Article Text

Abstract
Background Development of emphysema and vascular stiffness in chronic obstructive pulmonary disease (COPD) may be due to a common mechanism of susceptibility to pulmonary and systemic elastin degradation.
Objectives To investigate whether patients with COPD have evidence of systemic elastin degradation in the skin.
Methods The authors measured cutaneous elastin degradation using immunohistochemistry (percentage area of elastin fibres) in sun-exposed (exposed) and non-sun-exposed (non-exposed) skin biopsies in 16 men with COPD and 15 controls matched for age and cigarette smoke exposure. Quantitative PCR of matrix metalloproteinase (MMP)-2, -9, -12 and tissue inhibitor of metalloproteinase-1 mRNA and zymography for protein expression of MMP-2 and -9 were performed on homogenised skin. Arterial stiffness and emphysema severity were measured using carotid-femoral pulse wave velocity and quantitative CT scanning.
Results Skin elastin degradation was greater in exposed and non-exposed skin of patients with COPD compared with controls (exposed, mean (SD); 43.5 (12.1)% vs 26.3 (6.9)%, p<0.001; non-exposed 22.4 (5.2)% vs 18.1 (4.3)%, p=0.02). Cutaneous expression of MMP-9 mRNA and proMMP-9 concentrations was increased in exposed skin of COPD patients (p=0.004 and p=0.02, respectively) and was also associated with increased skin elastin degradation (r=0.62, p<0.001 and r=0.47, p=0.01, respectively). In the entire cohort of ex-smokers, cutaneous elastin degradation was associated with emphysema severity, FEV1 and pulse wave velocity.
Conclusions Patients with COPD have increased skin elastin degradation compared with controls, which is related to emphysema severity and arterial stiffness. Systemic elastin degradation due to increased proteolytic activity may represent a novel shared mechanism for the pulmonary, vascular and cutaneous features of COPD.
- COPD
- emphysema
- systemic effects
- elastin
- proteases
- COPD mechanisms
- lung proteases
- COPD epidemiology
- COPD exacerbations
- COPD pathology
- oxidative stress
- macrophage biology
- asthma
- cystic fibrosis
- imaging/CT, MRI, etc
- lung cancer
- primary pulmonary hypertension
- pulmonary embolism
- airway epithelium
- exhaled airway markers
- macrophage biology
- neutrophil biology
Statistics from Altmetric.com
- COPD
- emphysema
- systemic effects
- elastin
- proteases
- COPD mechanisms
- lung proteases
- COPD epidemiology
- COPD exacerbations
- COPD pathology
- oxidative stress
- macrophage biology
- asthma
- cystic fibrosis
- imaging/CT, MRI, etc
- lung cancer
- primary pulmonary hypertension
- pulmonary embolism
- airway epithelium
- exhaled airway markers
- macrophage biology
- neutrophil biology
Key messages
What is the key question?
Is systemic elastin degradation the mechanism of increased arterial stiffness and skin wrinkling in chronic obstructive pulmonary disease?
What is the bottom line?
Men with chronic obstructive pulmonary disease (COPD) have increased skin elastin degradation compared with controls matched for age and smoking history, and this was associated with arterial stiffness and emphysema severity.
MMP-9 was upregulated in the skin of COPD patients.
Why read on?
Systemic elastin degradation due to increased proteolytic activity may represent a novel shared mechanism for the pulmonary, vascular and cutaneous features of chronic obstructive pulmonary disease.
Introduction
Chronic obstructive pulmonary disease (COPD) is defined as a condition with extra-pulmonary effects and systemic consequences, including cardiovascular disease.1 ,2 Systemic vascular dysfunction is emerging as a specific mechanism that may contribute to the increased cardiovascular risk in COPD.3–5 We have demonstrated that systemic arterial stiffness correlates with the severity of emphysema in COPD patients.6 Thus, we postulated that elastin degradation in the lung parenchyma and systemic arterial walls is the mechanistic link between the pulmonary and systemic vascular manifestations of COPD.
