Article Text

Abstract
A review of the clinical manifestations of α1-antitrypsin (AAT) deficiency, including lung disease and liver disease, and risk factors affecting the rate of decline in lung function in AAT deficient patients.
- α1-antitrypsin deficiency
- lung function
- liver disease
- smoking
Statistics from Altmetric.com
It has long been recognised that α1-antitrypsin (AAT) deficiency is associated with an increase in clinical symptoms and diseases in several systems. In this review we discuss the pulmonary and hepatic conditions that may occur in patients with this deficiency and other less prevalent associated diseases, and explore factors that may influence the progression of the disease with particular reference to respiratory disease.
LUNG DISEASE
Clinical presentation
Patients may be diagnosed with AAT deficiency after presentation with symptoms or through family screening of an index case. The proportion of patients identified by screening is influenced by local and national practice, so studies of groups of patients may show some differences. In the UK registry of AAT deficiency, 76% of Pi Z patients have been identified through investigation of respiratory symptoms or disease and 19% through family screening. Similar proportions are reported in the National Heart Lung and Blood Institute (NHLBI) registry.1 A small number of subjects are identified from abnormal radiological, pulmonary, or blood tests or by the development of liver disease. In the UK registry only 3% of patients have been identified as a result of liver disease, although it is possible that greater numbers of such patients are identified elsewhere.
There can often be a delay in the diagnosis of AAT deficiency and one study has shown a duration of 7 years between the mean age at onset of symptoms and the mean age of subsequent diagnosis.2 Patients often present in the third or fourth decade with symptoms of breathlessness and other common symptoms include cough, phlegm, wheeze (with and without infections) and fatigue.1,3
One limitation of studies which use patient registries and cohorts is the ascertainment bias of the subjects studied. The groups of patients are not fully representative of the population of people with this deficiency, as recruitment through chest physicians means that symptomatic patients are more likely to be enrolled and studied. Many people with AAT deficiency will not have symptoms nor will they have significant lung function impairment, and currently these individuals are often only detected through family screening.4 Another type of selection bias that may affect studies involving patient registries is that due to survivorship, as patients who remain in the study population may retain a certain degree of health in order to continue their attendance.
A few studies have followed deficient individuals after detection through neonatal screening and these are more representative of the population with this condition. However, the largest cohort has only been studied for 25 years so far,5 providing few data on the natural history of the condition.
