Article Text

Abstract
BACKGROUND Pulmonary arteriovenous malformations (PAVMs) occur in over 25% of patients with the autosomal dominant disorder hereditary haemorrhagic telangiectasia (HHT). Mutations in two genes, endoglin andALK-1, are known to cause HHT. Each encodes a protein expressed on vascular endothelial cells and involved in signalling by members of the transforming growth factor (TGF)-β superfamily. To date, PAVMs have not been detected inALK-1 families. There is evidence from a single HHT family without pulmonary involvement that a third HHT gene may exist. To establish the existence of a further HHT gene responsible for PAVMs, linkage analyses were performed on an expanded PAVM-HHT family in which HHT did not result fromendoglin mutations.
METHODS Family members were assessed clinically to assign HHT disease status and were screened for PAVMs. DNA was extracted from blood obtained from 20 individuals of known disease status. Short tandem repeat polymorphic markers spanning the intervals containing the endoglin andALK-1 genes were amplified by the polymerase chain reaction using 33P-labelled oligonucleotide primers, separated by denaturing polyacrylamide gel electrophoresis (PAGE), and the resultant autoradiographs were examined for allele sizes. Linkage analyses were performed using MLINK and GENEHUNTER.
RESULTS Twelve members spanning four generations were affected with HHT. Two had proven PAVMs, one with a classical appearance, the other exhibiting microscopic PAVMs exacerbated by pregnancy. Two point lod and multipoint lod scores significantly excluded linkage to endoglinand ALK-1 in this pedigree.
CONCLUSIONS This study confirms the existence of a third HHT locus that accounts for disease in some HHT patients with pulmonary involvement.
- genetic linkage
- pulmonary arteriovenous malformations
- transforming growth factor β
Statistics from Altmetric.com
Pulmonary arteriovenous malformations (PAVMs) provide a direct communication between the pulmonary arterial and venous systems without an intervening capillary bed, either through complex vascular structures supplying and draining a bulbous aneurysmal sac1 or through smaller calibre telangiectatic vessels. The full picture of complications is still emerging.2 Most can be attributed to the right-to-left (R-L) shunt that may exceed 40% of the cardiac output in severe cases. Hypoxaemia is common but is usually well tolerated by the patient because of the attendant low pulmonary vascular resistance. However, cerebral abscesses, cerebrovascular accidents, and transient ischaemic attacks due to paradoxical embolism through the PAVMs result in considerable morbidity and mortality.3-6 Although individual PAVMs are amenable to surgical resection, in most individuals multiple lesions are likely with time and therefore the advent of the parenchymal-sparing embolisation techniques has transformed the outlook for patients.7 To reduce the incidence of paradoxical embolism, embolisation of all feasible PAVMs is recommended with antibiotic prophylaxis for dental and surgical procedures in all patients with residual shunts.2 ,8 As up to 50% of patients with PAVMs are asymptomatic before a cerebral event, this highlights the need for effective clinical screening methods for PAVMs.
More than 70% of PAVMs occur in patients with hereditary haemorrhagic telangiectasia (HHT, Rendu-Osler-Weber syndrome), a vascular disorder inherited as an autosomal dominant trait. HHT has a prevalence rate of about one in 10 000 in Europe9 and classically presents with epistaxis and telangiectasia, particularly on the lips, tongue, buccal mucosa, and digits.10 ,11 Gastrointestinal bleeding due to telangiectasia occurs in approximately one third of patients, especially in later life, and is well recognised. However, the significance of AVMs in the pulmonary (>25%), hepatic (8–16%), and cerebral (15%) circulations is often overlooked. Although all three can cause considerable mortality and morbidity, only PAVM treatment can be safely attempted in all patients. Thus, PAVM screening programmes have been targeted towards patients with HHT,12 ,13 with attempts made to identify genetic subgroups of HHT patients who are at particular risk of developing PAVMs.