Elastin is an essential structural protein in the lungs, maintaining airway patency and ensuring elastic recoil. Emphysematous changes are, in part, due to elastin fibre breakdown which causes parenchymal destruction, reduced lung compliance and airway collapse.7 An imbalance of proteases and antiproteases in COPD, with a net increase in elastolytic activity, is thought to play an important role in the pathogenesis of emphysema,8 and recent work has focused on the role of matrix metalloproteinases (MMPs) and their equivalent antiproteases.9 ,10 MMPs have also been implicated as a pathogenic mechanism for the increased arterial stiffness that occurs with ageing, caused in part by elastin breakdown in the arterial wall.11 ,12
The hypothesis that COPD may be associated with both local lung and systemic abnormalities in connective tissue was addressed by Smith et al in 1967. Having shown abnormalities in the dermis of patients with obstructive lung disease, primarily in collagen, the authors postulate that these changes may ‘reflect a primary defect of connective tissue in the body as a whole’.13 However, the smoking history of the subjects is not reported. More recent studies have suggested that elastin degradation may occur outside the pulmonary compartment in COPD. Lee et al have reported increased circulating elastin antibodies in the plasma of emphysematous compared with non-emphysematous subjects.14 Furthermore, Patel et al reported that skin wrinkling was associated with emphysema in smokers, and postulated that this may be due to changes in collagen and elastin.15
We hypothesise that the increase in elastin degradation and protease–antiprotease imbalance responsible for emphysema is present in systemic tissue, causing increased arterial stiffness and skin wrinkling. We therefore examined skin biopsies from patients with COPD and matched control ex-smokers for evidence of elastin degradation.
Methods
Thirty-five male ex-smokers were recruited for a study of vascular function from primary care and a hospital respiratory outpatient clinic.3 Of these, 16 men with COPD and 15 male controls agreed to undergo skin biopsies. Inclusion and exclusion criteria of these study subjects have been previously published.3 In brief, the subjects with COPD had a history consistent with the disease and postbronchodilator spirometric evidence of airflow limitation (FEV1 to FVC ratio<0.7), while the controls had normal spirometry and no symptoms of respiratory disease. All subjects were ex-smokers of at least 6 months and had a greater than 10 pack year smoking history. They had no history of vascular disease or any other systemic inflammatory condition, and were not prescribed statins, oral corticosteroids or long-term oxygen therapy. Prebronchodilator and postbronchodilator spirometries were measured (Alpha Spirometer; Vitalograph, Buckingham, UK) according to American Thoracic Society/European Respiratory Society standards.16 Serum C reactive protein concentrations were measured using a highly sensitive immunonephelometric assay (Behring BN II nephelometer; Global Medical Instruments, Ramsay, Minnesota, USA). All studies were conducted at the Wellcome Trust Clinical Research Facility, Royal Infirmary, Edinburgh, approved by Lothian Regional Ethics Committee and conducted with the written informed consent of all participants.
Skin biopsy processing and staining
Two 4 mm punch biopsies were obtained from the sun-exposed skin of the dorsal surface of the forearm and non-sun-exposed skin of the buttock. Tissue was fixed in formalin and embedded in paraffin for morphometric analysis, snap-frozen and stored at −80°C for protein extraction and stored in RNAlater solution (Applied Biosystems, Carlsbad, California, USA) at −20°C for RNA extraction.
Histological evaluations were initially made in 3 μm thick elastic van Gieson-stained sections. Immunohistochemistry was performed using a monoclonal primary antibody to elastin (anti-human, Novocastra, Newcastle, UK) and a dextran polymer secondary antibody (Envision, Dako, Cambridge, UK; figure 1). One sun-exposed sample from a subject with COPD was not suitable for analysis.

Immunohistochemistry was performed on 3 μm sections of paraffin embedded skin with a human elastin-specific primary antibody and a dextran polymer secondary antibody stained with diaminobenzidine (DAB) and lightly counterstained with H&E. The elastin fibres are stained brown and examples indicated using arrows. Panel A shows a slide with mild elastin degradation and panel B with severe elastin degradation at 10× magnification.
Image processing and analysis
A single section was used for each subject. Images of each section were obtained using 10× magnification and processed with QCapture Pro software (Media Cybernetics, Bethesda, Maryland, USA). Consecutive images were merged using Adobe Photoshop CS4 (San Jose, California, USA) to enable analysis of the entire section. As elastin fibres degrade, they thicken and occupy a greater relative area,17 which can be quantified to assess elastin degradation in the skin.18 ,19 This phenomenon is called elastosis and is extensively reported in ageing and in response to UV light exposure. In a review of photoageing, Gilchrest described the histological hallmark of dermal elastosis as ‘thickened, tangled and granular amorphous elastin fibres’.20 We measured the area covered by elastin fibres in the reticular dermis using semiautomated quantification with ImageJ (NIH, Bethesda, Maryland, USA) in a random order by an investigator blinded to the subject identities (JM). This was expressed as a percentage of the total area measured.21
In order to validate our semiautomated elastin degradation measurements, an investigator (JM) blinded to the identities of the subjects ranked each section from 1 (mild elastin degradation) to 4 (severe elastin degradation). This visual scoring was concordant with the semiautomated measurements (Spearman's rank; r=0.86, p<0.001). Using visual scoring of elastin degradation as the outcome measure, we again showed higher elastin degradation in the COPD group in comparison with the control subjects (p<0.001).