Emphysema
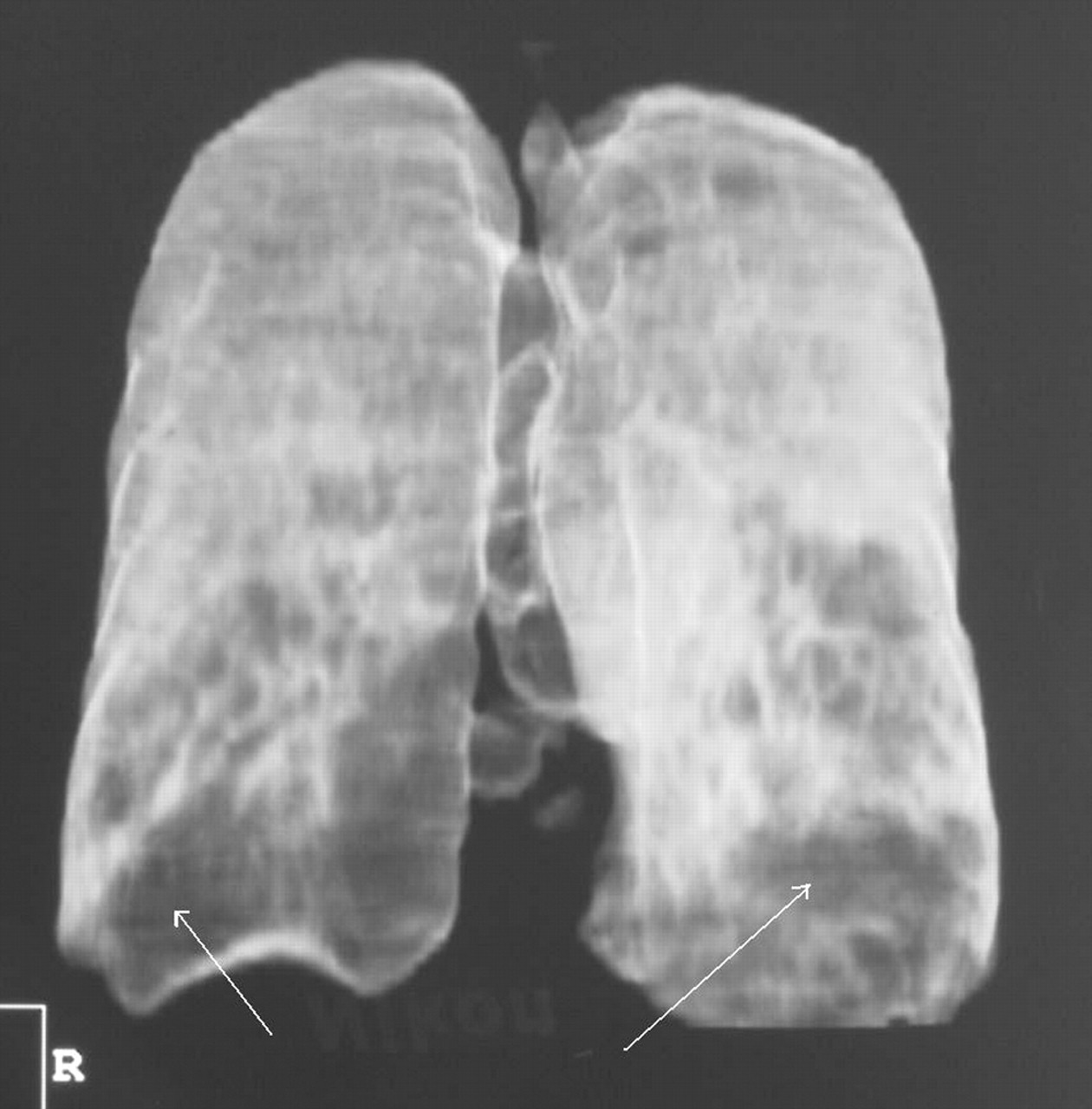
The close association between AAT deficiency and the development of emphysema was first described in 1963.6 The disease in these patients is usually of an earlier onset than in patients with chronic obstructive pulmonary disease (COPD) and often appears to be out of proportion to their smoking history. The typical pattern shows lower zone predominance (fig 1), although the emphysema may affect all zones. Plain chest radiographs may show evidence of hyperinflation with reduced lung markings, and bulla formation may also be associated with emphysematous lung.7 Lung function tests on symptomatic patients often show evidence of increased lung volumes and air trapping as well as impaired gas transfer. However, this picture is not universal and even patients with severe airways obstruction and prominent emphysema on the CT scan may have normal gas transfer.8

Three-dimensional reconstruction of a high resolution CT scan of a patient with severe AAT deficiency. The emphysema, represented by the darker areas, shows lower zone predominance, as highlighted by the arrows.
Airways disease and bronchodilator reversibility
Airflow obstruction, indicated by a reduced forced expiratory volume in 1 second (FEV1) and reduced FEV1 to forced vital capacity (FVC) ratio, may be seen in AAT deficient subjects. Although the amount of airflow limitation measured may fall anywhere in the range between no obstruction and severe obstruction, it is often out of proportion to the smoking history of the patient.
Airflow limitation is not always fixed and the symptoms and signs in AAT deficiency can be similar to features of asthma. Patients can often be given this diagnosis in childhood or early adulthood and 15% of patients in a Swedish cohort identified by neonatal screening had been diagnosed as having asthma by the age of 22.5 In the same cohort 29% of the patient group reported recurrent wheezing. In the larger NHLBI registry 21% of the total group and 12.5% of those with normal FEV1 had asthma as defined by reversible airflow obstruction, recurrent wheezing, and a reported diagnosis of asthma or allergy with or without elevated IgE levels.3 Studies on airway hyperresponsiveness in these patients are limited and group together patients of different phenotypes, but they suggest that airway hyperresponsiveness is not more prevalent in these patients than in control subjects.9
There is a wide variation in bronchodilator response in these patients. In the NHLBI registry 28% of patients showed reversibility of ⩾12% and >200 ml in FEV1 during the first visit, and this was seen in 49% over all visits in the study.3 In these patients the median increase in FEV1 was 330 ml (range 202–1492), which explains why many patients who present at a young age are given an initial diagnosis of asthma. Indeed, bronchodilator responses are also seen in patients without a reduced FEV1 below 80% predicted, and it may be that this is more common in the early stages of the disease.
