Article Text

Abstract
Background Indacaterol is a long-acting inhaled β2-agonist (LABA) for the treatment of chronic obstructive pulmonary disease (COPD). In previous studies, indacaterol provided 24 h bronchodilation on once-daily dosing with a fast onset of action. This study compared the efficacy and safety of indacaterol with the twice-daily LABA formoterol and placebo over 1 year.
Methods Patients with moderate to severe COPD were randomised to receive once-daily indacaterol 300 μg (n=437) or 600 μg (n=428), twice-daily formoterol 12 μg (n=435) or placebo (n=432) for 52 weeks in a double-blind double-dummy parallel group study. The primary efficacy variable was forced expiratory volume in 1 s (FEV1) measured 24 h postdose after 12 weeks (indacaterol vs placebo). Other outcomes included dyspnoea (transition dyspnoea index, TDI), use of as-needed salbutamol, symptom-based measures recorded on diary cards, exacerbations, health status (St George's Respiratory Questionnaire), BODE index (body mass index, obstruction, dyspnoea, exercise), safety and tolerability.
Results Indacaterol increased 24 h postdose FEV1 after 12 weeks by 170 ml (both doses) versus placebo and by 100 ml versus formoterol (all p<0.001). These significant differences were maintained at 52 weeks. Symptomatic outcomes were improved compared with placebo with all active treatments, and indacaterol was more effective than formoterol in improving TDI score and reducing the need for as-needed salbutamol. Indacaterol was well tolerated and had a good overall safety profile, including minimal impact on QTc interval and systemic β2-mediated events.
Conclusions Once-daily indacaterol is an effective 24 h bronchodilator that improves symptoms and health status and confers clinical improvements over a twice-daily 12 h LABA as a treatment for patients with moderate to severe COPD.
Trial registration number NCT 00393458.
- Pulmonary disease, chronic obstructive
- bronchodilator agents, indacaterol, formoterol, clinical trial, COPD pharmacology
Statistics from Altmetric.com
- Pulmonary disease, chronic obstructive
- bronchodilator agents, indacaterol, formoterol, clinical trial, COPD pharmacology
Current management guidelines for chronic obstructive pulmonary disease (COPD) provide a clear treatment algorithm for the maintenance treatment of COPD, recommending initial treatment with one or more long-acting bronchodilators for patients with moderate, severe and very severe disease, with the addition of inhaled corticosteroids (ICS) for patients who exacerbate frequently.1 The long-acting bronchodilators currently available include the twice-daily inhaled β2-agonists formoterol and salmeterol. It may be hypothesised that the sustained bronchodilation provided by a bronchodilator with a 24 h duration would reduce fluctuations in airway patency compared with twice-daily bronchodilators, and may improve clinical outcomes.
Indacaterol is a novel inhaled long-acting β2-agonist providing 24 h bronchodilation on once-daily dosing.2–4 This 1-year study was designed to provide efficacy and long-term safety information on indacaterol compared with placebo and formoterol. Our primary hypothesis was that indacaterol would have a greater effect than placebo on forced expiratory volume in 1 s (FEV1) at 24 h post-dose (‘trough’) after 12 weeks.
Methods
Patients
Subjects aged ≥40 years with a clinical diagnosis of moderate to severe COPD5 and a smoking history of ≥20 pack-years, with postbronchodilator (salbutamol 400 μg) FEV1 <80% and ≥30% predicted and ratio of FEV1 to forced vital capacity (FVC) <70% were enrolled in the study. Factors preventing study entry included respiratory tract infection or hospitalisation for COPD exacerbation within the previous 6 weeks, oral corticosteroids or change in ICS during the previous month, or history of asthma.
Study design
A 2-week run-in was followed by 52 weeks of double-blind treatment (see figure S1 in online supplement), with patients randomised to treatment (1:1:1:1) with stratification for smoking status (current/ex-smoker) using an automated interactive system. Data were collected from outpatient clinics and physicians' offices.
The study treatments were indacaterol 300 μg and 600 μg delivered once daily via single-dose dry powder inhaler (SDDPI), formoterol 12 μg twice daily (the standard therapeutic dose) via its proprietary SDDPI and matching placebos to indacaterol and formoterol taken each morning (08:00–10:00; both devices) and evening (20:00–22:00; proprietary SDDPI only). Fixed-dose combinations of ICS plus long-acting β2-agonist were replaced by monotherapy ICS at an equivalent dose and regimen plus salbutamol as-needed. Patients on ICS monotherapy continued treatment at a stable dose throughout the study. Patients were given salbutamol to use as-needed but were asked not to take it within 6 h before each visit. Other bronchodilators or corticosteroids were not allowed unless to treat a COPD exacerbation. Participating physicians were responsible for treating exacerbations.