Protein and RNA extraction
Samples of skin tissue were homogenised in lysis buffer and an EDTA-free antiprotease cocktail (Roche, Welwyn Garden City, UK) using the Qiagen tissuelyser (Crawley, UK). Protein quantification was performed using a bicinchoninic acid assay. RNA was isolated from skin following homogenisation using the RNeasy Mini Kit (Qiagen, Crawley, UK). Quantity and quality of RNA were calculated using a Nanodrop spectrophotometer (Nanodrop, Wilmington, Delaware, USA).
Gene expression
Total RNA (0.1 μg) was reverse transcribed in a reaction mix containing 4 μl of 5× reaction buffer, 2 μl dithiothreitol (0.1 M), 0.5 μl dNTPs (10 mM), 2 μl of random hexamers, 0.5 μl (200 U) Superscript II reverse transcriptase and 0.5 ml (40 U) RNase inhibitor (Amersham, Buckinghamshire, UK). A quantity of 0.1 μl of undiluted cDNA was used per reaction to assess expression of MMP-2, -9, -12 and tissue inhibitor of metalloproteinase (TIMP)-1; the primer and probe sets were custom probes (University of Edinburgh). These predesigned primers are tested and standardised for reproducible expression analysis. Primer and cDNA were added to the TaqMan universal PCR master mix (Applied Biosystems) containing all the reagents for PCR. Experiments were performed in duplicate. Quantification of the PCR products was performed with an ABI prism 7500 (Applied Biosystems) using a relative standard curve made up of cDNA of a known quantity. The mean quantity of target gene expression (MMP-2, -9 and TIMP-1) was determined from the generated standard curve. The correlations were r=0.97, r>0.99 and r>0.99, respectively, indicating good reproducibility of these measurements. All samples were normalised using an internal standard reference gene, 18S (r>0.99 for standard curve). All data are presented as a quotient relative to the control data.
Gelatin zymography
Sample separation was performed using precast 10% Tris-Glycine gels (7.5%), containing 0.1% gelatin (Invitrogen, Paisley, UK), in 2× non-reducing sample buffer at 120 V and Novex Tris Glycine SDS running buffer (Invitrogen, Paisley, UK). SDS was removed using zymogram renaturing buffer (Invitrogen, UK) for 60 min at room temperature. The gels were incubated overnight at 37°C in developing buffer (Invitrogen) and then stained with 0.1% Coomassie blue in 40% methanol and 10% acetic acid and destained until clear proteolytic bands appeared. Densitometry was performed using ImageJ (NIH). Normalisation between gels was performed using 10 ng of recombinant MMP-2 (R&D Systems, Abingdon, UK) before adjusting for total protein, allowing comparison between gels.
CT scanning
All 15 healthy controls and 13 subjects with COPD agreed to low-dose CT scanning performed at full inspiration using a 320-multidetector row CT scanner (Aquilion One, Toshiba Medical Systems, Nasushiobara, Japan). Non-contrast enhanced CT scans were obtained at 120 kV, 100 mAs during coached inspiratory breath-hold to total lung capacity. Images were reconstructed at 0.5-mm intervals with 0.5-mm thick slices using an FC-03 filter. The histogram of CT Hounsfield Units (HU) was corrected for the air offset in Toshiba CT scanners (about −985 HU instead of the nominal −1000 HU with the FC03 filter) using an extra-thoracic air calibration method.22 Emphysema was quantified using inhouse software as percentage low attenuation voxels below −910 and −950 HU (%LAA-910 and %LAA-950).23 While the latter, higher threshold distinguishes well between those subjects with COPD and healthy controls, using the former, lower cut-off allows more uniform distribution of emphysema across this cohort of former smokers. We therefore performed an analysis using both measures of lung density.
Arterial stiffness
We measured arterial stiffness using carotid-femoral pulse wave velocity as per the Expert Consensus Document on Arterial Stiffness24 in these cases and controls and presented the findings in a previous study.3 Carotid-femoral (aortic) pulse wave velocity is increased with increasing arterial stiffness.
Data analysis
COPD and control groups were compared using Student's unpaired t tests or an appropriate non-parametric alternative for variables not normally distributed. Associations among skin, postbronchodilator spirometry, emphysema severity using %LAA-910 and arterial stiffness measures were explored using Pearson correlations and associations with emphysema severity using %LAA-950 were explored using Spearman's rank correlation. C reactive protein was log-transformed for all analysis. All analyses were performed using SPSS V.18.0 and Graphpad prism (La Jolla, California, USA). Statistical significance was taken at p<0.05.