Linkage studies assigned the first HHT disease locus to chromosome 914 ,15 and the disease gene was subsequently identified as endoglin.16 The second HHT locus, HHT2, was mapped to chromosome 1217 ,18 and the mutated gene identified asALK-1.19 Linkage studies on one HHT family with an unusual predominance of hepatic AVMs (and no pulmonary involvement) indicated that this pedigree was not linked toendoglin orALK-1, suggesting the likelihood of a third HHT gene,20 although the exact locus and mutated gene have yet to be identified. Endoglin andALK-1 encode transmembrane proteins expressed predominantly on vascular endothelial cells and implicated in signalling by members of the transforming growth factor (TGF)-β superfamily.21 ,22 Numerous mutations have been described in endoglin 16 ,23-29 andALK-1,19 ,30 ,31 although no phenotypic differences have been ascribed to particular mutations within these genes. There appear to be differences betweenendoglin and non-endoglin families: PAVMs have been seen predominantly in endoglin-linked families32 and, to date, no ALK-1linkage or mutations have been described in patients with PAVMs (although only a small number have been screened). These data have led some to suggest that clinical screening for PAVMs should be restricted to endoglin-linked families.10
In this paper we extend previous findings indicating that a PAVM-HHT family is unlinked to endoglin and now show that, in this pedigree, HHT is also unlinked toALK-1. This confirms an earlier report that there is at least one further HHT locus, and extends this observation to HHT patients with PAVMs. A PAVM-HHT family linked toALK-1 is also described briefly. We discuss the implications for clinical practice and our understanding of the biology of the TGF-β signalling pathways and pathophysiology of HHT.
Methods
CLINICAL EVALUATIONS
These studies were performed with the approval of the Multicentre Research Ethics Committee of Scotland (MREC 98/0/42). With informed consent, both authors assessed additional and original members of families previously identified and screened by CLS.14 ,24Clinical evaluation included full history, physical examination for telangiectasia, cyanosis, and finger clubbing, oximetry measured by an Ohmeda 3800 oximeter for 10 minutes in both erect and supine postures and, when appropriate, full blood counts. Additional information was obtained from local physicians.
Disease status was designated using the current international consensus criteria.33 Affected status was assigned in the presence of any three of the following criteria: affected first degree relative; recurrent spontaneous epistaxis; mucocutaneous telangiectasia; and documented visceral manifestations. If only two of the four criteria were present (affected first degree relative and epistaxis), unknown status was assigned and the individual excluded from molecular analyses. However, if both a parent and child of the individual were affected, a diagnosis of obligate affected was made.
ASCERTAINMENT OF GENOTYPE
All affected and unaffected family members were venesected to provide blood for genetic analyses. DNA was extracted from whole blood using proteinase K digestion, phenol- chloroform extraction, and ethanol precipitation, with resuspension in Tris-buffered EDTA.34 Linkage studies were performed by analysis of short tandem repeat (STR) polymorphic markers as previously described.14 Markers of high heterozygosity were chosen on the basis of their position relative to the genes of interest and experience in previous molecular papers (endoglin,14 ,16 ALK-1 17 ,19). Primer sequences are published elsewhere.35 ,36 The STRs were amplified by the polymerase chain reaction (PCR) using approximately 25 ng DNA template, 20 ng of each PCR primer (forward primer labelled with33P) in 10 μl reactions with TaqDNA polymerase as previously described.14 Reaction times in a PTC-100 thermal cycler (MJ Research) were typically 30 cycles of 95 degrees for 20 seconds, primer specific amplification temperature for 40 seconds, and 72 degrees for 60–100 seconds according to product length. The products were size separated by denaturing polyacrylamide gel electrophoresis and detected by autoradiography. Allele patterns were assessed by eye by two observers blinded to disease status.
LINKAGE ANALYSES
Two point and multipoint analyses were performed using MLINK and GENEHUNTER37 via Human Genome Mapping Project (HGMP) computational resources. Based on previously published estimates9 and our previous studies in this population,14 penetrance was set at P = 0.8 for individuals aged 12–35 and P = 0.95 for those aged over 35, giving two liability classes. Apparently unaffected children under the age of 12 years were excluded as penetrance is less than 80% at this age.14 A disease gene frequency of 0.0001 was used and recombination rates were assumed to be equal in both sexes. Lod scores were calculated initially with equal allele frequencies, and subsequently according to the allele frequencies in the population.
The linkage analyses were targeted at the three HHT-PAVM families which we previously reported as unlinked toHHT1/endoglin, with no mutations in the coding regions of endoglin.24The first, family T,14 was insufficiently informative to obtain significantly negative lod scores across the entireALK-1 interval. The second, family V, in which one of six affected members had PAVMs presenting with clubbing and a 25% R-L shunt, was linked to ALK-1 (Z = 1.48, θ = 0.00, D12S368), although the exact mutation has not yet been identified. In this paper we present data on the third of these pedigrees.