Although airflow obstruction and parenchymal destruction can often be seen together in the same patient, this is not always the case. Some patients have marked emphysema with very little airways disease, and the reasons for this relative preservation of airway function are unclear. By contrast, some patients have severe airflow obstruction with little parenchymal disease and show preserved gas transfer (fig 2).

Baseline lung function data for each of 398 Pi Z patients on the UK registry are shown for forced expiratory volume in 1 second (FEV1) and carbon monoxide transfer factor (Tlco) as % predicted. Although a significant correlation is seen overall between FEV1 % predicted and Tlco (r = 0.67, p<0.001), some patients have discordant physiology (FEV1<80% predicted and Tlco >80% predicted and vice versa) as shown by the filled data points.
Chronic bronchitis
Registries of patients with AAT deficiency show that as many as 43% of patients have chronic sputum expectoration, as defined by Medical Research Council (MRC) criteria, even in non-smokers. Patients with chronic bronchitis tend to have more severe airflow obstruction and more extensive emphysema than those without, despite similarities in age and smoking history.10,11
Bronchiectasis
The presence of bronchiectasis in patients with AAT deficiency is well recognised and, in series using CT scanning for detection, the incidence varies from 41% to 43%.7,12,13 However, in the largest study the incidence of bronchiectasis was 26%,10 which is similar to that seen in usual COPD,14 suggesting that the incidence is not increased. In support of this, Cuvelier et al compared a group of patients with bronchiectasis, mainly diagnosed by CT scan, and a group of control subjects and found similar AAT phenotypes and gene frequencies in the two groups.15 A greater frequency of Pi Z alleles was seen in patients with bronchiectasis who also had emphysema, but the number of patients with the Pi Z phenotype was too small to provide firm conclusions. Although the bronchiectasis seen may be severe and associated with chronic sputum production, sputum expectoration itself is neither a sensitive nor a specific feature for detecting the presence of bronchiectasis and CT scanning remains the most reliable tool.10
Exacerbations
Exacerbations occur in around 50% of patients who attend the UK registry and the mean duration of episodes is approximately 15 days (unpublished data). Exacerbations occur more frequently in patients with chronic bronchitis,10 in index patients identified as a result of their lung disease, and in those with more severe disease as assessed by the GOLD (Global initiative for Chronic Obstructive Lung Disease) criteria.16 The episodes are associated with a greater degree of inflammation than in patients not deficient in AAT (fig 3).17

{kind=link}
{kind=link}
{kind=link}
Mean (SE) sputum concentrations of chemoattractants at the start of an acute exacerbation in non-deficient COPD control patients and patients with α1-antitrypsin deficiency (AATD). Significantly higher levels of IL-8 and leukotriene B4 were seen in patients with AAT deficiency (p = 0.01 and p = 0.02, respectively). Data derived from Hill et al.17
LIVER DISEASE
Only a small subpopulation of Pi Z patients develop liver injury that affects health and this may be due to genetic or environmental differences in the hepatocellular response to AAT Z protein accumulation in the hepatocytes.
Acute liver disease and neonatal disease
AAT deficiency is a common cause of neonatal cholestasis in countries with a high prevalence of AAT deficiency. One population based epidemiological study on 200 000 infants followed 127 Pi Z children from birth to the age of 18.18–20 Fourteen children had prolonged obstructive jaundice, eight had minimal biochemical abnormalities as neonates, and two of these 22 children died in early life with cirrhosis. Enzyme abnormalities were found to be common in early life and, by the age of 6 months, 60% of the healthy children had raised serum transaminases. By the age of 18, 12% of the Pi Z patients were found to have abnormal liver function tests but none of these patients had clinical evidence of liver disease.