Results
Men with COPD and controls were well matched for age and smoking history. Patients with COPD had a range of severity of airflow limitation (GOLD stage 1–4), were taking medication consistent with their disease severity and had considerably more emphysema than controls (table 1).
Subject characteristics
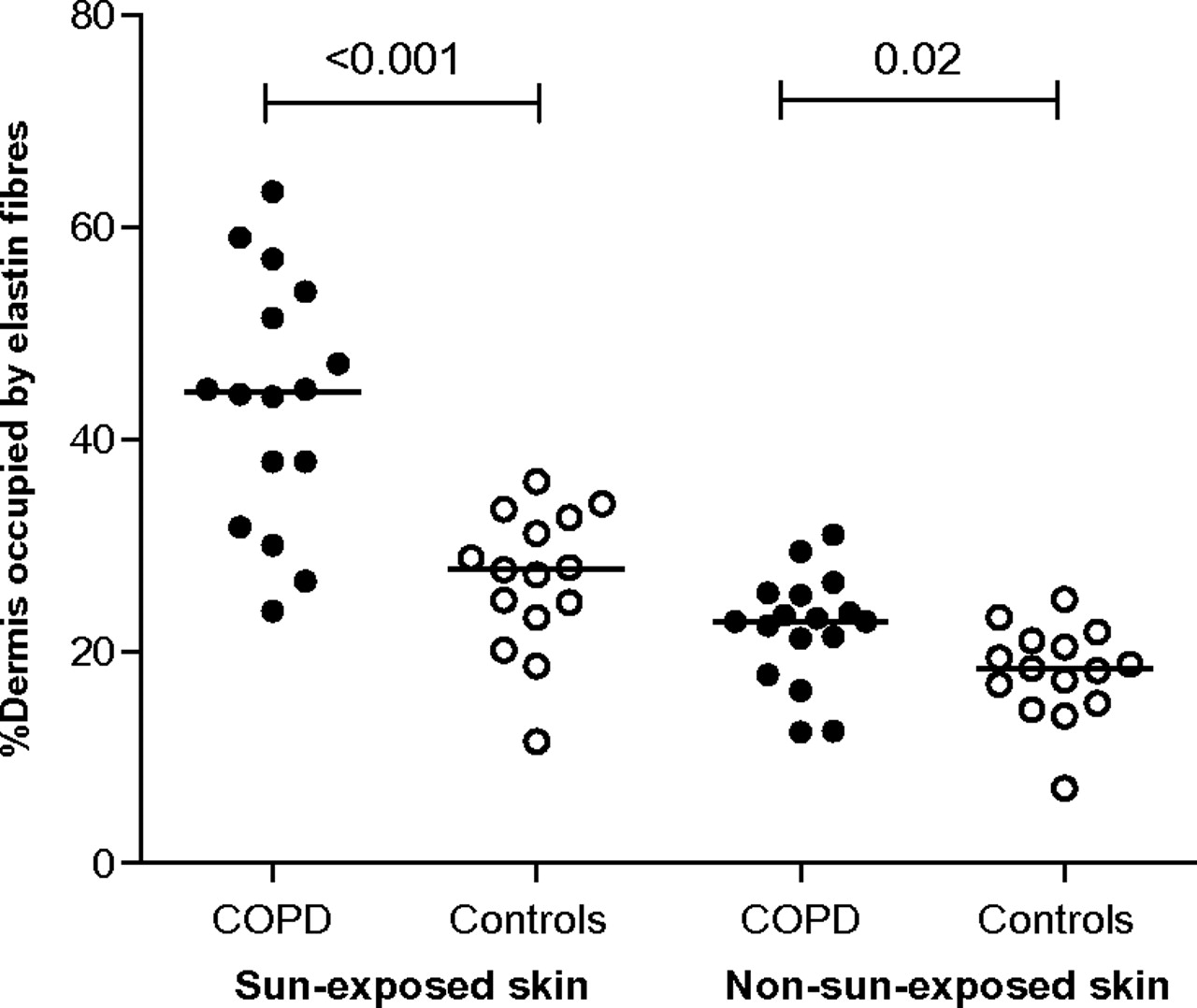
Elastin degradation was greater in sun-exposed skin (exposed) in patients with COPD in comparison with controls (mean (SD); 43.5 (12.1)% vs 26.3 (6.9)%, p<0.001). Elastin degradation was also greater in non-sun-exposed skin (non-exposed) in patients with COPD (mean (SD); 22.4 (5.2)% vs 18.1 (4.3)%, p=0.02; table 2; figure 2).