Assessments
Patients visited the clinics on days 1, 2, 15, 29, 84, 85, 113, 168, 197, 253, 364 and 365. The visits on days 1 and 2, week 12 and week 52 were on consecutive days in order to provide a precise trough FEV1 value which was based on the mean of two measurements taken 23 h 10 min and 23 h 45 min following the previous day's morning dose. At other visits the 15 min predose value was used as a ‘trough’ measurement. In a subset of patients, spirometry was performed serially over 24 h postdose. Patients were given a diary to record peak expiratory flow (PEF), symptoms, use of salbutamol, any change in concomitant medications and adverse events.
COPD exacerbations were defined as onset or worsening of more than one respiratory symptom (dyspnoea, cough, sputum purulence or volume or wheeze) for >3 consecutive days (based on diary cards or patients' reports of their health since the previous visit) plus documented proof of intensified treatment (eg, systemic steroids, antibiotics or oxygen) and/or hospitalisation or emergency room visit.
Health status (St George's Respiratory Questionnaire, SGRQ)6 and dyspnoea (baseline and transition dyspnoea indices, BDI/TDI7) were measured on day 1 and weeks 4, 8, 12, 24, 44 and 52. The modified Medical Research Council (mMRC) dyspnoea scale8 9 was measured on day 1, week 12 and week 52 followed by the 6-min walk test10 as components of the BODE (body mass index, obstruction (FEV1, % predicted), dyspnoea, exercise) index.11
Adverse events were recorded at each visit. ECG assessment, blood pressure and pulse rate measurements and blood sampling for haematology were performed at regular intervals.
Objectives and outcomes
The primary objective was to test the hypothesis that both indacaterol doses were superior to placebo in their effect on trough FEV1 after 12 weeks. The important secondary efficacy outcomes were days of poor COPD control (a composite measure used in formoterol registration studies,12 13 defined as the number of days with a score of ≥2 on a 0–3 scale for at least two symptoms out of cough, wheeze, production/colour of sputum and breathlessness), SGRQ total score and time to first exacerbation. Other secondary efficacy outcomes included other spirometry, TDI scores and percentage of responding patients, exacerbation rates, BODE index and end points derived from diary data. Safety outcomes included the incidence of adverse events and clinically notable values for plasma potassium (<3.0 mmol/l) and blood glucose (≥9.99 mmol/l). QTc interval was calculated using the Fridericia formula.
Statistical methods
Efficacy results are presented for the modified intention-to-treat (ITT) population including all randomised patients who received at least one dose of study drug but excluding patients from six sites owing to non-conformance with good clinical practice. Safety results are presented for all patients who received at least one dose of study drug. All analyses presented here were preplanned.
The primary variable was analysed using a mixed model analysis of covariance (ANCOVA) with treatment as a fixed effect and baseline FEV1 and FEV1 reversibility as covariates. The same model (with appropriate covariates) was used to analyse other efficacy variables. The results are presented as estimated adjusted treatment effects (least squares means) and treatment contrasts with 95% CIs and two-sided p values. For trough FEV1 at weeks 12 and 52, SGRQ total score, TDI score and daily salbutamol use, missing values were replaced using last observation carried forward. Data were carried forward for a maximum of 11 weeks, and for trough FEV1 no data were carried forward from before day 15. Exploratory analyses of week 12 trough FEV1 values were performed in subgroups defined according to baseline age, disease severity, smoking history or ICS use. A hierarchical testing procedure to control type I error for multiplicity was used for the comparisons of indacaterol and placebo for the primary end point, then in turn for days of poor COPD control, SGRQ total score at week 12 and time to first COPD exacerbation. Other analyses were not adjusted for multiplicity.
Time to first COPD exacerbation was analysed using a Cox regression model with the assumption of proportional hazards checked with Schoenfeld residuals of treatment effects plotted against time. The number of COPD exacerbations over 52 weeks was analysed using a Poisson regression model with a sensitivity analysis to assess the impact of premature discontinuations by imputing an additional one exacerbation for these patients. Neither the Cox nor the Poisson regression model assumes normality. Normal distribution was assumed for efficacy and safety variables where ANCOVAs were performed. For the primary and important secondary variables (excluding exacerbations), the normality assumption was checked with a quantile–quantile plot of residuals for each treatment group.
