Article Text

Abstract
Background: The interaction between neutrophils and platelets may be important in the modulation of pulmonary haemodynamics under systemic inflammatory conditions. A study was undertaken to examine whether antiplatelet agents inhibit platelet-neutrophil adherence and ameliorate the pulmonary haemodynamic response to fMLP by inhibiting thromboxane release in endotoxin primed lungs. fMLP stimulates neutrophils but not platelets; however, thromboxane synthesis in neutrophils is very low.
Methods: Rats were pretreated with either cilostazol (300 mg/kg) or aspirin (50 mg/kg) before endotoxin priming (5 mg/kg). Platelets in the lung were identified by fluorescent immunohistochemistry. Platelet-neutrophil adherence was analysed by flow cytometry of the lung vascular flush. The effect of fMLP (10−6 M) on thromboxane release, lung weight (an indicator of pulmonary vasoconstriction), and lung filtration coefficient was determined in an isolated lung system perfused at a constant pressure difference.
Results: Endotoxin induced platelet accumulation and platelet-neutrophil adherence in the lung capillary were completely inhibited by cilostazol and aspirin while neutrophil recruitment was not affected. The fMLP challenge caused a significant increase in thromboxane B2 levels in endotoxin primed lungs. The fMLP induced decrease in lung weight was enhanced by endotoxin priming (from −4.9 to −63.9 mg, 95% CI of mean difference −99.5 to −10.5 mg, p<0.05). The fMLP induced increase in the lung filtration coefficient was also enhanced by endotoxin priming (from 0.63 to 2.40 mg/min/cm H2O/g, 95% CI of mean difference 1.17 to 2.37 mg/min/cm H2O/g, p<0.05). Treatment with cilostazol and aspirin completely inhibited the enhanced pulmonary haemodynamic response to fMLP.
Conclusion: The neutrophil–platelet interaction is of critical importance in the modulation of pulmonary haemodynamics via thromboxane.
- endotoxin priming
- platelets
- neutrophils
- thromboxane
- pulmonary vasoconstriction
Statistics from Altmetric.com
It is well known that neutrophils are primed by inflammatory substances such as endotoxin and are ready in lung capillaries to respond to an enhanced release of a wide array of inflammatory mediators by second stimulation.1–3 Primed neutrophils may be identified in patients under systemic inflammatory conditions such as sepsis or following extensive trauma and surgery—indications with a high risk for the subsequent development of acute lung injury.4
Although neutrophils play an important role in the pathophysiology of acute lung injury, interaction with platelets may potentiate the production of eicosanoids such as thromboxane, thereby altering the pulmonary haemodynamic response to second stimulation. Voelkel et al5 reported that N-formyl-l-methionyl-l-leucyl-l-phenylalanine (fMLP), a neutrophil chemotactic peptide, causes strong vasoconstriction and oedema via thromboxane which is probably released from cells trapped in lung capillaries of endotoxin primed rats.6 They found cell sedimentation composed not only of neutrophils, but also of platelets, in the lung capillaries of endotoxin primed rats.6 However, neutrophils alone do not explain the production of high levels of thromboxane in the lung. fMLP stimulates neutrophils but not platelets;7,8 however, the activity of thromboxane synthesis in neutrophils is very low.9
fMLP stimulated neutrophils may produce thromboxane in coordination with platelets, the most important source in vivo. It has been shown that the neutrophil–platelet interaction is critical in the production of eicosanoids.8,10,11 Neutrophils and platelets are known to contact via platelet P-selectin and neutrophil β2 integrins (CD11b/CD18).12,13 In this study we sought to examine whether endotoxin priming enhances the adherence of neutrophils and platelets in the lung capillaries and, if so, whether interruption of the neutrophil–platelet interaction with antiplatelet agents ameliorates the haemodynamic response to fMLP by inhibiting thromboxane release.
METHODS
Animals
Specific pathogen free male Sprague-Dawley rats weighing 300–350 g were used in this study (n = 90). All animals received humane care in compliance with the guidelines from the University Committee on Animal Resources, Tohoku University, Sendai, Japan. All animal experiments were conducted in accordance with the “Principle of Laboratory Animal Care” formulated by the Institute of Laboratory Animal Resources and the “Guide for the Care and Use of Laboratory Animals” prepared by the Institute of Laboratory Animal Resources and published by the National Institute of Health.