Skin measurements

Patients with chronic obstructive pulmonary disease (COPD) have increased elastin degradation in both sun-exposed and non-sun-exposed skin. Symbols represent individual values and the horizontal lines the means.
MMP-9 mRNA expression and proMMP-9 protein concentrations were higher in the exposed skin of COPD patients in comparison with controls (table 2). There was little detectable MMP-9 activity present in any subject. There was greater MMP-2 and TIMP-1 mRNA expression in the exposed skin of COPD patients, but these differences were not statistically significant. MMP-2 and -9 mRNA expression were positively associated with elastin degradation in exposed skin (MMP-2 r=0.55, p=0.002; MMP-9 r=0.62, p<0.001; figure 3). Differences in MMP expression between COPD and controls were not seen in the non-exposed skin. There was little MMP-12 mRNA expression in the skin of any of the subjects.
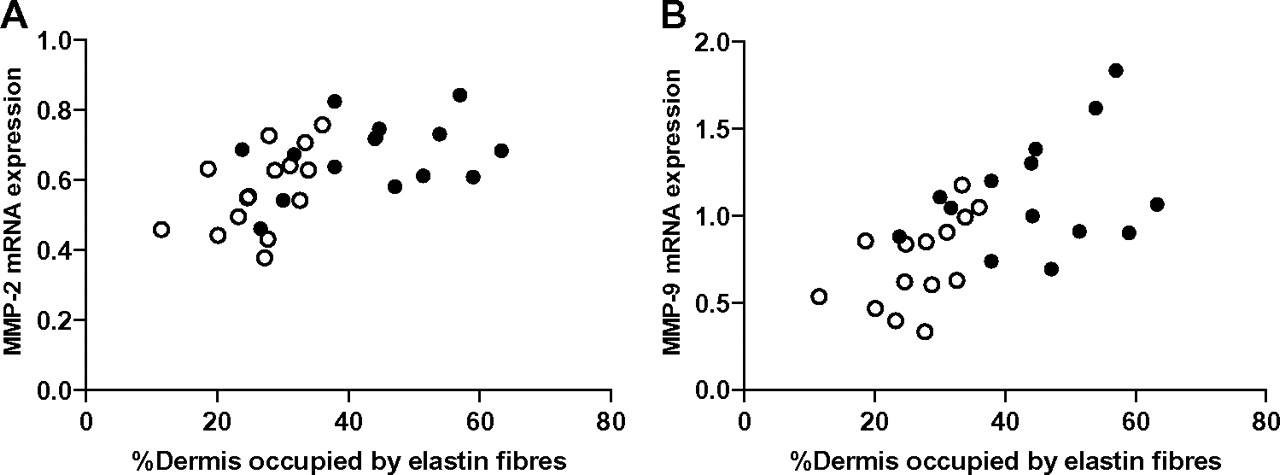
Sun-exposed skin elastin degradation was associated with both matrix metalloproteinase (MMP)-2 (panel A; r=0.55, p=0.002) and MMP-9 (panel B; r=0.62, p<0.001) mRNA expression. The y-axis represents relative expression based on an internal standard. Subjects with chronic obstructive pulmonary disease are represented by the closed circles (●) and the controls are represented by the open circles (○). MMP-2 expression was detectable in all subjects; however, MMP-9 expression was below the lower limit of detection in two subjects.
