. The most significant result on chromosome 6p23 implicates CD83, the gene encoding for the CD83 antigen expressed on dendritic cells and which may play a role in immune modulation in the airways.30 The SNP on chromosome 5q31 lies within the 3′ UTR of C5orf56, however it is downstream of the Th2 cytokine genes IL4 and IL13, and RAD50, a region previously reported for suggestive association with severe asthma by the TENOR study.17 The remaining two regions downstream of PRPS1L1 on 7p21 and intergenic on 13q31 are potentially novel with unknown function in asthma and warrant further study.
We list in online table E4 those loci not followed up in the AAGC study due to not meeting our strict predefined threshold for supporting SNPs in the region (online table E4). Interestingly, these loci include TSLP, which was identified as a potential asthma locus previously.16 We then undertook a MAGENTA31 pathway analysis on the full GWAS dataset to look for enrichment of association in known biological pathways from six databases (Gene Ontology, Ingenuity Pathway, KEGG, PANTHER Pathways, PANTHER Molecular Function and PANTHER Biological Processes). No pathway was significant after correction for multiple testing, although the Fc Epsilon RI Signalling pathway reached nominal significance (p=0.0014) and includes IL13.
We then went on to assess the contribution of previously identified asthma susceptibility loci in patients with severe disease reported by the recent GABRIEL study.14 As the GABRIEL study is currently the largest published association study investigating the genetic determinants of asthma, we aimed to determine if these signals also contribute to disease susceptibility in our severe asthma cohort. Individuals in AUGOSA were included in the total case subjects in the GABRIEL study. However, as they only constitute a relatively small proportion (8.9%), an association signal within GABRIEL for mild to moderate asthma would be unlikely to originate only from AUGOSA individuals.
For genome-wide significant results compared with mild to moderate asthma from GABRIEL, effect sizes for the risk allele are greater in our cohort of patients with severe asthma for rs3771166 on chromosome 2q21: the observed OR for the protective allele was 0.79 compared with 0.87 in GABRIEL for rs2305480 on chromosome 17q12-21; the observed OR for the protective allele was 0.80 compared with 0.85 for rs3894194 on chromosome 17q12-21; the OR for the risk allele was 1.25 compared with 1.17. Further to this, we show supporting evidence for loci reported by previous studies such as the GABRIEL study, including IL13 on 5q31 (p=9.30×10(−5)) and SMAD3 on 15q22.33 (p=2.88×10(−4)) containing weaker association signals which fell just below the threshold for significance chosen for our primary analyses.
The explanations behind the increased effect size of risk alleles in severe asthma are potentially twofold. The most likely explanation is that the contribution of genetic effects driven by variation in these genes is greater in patients with more severe asthma. However, it is possible that in populations with milder asthma misclassification of cases and controls may result in an underestimate of true effect sizes. Furthermore, differences in population sizes used in analyses may also reflect our varying ability to determine robust effect sizes.
A comparison of severe versus mild to moderate asthma was carried out. This identified a potentially novel locus on 4p32.1 meeting genome-wide significance which may be specific to the development of severe as opposed to milder forms of asthma. The sentinel SNP rs981516 is in an intergenic region but in linkage disequilibrium with rs17291045 (r2=0.423), a SNP previously reported to be strongly associated with progression in HIV-1 infection and may have functional effects on viral control.32 The identified SNP was not replicated in the AAGC study, however given the modest effect size seen in the discovery GWAS (OR=1.50), a much larger follow-up cohort would have been necessary to reliably assess replication.
Although the current study is the largest effort so far to determine genetic determinants of severe asthma, we are still limited by the numbers of subjects in being able to generate enough statistical power to detect all variants with modest effects. While we can probably exclude major effects being driven by a single gene as a specific risk for severe asthma, our data suggest there may be a number of loci which may be specific for severe asthma but with relatively small overall contributions to the risk of developing severe disease. The obvious solution to resolving this issue is to undertake further replication studies in much larger severe asthma populations. However, these populations by their very definition are hard to recruit: the current study included subjects recruited from eight major centres in the UK and replication in a second study consisting of subjects recruited from two major centres in Australia. Hence, obtaining suitable replication populations to take this work forward will require additional international efforts to establish appropriately large populations with severe disease.
In summary, we provide evidence to support an enhanced role for known genetic risk factors for asthma in the development of severe disease, and also have identified novel loci which may be specific to the development of severe as opposed to milder forms of asthma. These results potentially provide insight into the biological mechanisms that underlie the regulation of severe asthma and might help in the discovery of novel therapeutic targets for disease.
Acknowledgments
We would like to thank all members of study cohorts taking part in this study, particularly those who provided consent to use of their DNA for genetic epidemiologic analyses and the research staff who contributed to the successful completion of all field studies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
-
YIW and NRGS are joint first authors.
-
↵* A full list of collaborators is available in the online appendix.
-
Funding We acknowledge support of Asthma UK, the Wellcome Trust and the Medical Research Council. Specifically, we acknowledge use of phenotype and genotype data from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02 (http://www.b58cgene.sgul.ac.uk/). Genotyping for the B58C-WTCCC subset was funded by the Wellcome Trust grant 076113/B/04/Z. The B58C-T1DGC genotyping utilised resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), National Human Genome Research Institute (NHGRI), National Institute of Child Health and Human Development (NICHD), and Juvenile Diabetes Research Foundation International (JDRF) and supported by U01 DK062418. B58C-T1DGC GWAS data were deposited by the Diabetes and Inflammation Laboratory, Cambridge Institute for Medical Research (CIMR), University of Cambridge, which is funded by Juvenile Diabetes Research Foundation International, the Wellcome Trust and the National Institute for Health Research Cambridge Biomedical Research Centre; the CIMR is in receipt of a Wellcome Trust Strategic Award (079895). CEB is funded as a Wellcome Trust Senior Clinical Fellow. MDT has been supported by MRC fellowships G0501942 and G0902313. The Australian Asthma Genetics Consortium is funded by the National Health and Medical Research Council of Australia (613627).
-
Competing interests None.
-
Ethics approval Ethics approval was provided by individual study centres during study recruitment.
-
Provenance and peer review Not commissioned; externally peer reviewed.