All safety end points were summarised for the safety population.
Sample size
With a SD of 270 ml for trough FEV1,12 13 108 evaluable patients per group were needed to detect a 120 ml difference between indacaterol 300 μg and placebo as significant at the 5% level (two-sided) with 90% power, with 84% power (due to the hierarchical testing) for the indacaterol 600 μg versus placebo comparison. However, the plan for at least 300 patients per indacaterol dose group to provide a robust safety database provided 99% power for the primary comparisons.
Results
Patients
Patient disposition is shown in figure 1, which shows that the ITT population was modified to exclude 129 patients (7.4% of randomised) before unblinding because of non-conformance with good clinical practice irrespective of treatment group, the main reasons for non-conformance being substandard spirometry, unsatisfactory completion of diary cards and health status questionnaires and inability to verify source data. Baseline data for all patients (safety population) are shown in table 1. Approximately 5% of patients were enrolled before an early protocol amendment stipulating postbronchodilator (rather than prebronchodilator) spirometry values as entry criteria and had baseline spirometry outside specified limits.

Patient flow through study.
Patient demographic data and other characteristics at baseline (safety population)
Spirometry
Trough FEV1 at week 12 with both indacaterol doses was 170 ml higher than placebo (p<0.001) and 100 ml higher than formoterol (p<0.001) (table 2). Over the remainder of the study, significant differences versus placebo were maintained at a similar level for indacaterol, while the difference between formoterol and placebo diminished (table 2; figure 2). Analysis of trough FEV1 at week 12 for the ITT population (ie, including patients from the centres excluded from the modified ITT analysis) is provided in table S1 in the online supplement.
Primary and additional efficacy outcomes

Trough forced expiratory volume in 1 s (FEV1) after 1 day and at weeks 12 and 52 of treatment (modified intention-to-treat population). Data are least squares means±SE. Between-treatment comparisons are shown in table 2.
Supportive analysis of the primary variable excluding any patients with protocol deviations (13–15% of patients across the groups, including those not meeting the postbronchodilator spirometry entry criteria; see table 1) showed similar results (not shown). The group mean values and differences between treatment and placebo groups in trough FEV1 after 12 weeks were similar for subgroups of patients analysed according to age, COPD severity, smoking status and ICS use.
At 5 min postdose on day 1, compared with placebo FEV1 increased by 130 ml (95% CI 110 to 150) with indacaterol 300 μg, by 150 ml (95% CI 140 to 170) with indacaterol 600 μg and by 140 ml (95% CI 120 to 160) with formoterol (all p<0.001 vs placebo). Serial measurements of FEV1 showed a significant effect of indacaterol over placebo at each time point over 24 h postdose (figure 3). Other spirometry results are shown in the online supplement.

Serial measurements of forced expiratory volume in 1 s (FEV1) from −15 min to 24 h postdose measured in subset with serial spirometry measurements (12 h serial spirometry subset) at week 12. Data are least squares means±SE. Treatment differences: p<0.001 for indacaterol (both doses) vs placebo at each time point; p<0.05 for formoterol vs placebo at time points from 5 min to 11 h 45 min postdose; p<0.05 for indacaterol 300 μg vs formoterol at −15 min, 2 h and from 6 h to 23 h 45 min; p<0.05 for indacaterol 600 μg vs formoterol at all time points apart from 5 min postdose. Patient numbers at each time point are shown in table S3 in the online supplement.