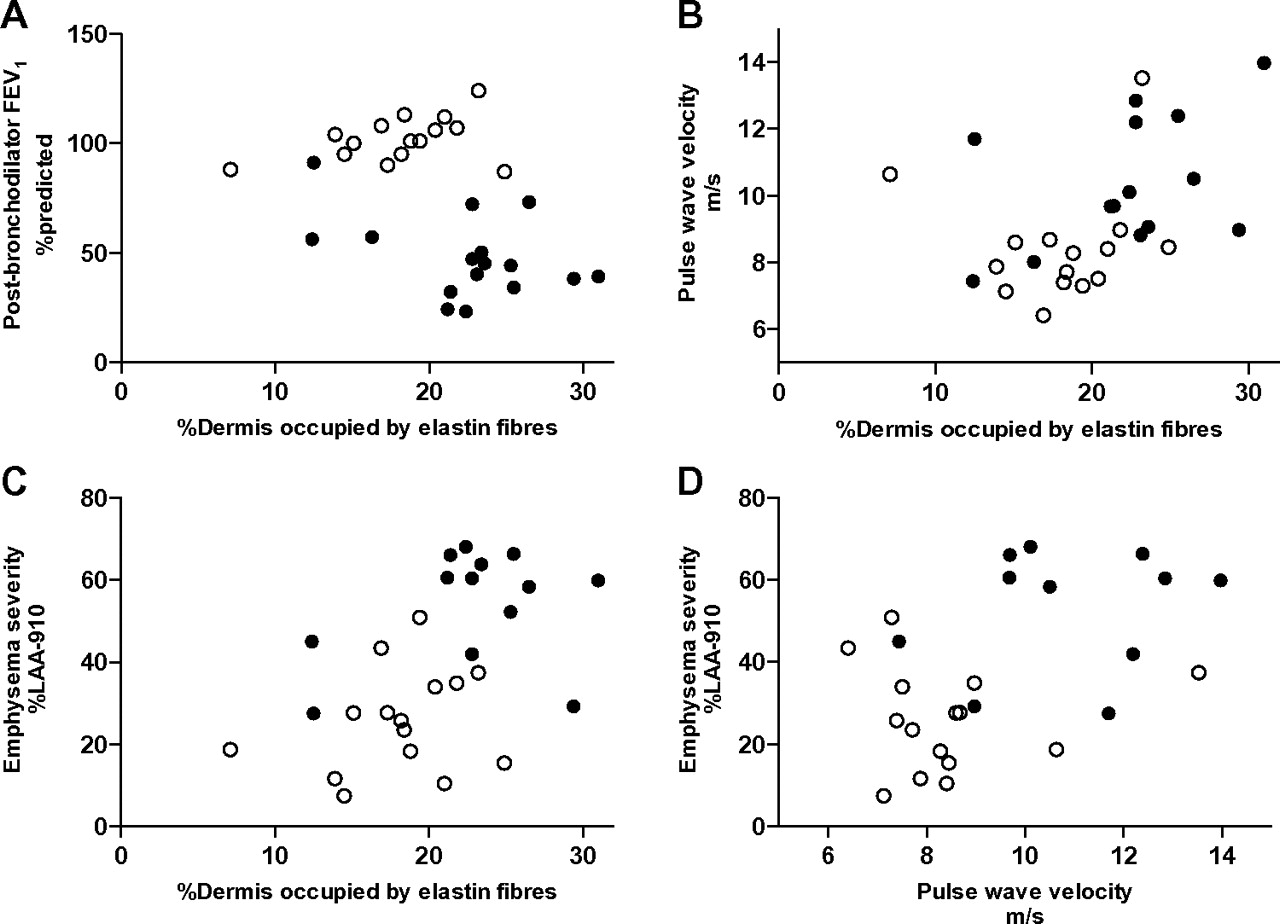
In exploratory analyses examining the whole cohort as a group of ex-smokers, we found that exposed and non-exposed skin elastin degradation were both related to FEV1 (exposed r=−0.56, p=0.001, non-exposed r=−0.45, p=0.01) and to emphysema severity (%LAA-910: exposed r=0.54, p=0.003, non-exposed r=0.50, p=0.007; %LAA-950: exposed r=0.46, p=0.01, non-exposed r=0.54, p=0.003; figure 4). Elastin degradation in non-exposed skin was associated with aortic pulse wave velocity (r=0.42, 95% CI 0.06 to 0.68, p=0.02). An association of a smaller magnitude but in the same direction was found for exposed skin but it was not statistically significant (r=0.19, 95% CI −0.19 to 0.53, p=0.32). Aortic pulse wave velocity was associated with emphysema severity (%LAA-910: r=0.47, p=0.01) and high-sensitivity C reactive protein (r=0.38, p=0.05); high-sensitivity C reactive protein was not associated with cutaneous elastin degradation or emphysema. Age was neither associated with emphysema severity nor skin elastin degradation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Non-sun-exposed skin elastin degradation (percentage of dermis occupied by elastin fibres) was associated with airflow obstruction (panel A: forced expiratory volume in one second (FEV1) %predicted; r=−0.45, p=0.01), arterial stiffness (panel B: aortic pulse wave velocity; r=0.42, p=0.02) and emphysema severity (panel C: percentage low attenuation voxels below −910 Hounsfield units (%LAA-910); r=0.50, p=0.007). Arterial stiffness was associated with emphysema severity (panel D: %LAA-910; r=0.47, p=0.01). Pearson's correlations were used across all subjects. Subjects with chronic obstructive pulmonary disease are represented by the closed circles (●) and the controls are represented by the open circles (○).
Discussion
Patients with COPD have evidence of greater cutaneous elastin degradation than age- and smoking-matched controls. Our findings represent evidence of enhanced degradation of elastin outside of the lung in COPD, and we believe that systemic elastin degradation may be a novel systemic feature of COPD. Furthermore, we have found increased expression of MMP-9 in the skin of patients with COPD, a protease implicated in the pathogenesis of emphysema, and these data suggest a role in systemic elastin degradation in COPD.