There may be other genetic factors that predispose to the development of childhood liver disease as certain families appear to have a greater risk. In one study significant liver disease was seen in 21% of “at risk” siblings.21 Perlmutter et al have shown that an increase in the synthesis of stress proteins occurs in a subset of Pi Z individuals with liver disease,22 but it remains unclear whether this is a cause or effect. Breast feeding may provide some protection against the development of severe liver disease and early death in childhood,23 although other studies have shown no difference in outcome compared with bottle fed infants.21
Chronic liver disease and fibrosis
AAT deficiency accounts for a high proportion of liver transplants in children with chronic liver disease when biliary atresia is excluded.24 Two case-control necroscopic studies looked at the risk of liver disease in adult homozygotes and found an odds ratio for cirrhosis of 8.3 (95% CI 3.8 to 18.3),25,26 although some of these patients were asymptomatic. The presentation of patients with chronic liver disease due to AAT deficiency is indistinguishable from that due to other causes and, although this can occur at any time of life, it is increasingly seen with advancing age. An association with chronic liver disease is seen in patients who have never smoked, but this may be due to longer survival in these patients who have not developed severe lung disease. Apart from a higher incidence in men than in women, no host or environmental factors (including viral hepatitis or high alcohol use) have been associated with the development of liver disease in adult homozygous patients studied.
Hepatocellular carcinoma
Necroscopic studies have shown an odds ratio of developing primary liver cancer of 5.0 (95% CI 1.6 to 15.8) in patients with AAT deficiency. Primary liver cancer occurs more commonly in association with cirrhosis, although it has been seen in patients without cirrhosis.27
OTHER CLINICAL MANIFESTATIONS
Panniculitis
Many cases of AAT deficiency associated panniculitis have been reported.28–31 Commonly, the condition leads to painful red nodules on the thighs of an adult with AAT deficiency and these lesions may then ulcerate and drain clear sterile fluid. Fat necrosis may be seen in association with the lesions and relapsing episodes may occur. Treatment options for the panniculitis include corticosteroids, dapsone and tetracyclines, although AAT replacement seems particularly efficacious.32–34
Vasculitis
A higher incidence of AAT deficient phenotypes has been seen in groups of patients with systemic vasculitis, particularly in those with anti-proteinase 3 antibodies and Wegener’s granulomatosis.35–39 However, the incidence of anti-proteinase 3 antibodies in AAT deficient patients is low, and many do not develop features of systemic vasculitis.37,40 It therefore appears that AAT deficiency is only a minor risk factor for the development of vasculitis.
Pancreatitis
An association between AAT deficiency and pancreatitis has been the subject of some case reports and case-control studies,41–43 but other studies have found no difference in AAT phenotype in patients with pancreatitis compared with controls.44 Witt et al recently found no difference in allele frequency between a group of patients with chronic pancreatitis and control subjects.45
Cardiovascular disease
There is a theoretical association between aortic aneurysmal disease and AAT deficiency due to uninhibited elastase activity on the elastic tissue of arterial walls, but this has not been supported in published data46 nor has an association been shown with intracranial aneurysms. In fact, both the Pi Z and Pi MZ phenotypes have been associated with lower blood pressure in men and Pi MZ may be associated with a reduced risk of ischaemic cerebrovascular and ischaemic heart disease.47 The risk of cardiovascular disease in patients with severe AAT deficiency has not been sufficiently investigated to formulate firm conclusions.
Renal disease
Numerous case reports have described individuals with AAT deficiency and glomerulonephritis, particularly in children and young adults.48–51 Membranoproliferative glomerulonephritis has been described most frequently although other forms of glomerulonephritis have also been seen.52 However, most cases report individuals who also have liver complications, and it may be that the renal disease is a consequence of the liver disease. One study on groups of Pi MZ and Pi Z patients without liver disease did not identify a difference in nephropathy between the two groups, although the number of patients studied was small.53 Patients with deficiency and anti-neutrophil cytoplasmic antibody (ANCA) positive vasculitis may also develop renal disease.
RISK FACTORS AFFECTING RATE OF CHANGE IN LUNG FUNCTION
The degree of lung function impairment can vary greatly among patients with the same phenotype for AAT deficiency,54 and can be significantly different in siblings with the same phenotype. Some environmental factors have been shown to affect the development and progression of disease in these patients, but it is likely that other host factors are also important.