Clinical outcomes
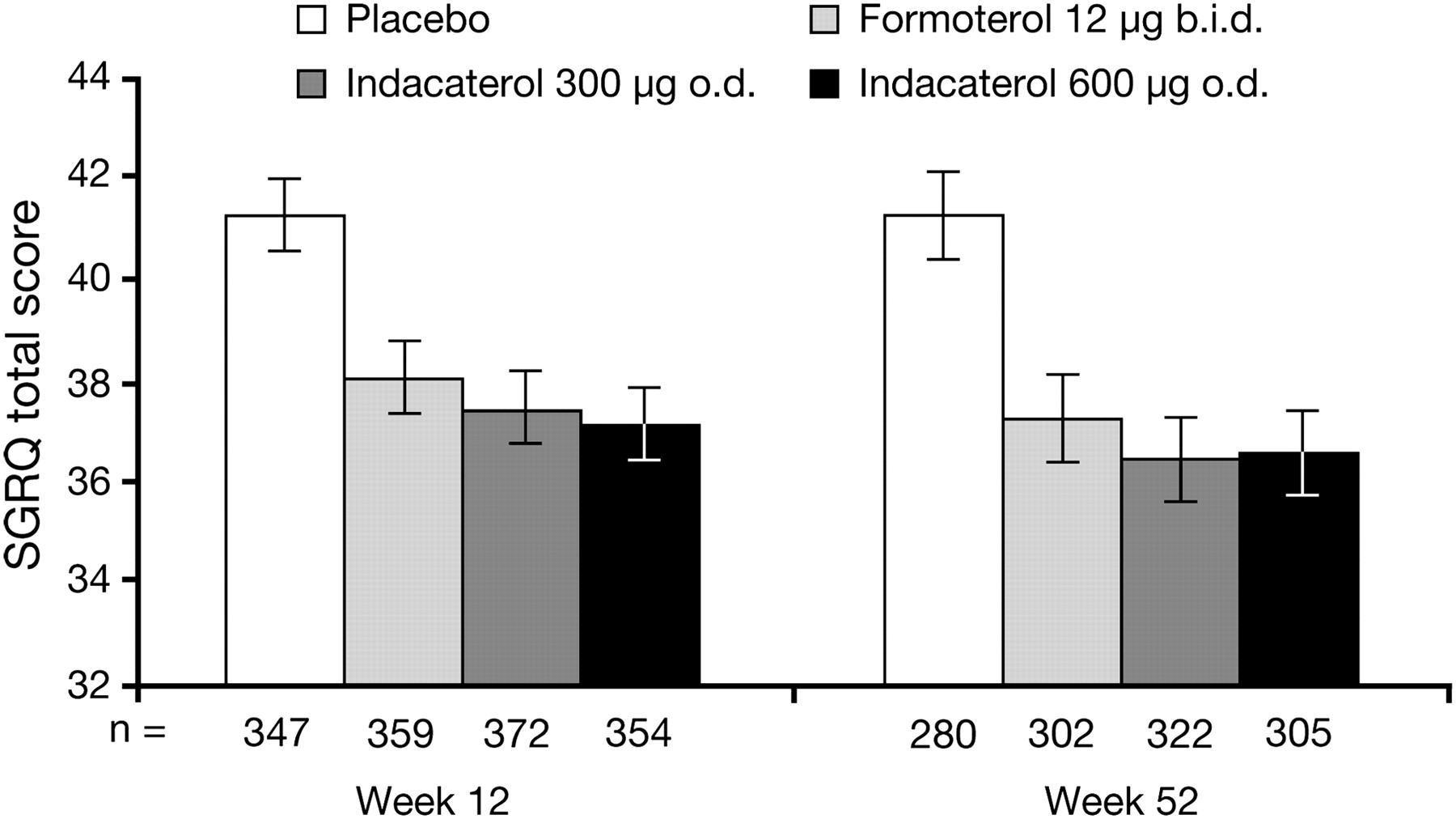
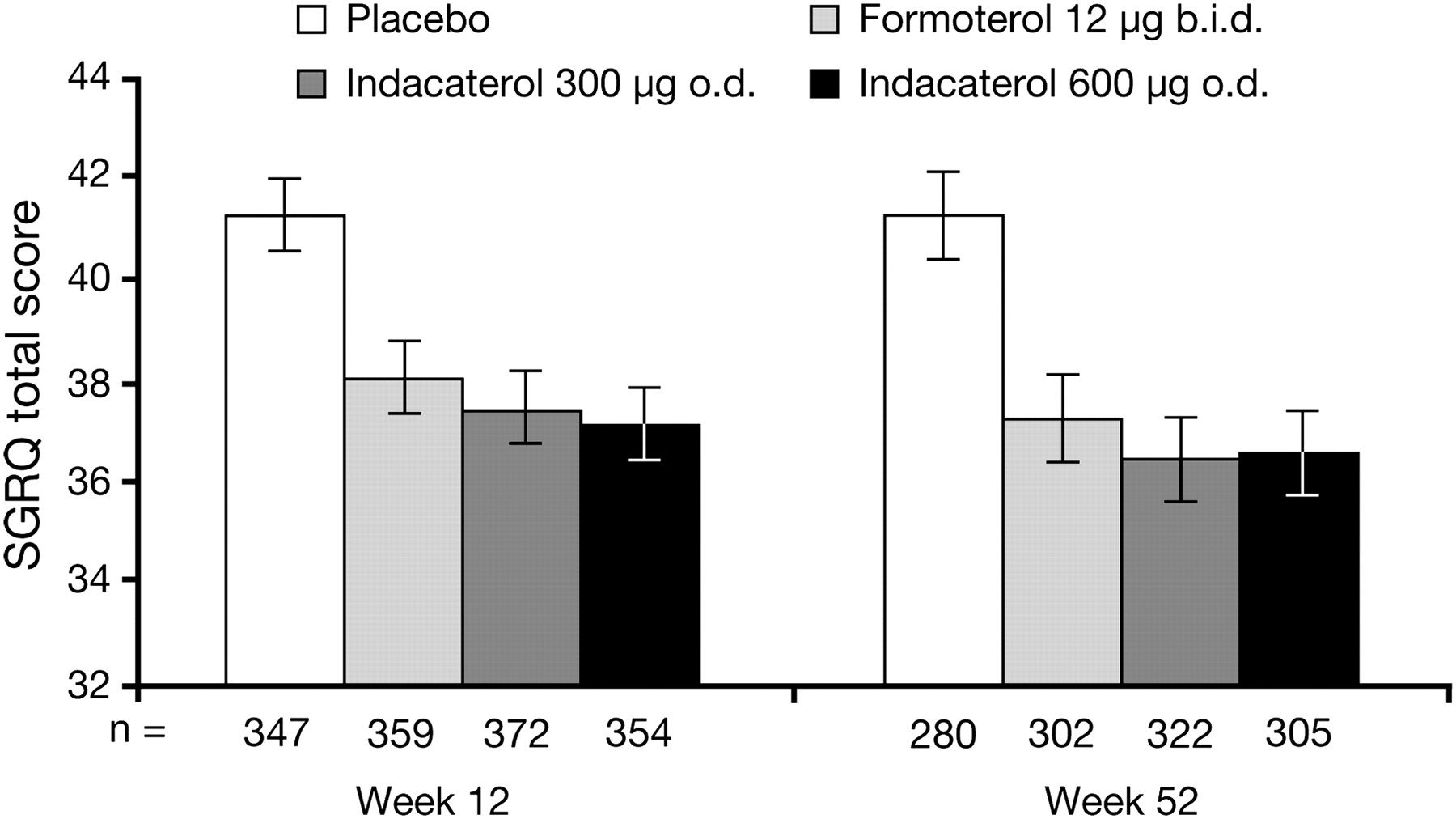
Both indacaterol doses were superior to placebo for all secondary end points controlled for type I error (ie, days of poor control, SGRQ and time to first exacerbation; differences between indacaterol and formoterol for these end points were not statistically significant; tables 2 and 3, figure 4). Time to first COPD exacerbation was improved with all treatments, as shown by the HRs in table 3, although there were too few events to calculate event-free time. Exacerbation rate ratios were similar without imputation (table 3) and with imputation (not shown).
Analysis of time to first COPD exacerbation and exacerbation rates
{kind=link}
{kind=link}
{kind=link}
{kind=link}
St George's Respiratory Questionnaire (SGRQ) total score at weeks 12 and 52. Data are least squares means±SE. Between-treatment comparisons are shown in table 2.
Both indacaterol and formoterol increased TDI scores, with the differences between indacaterol and placebo close to or exceeding the 1-point minimum clinically important difference14 15 (table 2). TDI scores were significantly higher with indacaterol than with formoterol at week 12 but not at week 52. At week 12, 63% and 58% of patients treated with indacaterol 300 and 600 μg, respectively, had a clinically important increase of ≥1 in TDI score compared with 40% of patients given placebo (p<0.001) and 53% of patients treated with formoterol (p<0.01 for indacaterol 300 μg vs formoterol). Efficacy outcomes based on diary data and BODE index (table 2) were generally significantly improved with indacaterol and formoterol versus placebo, with significant differences between indacaterol and formoterol for use of as-needed salbutamol, nights without awakenings and PEF.