Elastin degradation in non-sun-exposed skin was associated with both emphysema severity and aortic pulse wave velocity. We speculate that the systemic elastin degradation demonstrated in the skin may also occur in other elastin-rich structures, such as the walls of large conduit arteries, and is an explanation for the increased arterial stiffness in COPD.
Development of emphysema, arterial stiffness and elastin degradation are features of normal ageing, reinforcing the importance of matching our groups for age. Our findings support the hypothesis that COPD is a disease characterised by accelerated ageing. Recent manuscripts have suggested that the arterial stiffness associated with COPD may in part be due to increased vascular calcification and that vascular calcification is associated with emphysema severity.25–27 Indeed, this mirrors the medial elastocalcinosis seen in large vessels as a feature of normal ageing.28 As the elastin in the vessel wall is degraded, the calcium content is thought to increase. Thus, measures of vascular calcification in the aorta may also be a reflection of elastin degradation. In addition, as malnutrition is associated with elastin degradation in the lungs and vasculature29 it is noteworthy that subjects with COPD had a similar mean body mass index to the controls.
Both the ATS/ERS consensus definition and the GOLD guidelines classify COPD as a disease with systemic consequences and extra-pulmonary effects.1 ,2 However, it is difficult determine whether this relationship is causal—that is, due to a direct consequence of COPD—or results from a combination of shared risk factors for COPD and cardiovascular disease such as smoking, social deprivation and a sedentary lifestyle. We have performed carefully controlled studies comparing patients with COPD and controls matched for age and smoking history that suggest arterial stiffness is an independent systemic manifestation of COPD.3 Furthermore, emphysema severity is closely related to arterial stiffness independent of cigarette smoking in patients with COPD.6 Together, these findings suggest a shared susceptibility to elastin degradation in the pulmonary and systemic tissues of COPD patients.
Vascular samples from conduit arteries are not readily available; however, previous research on the dermatological manifestations of COPD suggested that skin may be a suitable surrogate for assessing systemic elastin degradation. Using visual categorisation of facial wrinkling, skin wrinkling has been associated with emphysema severity and lung function in a cohort of patients with COPD.15 Our study provides the first direct evidence of increased elastin degradation in the skin in COPD patients, and importantly we are able to demonstrate that this observation is independent of cigarette smoke exposure. Two previous studies have reported increased cutaneous elastin degradation using biopsy specimens in cigarette smokers18 ,30 and have shown relationships with lung function.30 In the study by Francès et al, lung function was not measured and thus the potential contribution of lung disease to elastin degradation was not assessed in these smokers. Just et al reported an association with airflow limitation that was thought to be secondary to cumulative cigarette smoke exposure rather than a manifestation of lung disease.
In order to investigate potential mechanisms for increased systemic elastin degradation in COPD, we measured expression of mRNA coding for MMP-2, -9, -12 and TIMP-1 (the endogenous inhibitor of MMP-9). We selected these proteases specifically as they have been implicated in the pathogenesis of both COPD and arterial stiffness.31–35 MMP-9 mRNA expression was increased in patients with COPD, and this finding was supported by the presence of increased proMMP-9 concentrations (an inactive precursor of MMP-9) in the skin in COPD patients. Expression of MMP-2 and TIMP-1 was also higher in patients with COPD, but the differences were not statistically significant. Previous work has shown increased MMP-2 in the peripheral lung of patients with early emphysema.31 In addition, Aldonyte et al reported increased basal and LPS-stimulated release of MMP-9 from peripheral blood monocytes isolated from individuals with COPD in comparison with controls.35 Both MMP-2 and MMP-9 have also been implicated in the pathogenesis of arterial stiffness and atherosclerosis. MMP-2 is upregulated in the arteries of patients with chronic kidney disease, a condition associated with increased arterial stiffness, in comparison with matched donors.32 In a healthy population, circulating MMP-9 levels are associated with arterial stiffness and polymorphisms in the MMP-9 gene predispose to arterial stiffness.33 ,34
Expression of MMP-2 and MMP-9 mRNA were increased in the skin of patients with COPD. Taken together with previous work showing increased MMP-9 release from circulating monocytes,35 these observations suggest systemic upregulation of proteases in COPD. The persistence of pulmonary and systemic inflammation long after the cigarette smoke stimulus has been removed has raised the possibility of the pathogenesis of COPD having an autoimmune component.36 Thus cell-mediated immunity may drive both pulmonary and systemic elastin degradation in emphysema.14 An alternative hypothesis is that the systemic inflammation associated with COPD may increase production of MMPs from local inflammatory cells. Inflammatory mediators known to be increased in COPD, such as interleukin-8 and tumour necrosis factor-alpha, stimulate increased production of MMPs from neutrophils.37 ,38
Protease–antiprotease imbalance is thought to play a key role in the pathogenesis of COPD and TIMP-1 is the antiprotease that inhibits MMP-9. However, although TIMP-1 expression was increased in COPD, the differences were not significant suggesting a protease–antiprotease imbalance causing elastin degradation is due to upregulation of MMP-9.