Results
CLINICAL RESULTS
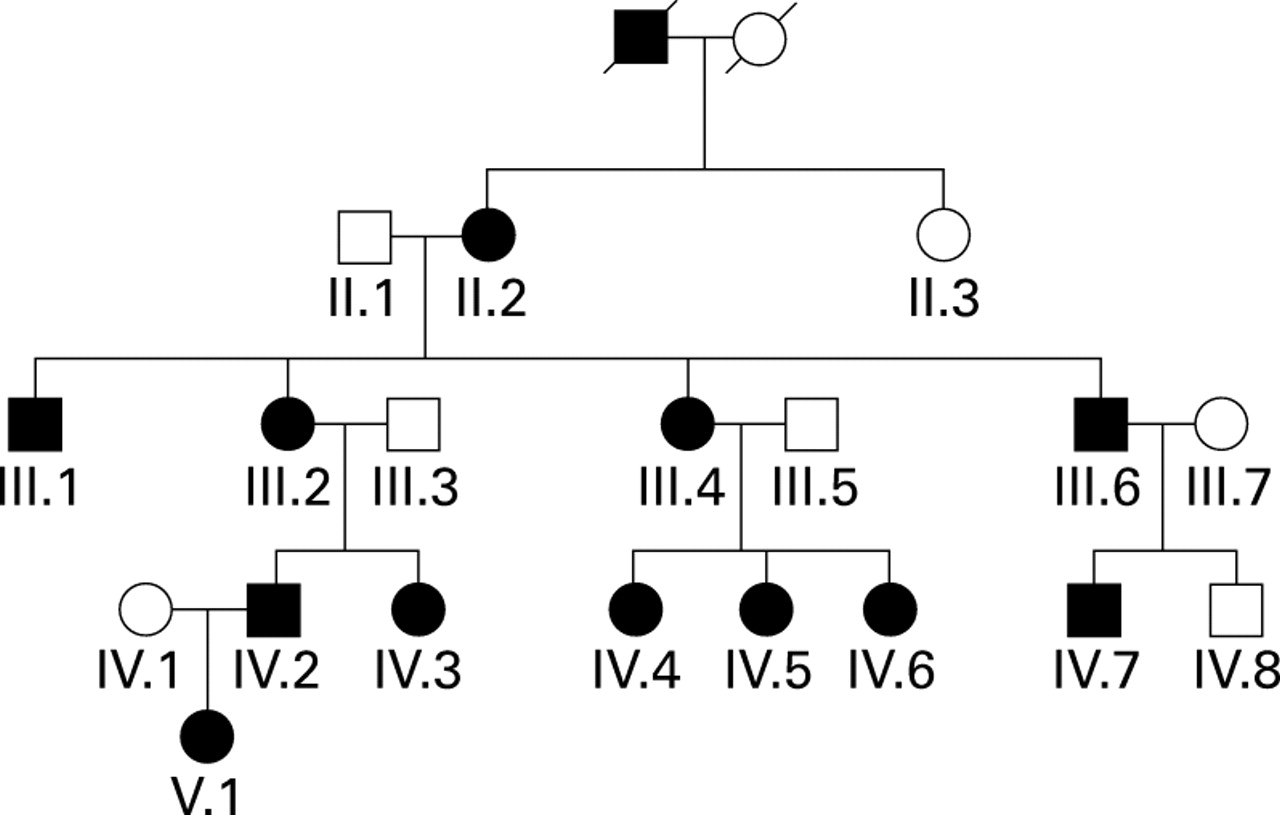
The expanded pedigree of family S, previously briefly described by Shovlin et al,24 is illustrated in fig 1. The results of the clinical investigations are summarised in table 1. Twelve members spanning four generations were affected by HHT as defined by positive family history, epistaxes, and telangiectasia. Individual III.2 was classified as an obligate affected as she had an affected parent and two affected children, although personally fulfilled only two diagnostic criteria. Three further individuals who were offspring of affected family members had epistaxis alone and no affected children; they were therefore classified as being of unknown status and are not illustrated here, nor are the five offspring of affected family members who were unavailable for review.

Pedigree of family S. Affected individuals are indicated by black symbols, unaffected by white symbols. Squares represent males, circles represent females, and a slashed symbol indicates a deceased individual. Individuals of unknown status are not illustrated.