Safety
COPD worsening and nasopharyngitis were the only adverse events reported by >10% of patients in any treatment group (table 4). Most cases of COPD worsening were mild or moderate in severity (indacaterol 300 μg, 92%; indacaterol 600 μg, 91%; formoterol, 84%; placebo, 87%). All cases of nasopharyngitis were mild or moderate with indacaterol and formoterol.
Number (%) of patients with adverse events
Eight patients died during treatment and four during follow-up. Of the deaths during treatment, two were due to cardiac arrest (indacaterol 300 μg; placebo), one to multiorgan failure (formoterol), one to respiratory failure (formoterol) and four to sudden death (one formoterol; three placebo). The indacaterol-treated patient who died had discontinued treatment 3 days earlier owing to dyspnoea (suspected as related to treatment). The three deaths during formoterol treatment were considered unrelated to formoterol.
Tremor was reported in 0.2%, 1.9%, 1.2% and 0.5% of the indacaterol 300 μg, 600 μg, formoterol and placebo groups, respectively, and tachycardia in 0.9%, 0.7%, 0.5% and 1.2%, respectively. Two cases were severe (tremor in one patient receiving placebo and atrial tachycardia in one patient receiving indacaterol 600 μg).
In addition to patients reporting cough as an adverse event (table 4), investigators were asked to record any instances of cough occurring within 5 min of drug administration during clinic visits regardless of whether they considered it an adverse event. This was observed in an average of 19.1% of patients in both indacaterol groups, 0.8% of the formoterol group and 1.8% of the placebo group. The cough typically started within 15 s of inhaling indacaterol, had a median duration of 12 s or less, and was not associated with bronchospasm. Importantly, the presence of this cough was not associated with any increase in discontinuation rates.
An increase in QTc interval of >60 ms from baseline occurred in one patient (0.2%) in each of the indacaterol and formoterol groups and in no patient in the placebo group. No patient had an absolute value >500 ms. Serum potassium concentrations <3.0 mmol/l were recorded for two patients (0.5%) in each of the indacaterol 300 μg and placebo groups and not in the other two groups. Group mean values of serum potassium 1 h postdose at week 52 were not significantly different. Blood glucose concentrations of >9.99 mmol/l were recorded for 8.0%, 9.0%, 6.5% and 7.5% of the indacaterol 300 μg, 600 μg, formoterol and placebo groups, respectively. Group mean values of blood glucose 1 h postdose at week 52 were statistically significantly different for indacaterol 600 μg compared with placebo (5.86 vs 5.61 mmol/l, p=0.033).
Discussion
This study provides clear evidence of effective 24 h bronchodilation with once-daily administration of indacaterol 300 or 600 μg. The effect of indacaterol on trough FEV1 relative to placebo exceeded both the 120 ml level versus placebo that was prespecified as clinically significant for this study and the 100–140 ml range proposed recently as a minimal important difference.17 Bronchodilator efficacy was independent of age, severity of airways obstruction and smoking status. Results for trough FEV1 at week 12 were very similar when analysed for the ITT population, suggesting that the exclusion of patients from six sites for non-conformance with good clinical practice had little impact on efficacy outcomes.
The bronchodilator effect of indacaterol was accompanied by significant improvements compared with placebo in dyspnoea and health status (both by a clinically meaningful margin15 16), time to first exacerbation, BODE index, use of as-needed salbutamol and symptom-based variables derived from diary data. Although this study was not primarily powered to detect significant differences between indacaterol and formoterol, indacaterol was more effective for several outcomes including bronchodilator efficacy. The 100 ml difference in trough FEV1 between the two treatments after 12 weeks occurs in the early morning, a time that patients describe as worst for COPD symptoms.18 Among the clinical outcomes, differences favoured indacaterol over formoterol in dyspnoea score, use of as-needed salbutamol and nights without awakenings. Indacaterol had a greater effect than formoterol on SGRQ total score, although the difference did not reach statistical significance.
Indacaterol increased the time to first exacerbation and reduced the exacerbation rate relative to placebo; similar effects were seen with formoterol. Both treatments improved the BODE index, which correlates with risk of exacerbations.19 The low apparent rate of exacerbations and the effects of treatment need to be viewed cautiously since it is conceivable that some episodes were missed, owing perhaps to incomplete diary card reporting when patients were feeling particularly ill. A definition based only on documented treatment and/or hospitalisation, as used elsewhere,20 might have increased the frequency of reporting. Additionally, we recruited patients with relatively stable disease who were not required to have a history of frequent exacerbations. Appropriately designed and powered studies are under way to scrutinise the effect of indacaterol on exacerbations.