The novel finding of increased systemic elastin degradation in COPD is important for a number of reasons. First, these observations provide further evidence of a role for proteases in the pathogenesis of COPD and that they may contribute to extra-pulmonary manifestations of the disease. Second, the upregulation of MMP-2 and -9 specifically is interesting as these proteases, known as gelatinase A and B, are zinc-dependent endopeptidases with similar structures and therefore may be potential targets for treatment.39 Indeed, statins are reported to reduce MMP-2 and -9 production by human vascular smooth muscle cells40 and MMP-9 secretion by lung fibroblasts41 and macrophages.42 Novel anti-inflammatory compounds such as p38 mitogen-activated protein kinase inhibitors specifically target activation and production of MMPs and have been shown to reduce MMP-9 release.43 Finally, the skin is a readily accessible tissue that may reflect the cumulative effects of COPD and may therefore provide further insights into the pathogenesis of the systemic manifestations of this complex disease.
Study limitations
Although there were differences in elastin degradation in both sun-exposed skin and non-sun-exposed skin, in non-sun-exposed skin there were no differences in the expression of proteases. There was generally reduced expression of MMPs in non-sun-exposed skin in comparison with sun-exposed skin and production of cutaneous MMPs in response to ultraviolet radiation is well described.44 Thus, it may be that sunlight exposure is a complementary factor which increases elastin degradation in susceptible skin tissue, unmasking differences in protease production between the patients with COPD and the healthy subjects. However, the pathophysiology of both COPD and vascular disease is complex and the exact inflammatory mechanisms are as yet unclear. While we have specifically investigated cutaneous MMPs, it is important to acknowledge that other proteases may have a role in the pathogenesis of both vascular disease and the development of emphysema. Serine proteases (eg, neutrophil elastase) have been implicated in both atherosclerosis and COPD in humans45 ,46 as have cysteine proteases (eg, cathepsin K) in animal models.47 ,48 Thus, these other proteases may be involved in systemic elastin degradation in COPD.
We did consider measuring elastin degradation products, but our protein extraction methods were suitable to extract MMPs and not the relatively insoluble desmosine/isodesmosine from samples. In any case, a consistent and reliable methodology for assessment of these proteins is yet to be established. Indeed, desmosine levels in the lung parenchyma are similar in patients with severe COPD and healthy donor lungs.49 Future studies could specifically assess desmosine levels, its relationship with elastin degradation and its prospective use as a local tissue biomarker.
Although we did exclude individuals on regular oral steroids, we were unable to adjust for inhaled corticosteroid use, taken by the majority of patients with COPD. Skin thinning, bruising and atrophy are recognised side effects of corticosteroid therapy. A small biopsy study suggested that inhaled corticosteroids may affect collagen synthesis,50 but in the largest study assessing the effects of long term inhaled corticosteroids on skin there was no effect on skin collagen or thickness.51 No specific effect of inhaled corticosteroids on elastin fibres has been described.
Conclusions
Patients with COPD have increased skin elastin degradation in comparison with age- and smoking-matched controls and cutaneous elastin degradation was related to both emphysema severity and arterial stiffness. Furthermore, we identified upregulation of MMP-9 in the skin of COPD patients, which is associated with elastin degradation. Systemic elastin degradation due to increased proteolytic activity may represent a novel shared mechanism for the pulmonary, vascular and cutaneous features of COPD.
Acknowledgments
The authors would like to thank the respiratory research nurses, Andrew Deans and Joyce Barr, staff at the Wellcome Trust Clinical Research Facility and ELEGI laboratory for their help with these studies.
References
Footnotes
Funding This research was funded primarily by a grant from the Scottish Society of Physicians and a Chief Scientist Office Project Grant (CZB/4/424). DM is supported by a Chest, Heart and Stroke Scotland Research Fellowship (R40329). NLM is supported by British Heart Foundation Intermediate Clinical Research Fellowship (FS/10/024).
Competing interests None.
Ethics approval Ethics approval was given by Lothian Regional Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.