Smoking
The most important risk factor for the development of emphysema and airflow obstruction in AAT deficiency is active smoking. Piitulainen et al found an accelerated decline in FEV1 over 12 months in current smokers (70 ml/year, CI 58 to 82) compared with ex-smokers (41 ml/year, CI 36 to 48) and never smokers (47 ml/year, CI 41 to 53).55 Other studies have reported even higher rates of decline in lung function in smokers.56,57 Furthermore, there appears to be a dose-response relationship between cigarette consumption and change in FEV1 over time. Active smoking can affect lung function as early as the age of 18, with a significantly lower FEV1 and FEV1/VC in smokers than in non-smokers.58 However, active smoking does not explain all of the variability and patients who have never smoked still show variation in their clinical course.59
Passive smoking with an exposure of more than 10 years has been associated with chronic bronchitis in non-smoking individuals,60 but there has been no evidence to support an association between passive smoking in adulthood and lung function decline. However, parental smoking has been associated with some changes in lung function in adolescents with this condition,58 and it may be that passive smoking in childhood reduces the potential maximum lung capacity in early adulthood.
Exacerbations
Lower respiratory tract infections may also affect the clinical course of the disease4 and prior infections are associated with symptoms of cough and wheeze.61 The effect of exacerbations may be more apparent in patients with mild to moderate disease and an increasing number of exacerbations has been shown to correlate with the decline in gas transfer.62 It is therefore likely that interventions that reduce the frequency of exacerbations may also reduce this decline. Indeed, augmentation therapy, which may moderate lung function decline in some patients,63 may also be associated with a reduction in the frequency and severity of exacerbations,64 although prospective clinical trials are required to support this assumption. However, it has been shown that infusions of AAT reduce airway leukotriene B4 concentrations65 and this neutrophil chemotactic factor is thought to be central to the exacerbation episodes.
Environmental factors
Domiciliary use of kerosene heaters and working in agriculture for at least 10 years were shown to be associated with increased symptoms and decreased lung function in non-smoking Pi Z patients in the Swedish registry.60 Self-reported occupational exposure to gas, fumes, or dust was found to be an independent risk factor for lung function impairment in older patients who had never smoked.66 Mineral dust exposure, as detected by self-reported questionnaires, was also shown to be independently associated with chronic cough and with airflow limitation after adjusting for age and smoking in a group of American patients with more severe disease.61 However, it is still not clear if this is due to high total inhalational exposure to many agents in these patients or to a specific effect of mineral dust. Taken together, these studies suggest that environmental exposures may be associated with the development of respiratory symptoms in these patients and may also be a contributory factor to lung function decline. Further studies are needed to isolate the effects of individual agents and to identify groups of individuals who may be more susceptible to these effects.
Bronchodilator reversibility
Bronchodilator reversibility has been shown to be associated with a more rapid decline in FEV1 and is an independent predictor of decline after accounting for age, sex, and smoking status.3,62,63 Other features of asthma, such as attacks of wheeze and raised IgE, have not been associated with a greater decline in lung function.
Age
Age, male sex, and previous symptoms of wheezing were found to be independent predictors for lung function impairment in 225 Pi Z patients who had never smoked,66 but the relationship between age and the rate of decline in lung function is less well understood due to other confounding factors. Eden et al found that patients aged between 30 and 44 years had the most rapid decline in lung function, and age remained an independent predictor of lung function decline after adjustment for other confounders.3
Base lung function
In a study on Pi Z patients from the UK registry there was a correlation between the initial FEV1 and its subsequent decline, with the more severely affected patients showing the least change.62 Although this accelerated decline in less severely affected patients may illustrate a change in the disease course over time, it may be that survivorship bias “selects” some severe patients whose lung function is declining more slowly.
Genetic factors
Other unidentified genetic factors may lead to a predisposition to an accelerated decline in lung function or to increased susceptibility to the effects of smoking.67,68 These are discussed elsewhere in this review series.
CONCLUSIONS
AAT deficiency is a risk factor for the development of respiratory symptoms, early onset emphysema, and airflow obstruction in adult life. Environmental factors such as cigarette smoking, exacerbations, and dust exposure are additional risk factors and have been linked to an accelerated decline in this condition. Host factors such as age, bronchodilator reversibility, and other genetic factors also influence the development of the disease. Patients with lung disease have symptoms in common with usual COPD, with a high prevalence of chronic bronchitis. AAT deficiency may also lead to the development of acute or chronic liver disease in childhood or adulthood and has been linked to other diseases. Greater understanding of the processes that lead to the expression of clinical disease in this condition will help us to design new and earlier interventions to improve symptoms and to alter its course.
REFERENCES
Footnotes
-
R A Stockley is a member of AIR (The Alpha1 International Registry).