The 12-week time point was chosen for the primary end point since this is the minimum period required to demonstrate efficacy on FEV1 for purposes of formal drug registration, as well as to minimise the impact of patient dropout during the later stages of a 52-week study. Nevertheless, indacaterol maintained its significant bronchodilator effect during the study, while formoterol suffered from some loss in efficacy over 52 weeks. A previous 1-year study showed some loss of bronchodilator effect with formoterol at a higher dose of 24 μg twice daily,13 although other results showed no tolerance either at the same dose used here13 or with nebulised formoterol 50 μg once daily.21 Compared with previous studies,12 13 formoterol had a lesser bronchodilator effect in the present study, perhaps because the older studies were in patients with higher mean FEV1 reversibility of 17%. Nevertheless, the present double-blind comparison shows that indacaterol is the more effective bronchodilator and is similar or more effective for clinical outcomes. In addition, patient adherence to a once-daily dosing regimen may be better than with a twice-daily regimen since frequency of dosing has been shown to influence adherence.22
The choice of indacaterol doses was based on previous short-term study results of bronchodilator efficacy and safety.2–4 Evaluating the two doses over 1 year was felt sufficient to determine if the higher dose had a better effect on the secondary efficacy outcomes. Subsequent clinical investigation has shown that doses of 150–300 μg once daily are effective.23 However, it is important to know that a dose of 600 μg once daily (2–4 times the therapeutic dose) presented no safety concerns when given for a year. The overall incidence of adverse events was similar across the treatment groups and typical β2-mediated adverse events were rare. Signs of systemic β2-adrenoceptor activity were minimal, with no clinically meaningful differences between groups in mean serum potassium or blood glucose. Notable increases in QTc interval were observed very rarely and were not more common at the higher dose, suggesting that indacaterol has a very low potential for cardiac arrhythmogenicity. These properties point towards a good therapeutic ratio between efficacy and adverse event outcomes.
Once-daily indacaterol, with its 24 h bronchodilator effect, improved several clinical outcomes to a greater extent than a twice-daily long-acting β2-agonist in patients with moderate or severe COPD who are candidates for long-acting bronchodilator therapy.1 Indacaterol may prove useful as an alternative to established bronchodilators as initial regular therapy or, for those patients who require the added efficacy from combining two different agents, when added to a long-acting anticholinergic bronchodilator.1 It can be concluded that once-daily indacaterol has significant value for patients with COPD, providing clinical benefits over twice-daily formoterol for 1 year without loss of effect and with a favourable safety profile.
Acknowledgments
The authors thank the patients who took part and the staff at the participating clinical centres. Sarah Filcek (ACUMED) and David Young (Novartis) assisted in the preparation of the manuscript, including preparation of an initial draft; this support was funded by the study sponsor.
References
Supplementary materials
Web Only Data thx.2009.125435
Files in this Data Supplement:
Footnotes
Linked articles 130211.
Funding The study was funded by Novartis Pharma AG. Novartis was responsible for the conception and design of the study and analysis and interpretation of data. All authors had access to the study data, reviewed and revised the initial draft and subsequent versions of the manuscript, had final responsibility for the decision to submit for publication and approved the version submitted.
Competing interests DJ, PB, RO, MH and BK are employees of Novartis. RD has participated in advisory boards for Novartis, AstraZeneca, Boehringer-Ingelheim, Pfizer, ALK-Abello, UCB, Nycomed, Dainippon and ONO. KFC has received honoraria and research support from Novartis and has served on Novartis Advisory Boards. He also has similar relationships with GSK, Gilead, Merck and Boehringer-Ingelheim. RB has received reimbursement for attending scientific conferences and/or fees for speaking and/or consulting from Novartis. He also has similar relationships with AstraZeneca, Boehringer Ingelheim, GSK, Nycomed and Pfizer. The Pulmonary Department at Mainz University Hospital received financial compensation for services performed during participation in clinical trials organised by Novartis. HM has received fees for membership of advisory boards of various pharmaceutical companies (Altana, Boehringer Ingelheim, AstraZeneca) and for speaking (Boehringer Ingelheim, AstraZeneca, Chiesi, Altana). VN has no conflict of interest.
Ethics approval The study protocol was approved by the institutional review boards or independent ethics committees of participating centres. All patients provided their written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.