Experimental protocols
Rats were pretreated with two different types of antiplatelet agents—cilostazol, a selective inhibitor of the type III phosphodiestalase cilostazol,14 and aspirin. Cilostazol (300 mg/kg; Otsuka Pharmaceutical, Tokyo, Japan) was dissolved in 1 ml water and administered directly into the stomach via a small catheter 2 hours before endotoxin priming. Aspirin (50 mg/kg; Wako Chemical, Osaka, Japan) was dissolved in 0.5 ml sterilised saline and intraperitoneally injected 18 hours before endotoxin priming. Rats were then primed by an intraperitoneal injection of E coli endotoxin (5 mg/kg; Difco Laboratories, Detroit, MI, USA) dissolved in 1 ml sterilised saline. For control experiments, the same amount of saline vehicle was injected.
Myeloperoxidase (MPO) assay
At 18 hours following endotoxin priming, rats were anaesthetised with pentobarbital sodium (50 mg/kg intraperitoneally) and ventilated manually using a 10 ml syringe via a tracheostomy. After a median sternotomy, heparin sodium (200 U) was administered directly into the right ventricular cavity. The abdominal aorta and inferior vena cava were then cut at the diaphragm. The pulmonary artery was cannulated and both lungs were cleared of blood by flushing them with 50 ml saline at a pressure of 15 cm H2O at room temperature. The right lung was weighed and homogenised in 0.02% cetyltrimethylammonium bromide (CTAB) solution (Sigma, St Louis, MO, USA) in 50 mM potassium phosphate buffer at pH 6.0 on ice. The homogenate was centrifuged at 12 000g for 15 minutes at 4°C, the supernatant decanted, and the pellet weighed and resuspended in CTAB solution. The suspension was freeze thawed three times, followed by repeated sonication for 30 second bursts with an output of 30 Watts. The lung suspension was centrifuged at 12 000g for 15 minutes at 4°C and the supernatant (S1) collected. The pellet was further resuspended in CTAB solution, freeze thawed, sonicated, and centrifuged to obtain the second supernatant (S2). In our preliminary experiments we found that our samples had very low MPO activity following further permeabilisation steps (S3, S4 and S5), whereas the mixture of S1 and S2 recovered more than 85% of total MPO activity. A mixture of both S1 and S2 served as a sample for the lung MPO assay. MPO activity was determined by chemiluminescence using a cypridina luciferin analogue, 2-methyl-6-(p-methoxyphenyl)-3,7-dihydroimidaz[1,2-a]pyrazin-3-one hydrochloride (MCLA; Tokyo Kasei Kogyo, Tokyo, Japan) as a luminescence reagent.
Immuohistochemistry
The flushed left lung was immediately frozen fixed in liquid nitrogen and 3 μm tissue blocks were sliced. The tissue sections were preincubated with 10% non-immunised goat serum for 20 minutes and incubated with fluorescein isothiocyanate (FITC) labelled rabbit polyclonal anti-rat platelet antibody (Accurate, Westbury, NY, USA) at a dilution of 1:50 for 30 minutes at room temperature. The antigen-antibody complex was visualised via a fluorescent microscope at 450–490 nm of the emission wavelength. For a negative control, FITC labelled normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used instead of the antibody and no specific immunoreactivity was detected in any lung section.
Flow cytometry
Flow cytometry was performed on the cells obtained by flushing the lungs. The pulmonary artery and the left ventricle were cannulated. Both lungs were cleared of blood by flushing with 50 ml Earle’s Balanced Salt Solution (EBSS) at 37°C via the pulmonary artery, then further flushed in a retrograde fashion with 200 ml prewarmed calcium free Hank’s Balanced Salt Solution (HBSS) containing 1 mM EDTA. The lung vascular flush was centrifuged at 600g for 5 minutes. The cells were resuspended in calcium free HBSS at 105 cells/ml and reacted with FITC labelled anti-rat platelet antibody at a dilution of 1:20 for 20 minutes at 4°C. Fluorescence intensity was analysed using a flow cytometer (Ortho Clinical Diagnostic, Raritan, NJ, USA). For a negative control, FITC labelled normal rabbit IgG was used instead of the antibody, and there was no significant difference in the very low levels of non-specific fluorescent intensity between each experimental group.