Clinical features of family members
Two members of the family had PAVMs. The proband IV.2 presented with haemoptysis, clubbing, polycythaemia (Hb 18.9, haematocrit 0.55), hypoxaemia (Pao 2 8.2 kPa), and orthodeoxia (supine Sao 2 92%, erect 87%). The R-L shunt measured by 99mtechnetium-labelled albumin macroaggregates (radionucleotide scanning) was 24% of the cardiac output (normal range in this institution <3.5%).12 Two large PAVMs were successfully embolised with steel coils (fig 2A). Although individual IV.3 was asymptomatic with normal oxygen saturations (Sao 2 100% in supine and erect postures) when first evaluated at the age of 21 years, following pregnancy at the age of 26 she developed monthly epistaxes, frequent dizzy spells, and significant orthodeoxia (Sao 2 99% supine, 94% erect). Microscopic PAVMs were diagnosed on the basis of a positive contrast echocardiogram (15 months post partum), a R-L shunt of 7.2% six months later (fig 2B), and a normal thoracic axial computed tomographic (CT) scan at 5 mm intervals. She remains on prophylactic antibiotic therapy and under regular respiratory review.

PAVM patient data. (A) Post embolisation chest radiograph of case IV.2 indicating the presence of PAVM embolisation coils. (B) Perfusion scan using 99mtechnetium-labelled albumin macroaggregates on case IV.3. The significant cerebral activity indicates the presence of a right-to-left shunt quantified as 7.2% of the cardiac output (normal in this institution <3.5%12). In view of the recent suggestion that contrast echocardiography may be used in isolation as a first line screen for PAVMs in patients with HHT,13 it is worth noting that, in this patient, previously positive contrast echocardiography was negative on the day of the perfusion scan, a discrepancy which may be attributable to altered posture before and during the relevant examinations.
EXCLUSION OF ENDOGLIN
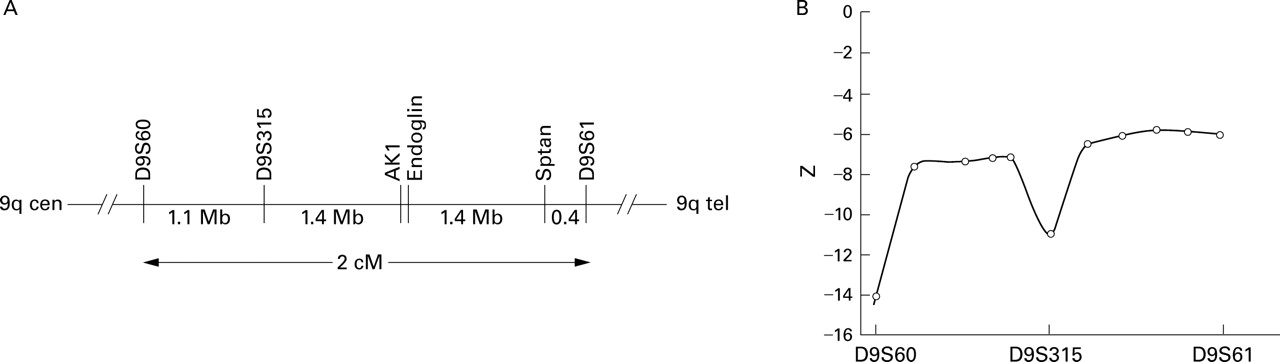
Linkage studies were first performed to confirm exclusion of linkage to endoglin, as previously reported in a subsection of the pedigree.24 TheHHT1 locus was originally mapped to a 2 cM interval on chromosome 9 flanked by D9S60and D9S61. 14 (The adjacentD9S61-D9S63 interval described by McDonald et al 15 was subsequently revised by McAllister et al 16 to between D9S60and D9S61.) Theendoglin gene has been mapped physically as equidistant between D9S315 and the non-erythrocytic α-spectrin gene SPTAN, which both lie within the interval flanked byD9S60 and D9S61(fig 3A, unpublished data).24 MarkersD9S60, D9S315, and D9S61 were used for linkage studies as they are more polymorphic than STRs associated withendoglin andSPTAN.24

Exclusion of endoglin as the disease gene. (A) Position of endoglin gene relative to flanking markers in the 2 cM interval between D9S60 and D9S61. Relevant physical distances (in Mb) are indicated below the horizontal line. The relative positions of endoglin and AK1 have not been resolved. (B) Multipoint lod scores across the HHT1 (endoglin) interval calculated using LINKMAP (similar figures were obtained between D9S60 and D9S61 using GENEHUNTER, data not shown).