In addition, a smear of lung vascular flush was fixed with ethanol, reacted with the antibody, and visualised via a fluorescent microscope. The population of neutrophils in the lung vascular flush was counted by staining with a Giemsa solution.
Pulmonary haemodynamic response to fMLP
The pulmonary artery and left ventricle were cannulated and the lungs were cleared of blood with EBSS as described above. Heart-lung blocks were then removed from the thorax and suspended via a force displacement transducer (Nihon-Koden, Tokyo, Japan) to monitor the lung weight continuously. The lungs were perfused with Krebs-Henseleit solution (pH 7.4) containing 6% bovine albumin and saturated with a mixed gas (N2: 74%, O2: 20%, CO2: 6%) at 37°C at a constant pressure difference where lung weight was stable during perfusion. In most of our experiments a stable lung perfusion was obtained at 12 cm H2O pressure on the pulmonary artery side when the pressure in the left ventricle side was initially fixed at 3 cm H2O. The pressures in the pulmonary circulation were adjusted by raising or lowering the reservoirs connected to the pulmonary artery and left ventricle. The lungs were maintained in an inflated state with an airway pressure of 2 cm H2O.
The pulmonary filtration coefficient was estimated by the isogravimetric method15 with modifications. After 30 minutes of baseline monitoring the pressure of the pulmonary artery and left ventricle were simultaneously increased by raising the reservoirs on both the pulmonary side and the left ventricle side by 3 cm H2O for 10 minutes. Changes in lung weight were continuously monitored during the study. Once lung weight had returned to baseline levels, fMLP (Sigma) was introduced into the perfusate at a final concentration of 10−6 M. The fMLP challenge caused a transient decrease in lung weight which lasted for up to 15 minutes. Since the lungs were perfused at a constant pressure difference, the observed decrease in lung weight may possibly represent a change in lung capillary volume by vasoconstriction. The lung weight returned to baseline levels within 90 minutes of the fMLP challenge, and again the pressure in the pulmonary artery and left ventricle were raised by 3 cm H2O for 10 minutes. The initial filtration rate of the lung capillaries was estimated by extrapolating the linear regression of the lung weight gain on a semilogarithmic scale to time zero. The filtration coefficient was calculated by dividing the initial filtration rate by the applied lung capillary pressure increment (3 cm H2O) and standardised to 1 g wet lung weight. To inhibit thromboxane action, 10−6 M of seratrodast (Takeda Pharmaceutical, Tokyo, Japan), a thromboxane A2 receptor antagonist,16 and 10−5 M of mecrofenamate (Cayman Chemical), a cyclooxygenase inhibitor, were introduced into the lung perfusate 10 minutes before the fMLP challenge. Thromboxane B2 levels in the lung perfusate 2 hours after the fMLP challenge were determined with a thromboxane B2 ELISA kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s instructions.
Data analysis
Data are presented as mean (SE) and statistical analyses were performed initially by using analyses of variance (ANOVA) across all groups excepting thromboxane B2 levels in the lung perfusate, because they did not show symmetrical distribution theoretically. Only where significant differences were found by ANOVA was it followed by a Bonferroni adjustment to identify which groups were significantly different. The size of effects was provided as mean differences with 95% confidence intervals (CI). For thromboxane B2 levels, data are presented as median with 95% CI, and statistical analyses were performed with the Kruskal-Wallis non-parametric test. When overall differences were identified, the Steel test was performed to identify which groups were significantly different. Statistical significance was defined as p<0.05.
RESULTS
Low levels of MPO activity were detected in the lungs of control animals. Endotoxin priming increased lung MPO activity significantly (from 10.2 (1.3) to 46.6 (5.6) × 106 cpm/right lung, p<0.05, fig 1). However, pretreatment with cilostazol or aspirin did not ameliorate the increased lung MPO activity in endotoxin primed rats (49.1 (3.1) and 43.7 (2.8) × 106 cpm/right lung, respectively; fig 1).

Effect of cilostazol and aspirin on lung myeloperoxidase (MPO) activity. Pretreatment with cilostazol or aspirin did not ameliorate the increased lung MPO activity in endotoxin (ETX) primed rats. Cilostazol and aspirin showed no significant influence on lung MPO activity in rats without endotoxin priming (not shown). Mean (SE) group data are shown (n = 5). *p<0.05 v control (ANOVA followed by a Bonferroni adjustment).
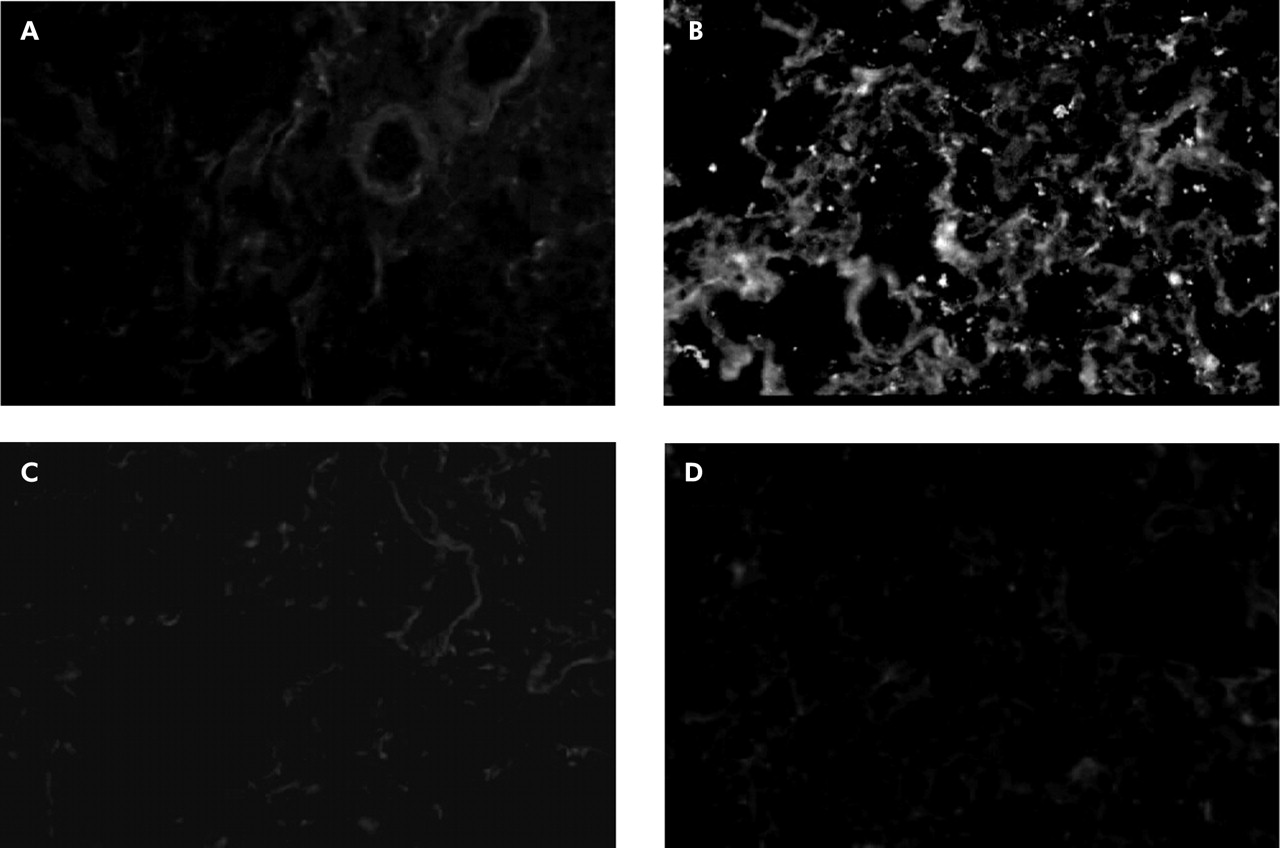
FITC labelled antiplatelet antibody identified no platelets in control specimens (fig 2A) but a number of platelets were found in lung sections obtained from endotoxin primed rats (fig 2B). When rats were pretreated with cilostazol and aspirin, no fluorescence immunoreactivity of platelets was detectable (fig 2C and D). Vehicle controls for cilostazol and aspirin did not inhibit platelet accumulation in endotoxin primed rats.