Two point lod score calculations using age related penetrance indicated that HHT was not linked to markers in this interval (table 2). Lod scores were not significantly different when calculated using altered allele frequencies (results not shown). Multipoint linkage calculations using the three loci also indicated that the HHT gene does not map to the interval, and excludes endoglin as the disease gene in this family (fig 3B).
Pairwise lod scores between chromosome 9 loci and HHT
EXCLUSION OF ALK-1
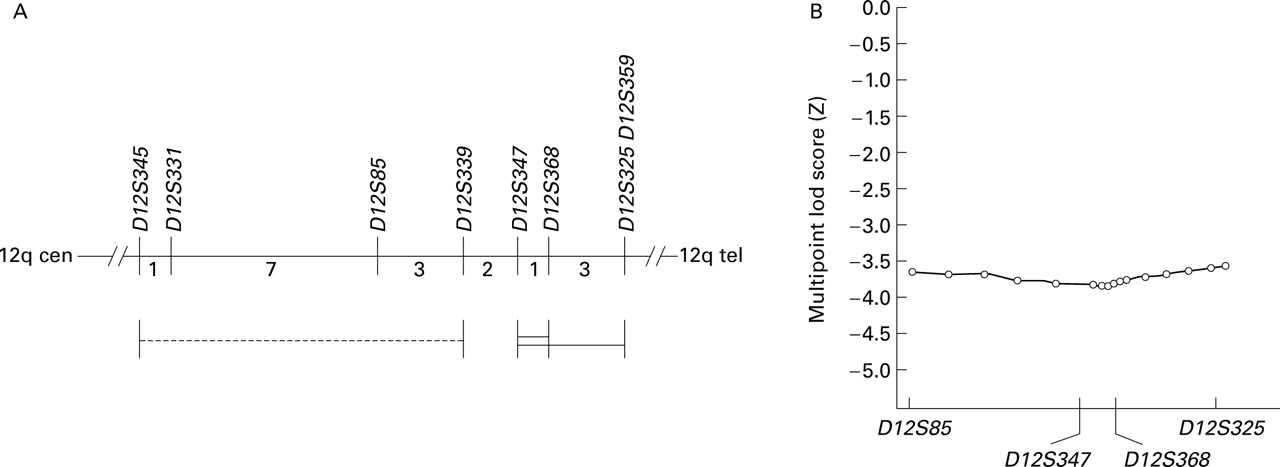
We then performed analyses to assess linkage toALK-1. Two original assignments of theHHT2 locus were given, one betweenD12S339 andD12S359 17 and the other in a neighbouring interval between D12S345 andD12S339 18 (fig 4A). The latter was revised on subsequent analyses, and the ALK-1gene is now known to lie in the 4 cM interval betweenD12S347 andD12S359, most probably in the 1 cM interval between D12S347 andD12S368.19 Nevertheless, we considered it prudent to examine both disease intervals.

Exclusion of ALK-1 as the disease gene. (A) Position of ALK-1 gene relative to flanking markers. All distances are indicated in cM according to Gyapar et al.35 The probable ALK-1 site is indicated by a double bar, with possible extension indicated by a single bar.19 The dotted line indicates one of the original intervals18 which has now been revised..19 (B) Multipoint lod scores across the HHT2 (ALK-1) interval calculated using LINKMAP (similar figures were obtained between D12S331 and D12S325 using GENEHUNTER, data not shown).
Two point lod score calculations using age related penetrance indicated that HHT was not linked to markers spanning the entire region (table 3). Lod scores were not significantly different when calculated using altered allele frequencies (results not shown). Multipoint linkage calculations using four loci spanning the disease interval also indicated that the HHT gene does not map to the region, and excludesALK-1 as the disease gene in this family (fig 4B).
Pairwise lod scores between chromosome 12 loci and HHT
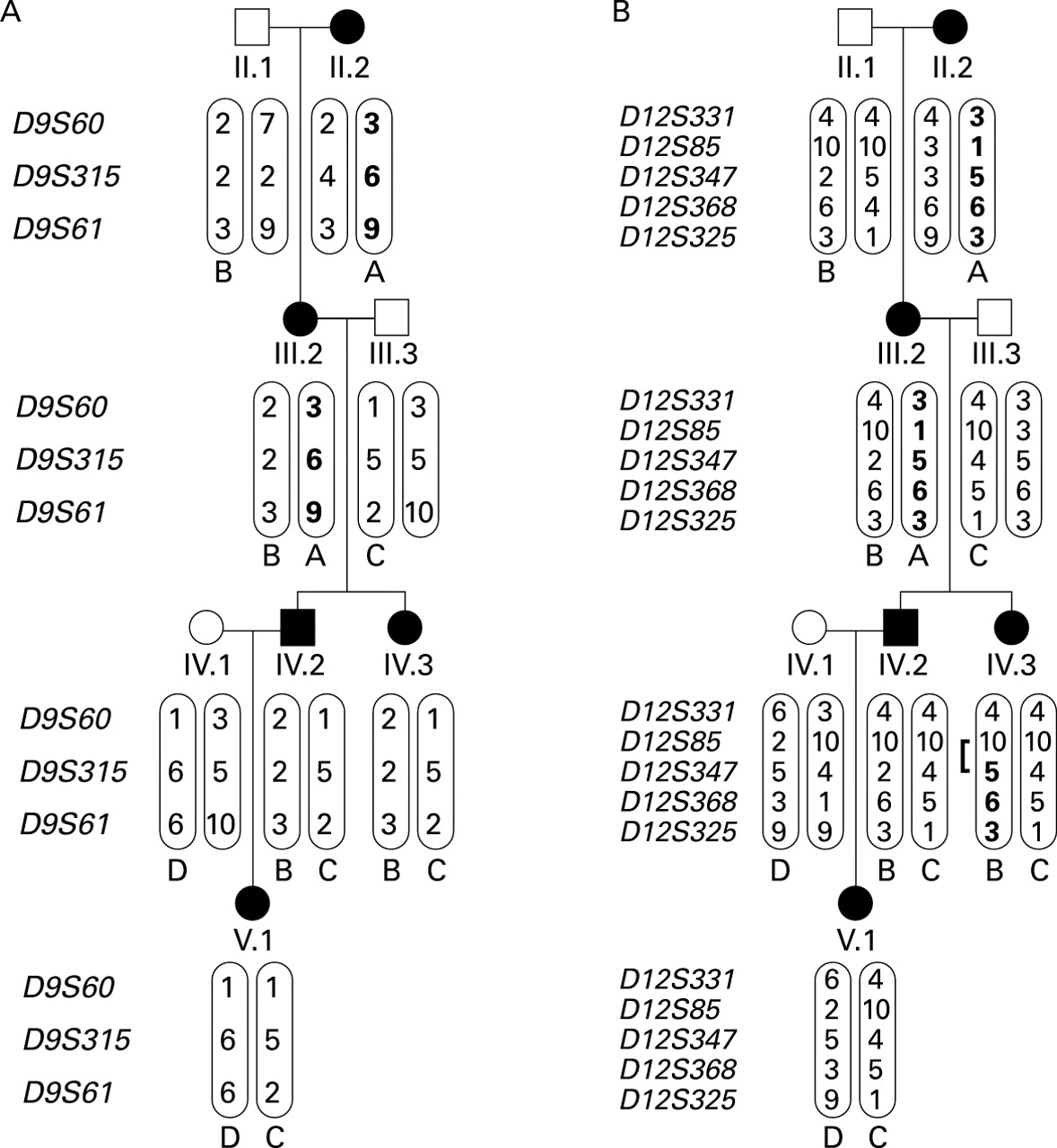
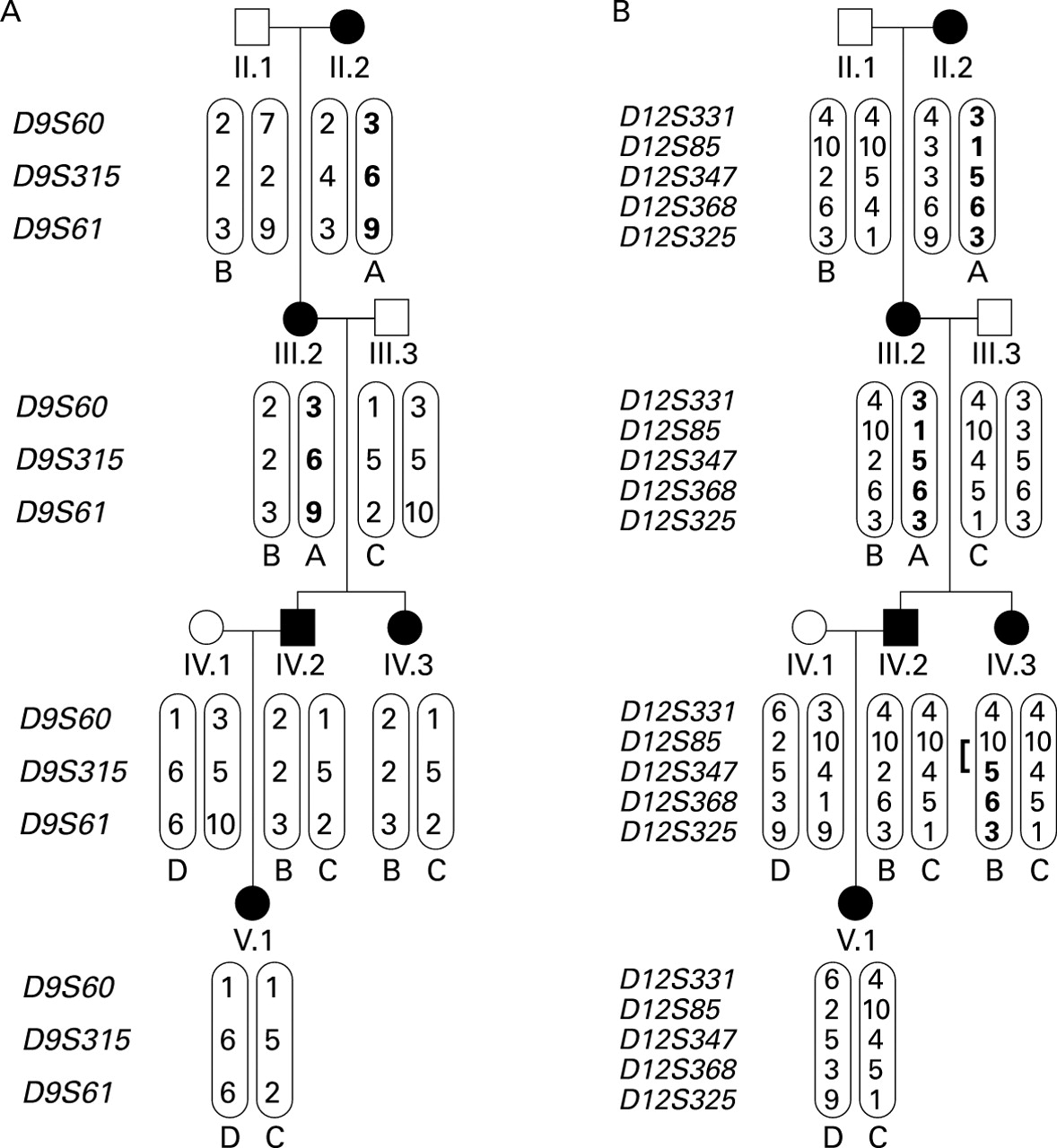
Limited haplotype analyses on the subsection of the pedigree involving the PAVM patients provided further confirmation of exclusion of both known HHT genes (fig 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Limited haplotype analyses. Haplotypes in a section of the pedigree including the two PAVM patients. (A) Chromosome 9 markers spanning endoglin. In affected individuals IV.2, IV.3, and V.1 all haplotypes (B, C and D) are derived from unaffected spouses. (B) Chromosome 12 markers spanning ALK-1. In affected individuals IV.2 and V.1 all haplotypes (B, C and D) are derived from unaffected spouses. Note the recombination event which has occurred in IV.3.
Discussion
We have shown by two point and multipoint linkage analyses that a large HHT pedigree with proven pulmonary involvement is significantly unlinked to the two known HHT genes,endoglin andALK-1. This confirms an earlier report that there is at least one further HHT locus20 and extends this observation to HHT patients with PAVMs.
Since incorrect assignment of the HHT phenotype could result in false negative lod scores, it is important to re-examine the phenotypes in all members of family S (table 1). All phenotypes were assigned before the molecular studies and only DNA from unequivocally affected or unaffected individuals was analysed. On both chromosome 9 and chromosome 12, of the four affected individuals in generation III (three of whom were parents or grandparents of affected individuals), two displayed one haplotype which could be traced to their affected parent II.2, and two the alternative. Analyses of the haplotypes transmitted to and by the PAVM patients were particularly instructive (fig 5) and further confirmed that HHT in this family cannot result from mutations in either endoglinor ALK-1.
Could this have been suspected before the molecular studies? There is a wide variation in disease expression between individuals with HHT. Most patients experience nose bleeds of varying severity and display mucocutaneous telangiectasia which increase with age, but the disease spectrum ranges from non-penetrance in 3% of carefully examined patients9 to significant involvement of visceral vascular beds. As in most HHT families, the affected individuals in this pedigree span all categories. We cannot comment on the incidence of cerebral or hepatic manifestations in this family as asymptomatic screening for these complications is not performed routinely in the United Kingdom. However, before this study, based on the behaviour and incidence of the PAVMs in two of 12 affected individuals, the literature on HHT might have suggested family S to be anendoglin family. The clinical presentation and embolisation response of family member IV.2 reflect most patients with PAVMs who present symptomatically. Family member IV.3 represents one of the more recently recognised cases of microscopic PAVMs detected essentially by a screening programme. The apparent precipitation of her symptoms and PAVMs by pregnancy is well recognised.38Although there is only one report illustrating a partial spontaneous improvement post partum,39 we believe this pattern is likely to reflect the physiological behaviour of PAVMs rather than her underlying HHT genotype.
The pulmonary involvement present in members of non-endoglin families has significant implications for clinical PAVM screening which is critical since patients are often asymptomatic at the time of cerebrovascular events and, particularly, cerebral abscesses.2 Available data still suggest that the detection rate for PAVMs will be higher in theendoglin-linked families. However, our data support the view that PAVM screening should be performed in all HHT patients as PAVMs may be present in individuals from families linked toALK-1 (family V) or families unlinked to either known locus (current data). The development of PAVMs in individual IV.3, who is one of the few reported cases in whom serial screening has been undertaken, highlights the importance of rescreening, particularly after pregnancy.
As well as the clinical implications, this family should provide an additional means of dissecting the complexities of signalling by the TGF-β superfamily by the ultimate identification of the mutated gene.Endoglin andALK-1 are both involved in the regulation of the response to ligands of the TGF-β superfamily, but the natural ligands and important functional signalling complexes for each have yet to be determined. Endoglin is an accessory protein for several receptor complexes of the TGF-β superfamily; in addition to its relatively well recognised involvement in signalling by TGF-β1 and TGF-β3, recent data suggest that endoglin might also be involved in signalling by bone morphogenetic proteins (BMPs) or activins.22Similarly, while ALK-1 is currently an orphan type I TGF-β receptor with no assigned natural ligand, there is evidence that it too may bind TGF-β ligands from several different subfamilies.40 It seems reasonable to speculate that the gene mutated in the pedigree described here may encode another component of the TGF-β signalling complex or downstream effectors, and its identification should significantly enhance our understanding of signalling by TGF-β family members. Many of these are implicated in vascular development, homeostasis, and repair (references cited by Shovlinet al 2 and Pece-Barbara22).
However, we should note that, for endoglin, identification of the HHT mutations, insights into biochemical functions relevant to the vasculature,41 and now the availability of murine models of HHT42 ,43 have yet to translate into a clear understanding of the pathophysiology of HHT. Thus, even following the identification of the gene(s) responsible for HHT in this and the pedigree previously described,20 there may remain a significant gap between our knowledge of the molecular abnormalities of HHT and the means by which these precipitate the development of abnormal vascular structures and, particularly, PAVMs.
Acknowledgments
GMFW is a British Heart Foundation Junior Fellow and CLS is supported by the Wellcome Trust. The authors would like to acknowledge the contributions of Dr H R Gribben, Dr D J M Sinclair, and Professor J M B Hughes to the clinical management of cases IV.2 and IV.3, and Dr J E Jackson who performed the angiography and embolisation of case IV.2. D Glass performed the radionucleotide shunt measurements, Dr M Blomley and Dr P Nihoyannopoulos performed echocardiography assessments, and C I Mordin and J Curtis carried out pulmonary function testing. We would also like to thank the family members and their general practitioners for their cooperation in these studies.