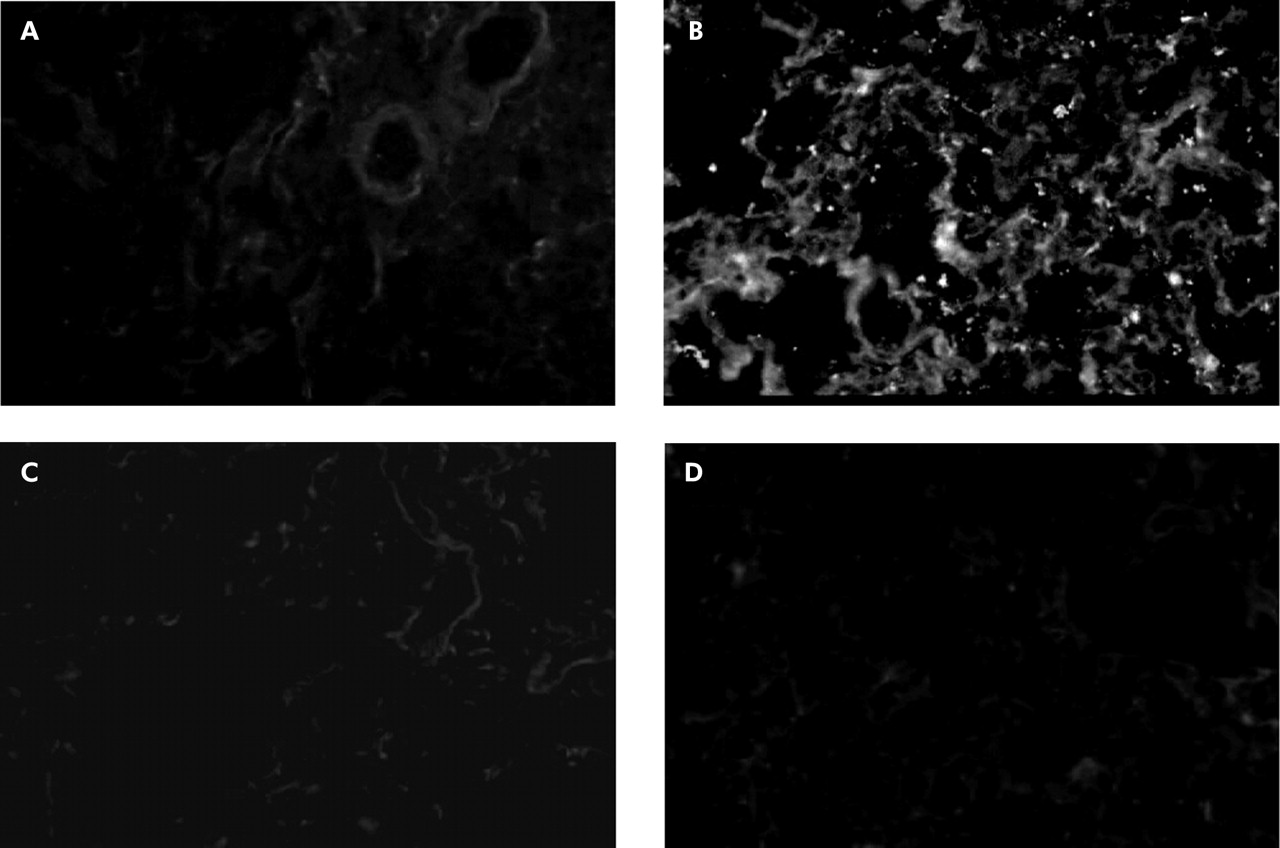
Immunohistochemistry of platelets in lung sections. FITC labelled anti-rat platelet antibody identified no platelets in control experiments, although very low levels of non-specific background staining outlined the alveolar wall structures (A). In contrast, a number of immunofluorescence spots of platelets were found in lung sections obtained from endotoxin primed rats (B). Pretreatment with antiplatelet agents completely inhibited platelet accumulation in the lung capillaries. There was no detectable platelet immunoreactivity in lungs pretreated with cilostazol (C) or aspirin (D). For immunohistochemical examinations, the flushed left lungs were frozen fixed in liquid nitrogen. Frozen lung sections were then reacted with FITC labelled polyclonal rabbit anti-rat platelet antibody and visualised via a fluorescence microscope at 450–490 nm of the emission wavelength. For negative controls, FITC labelled normal rabbit IgG was used instead of anti-rat platelet antibody, and no specific immunoreactivity was detected in any lung sections. Original magnification ×200.
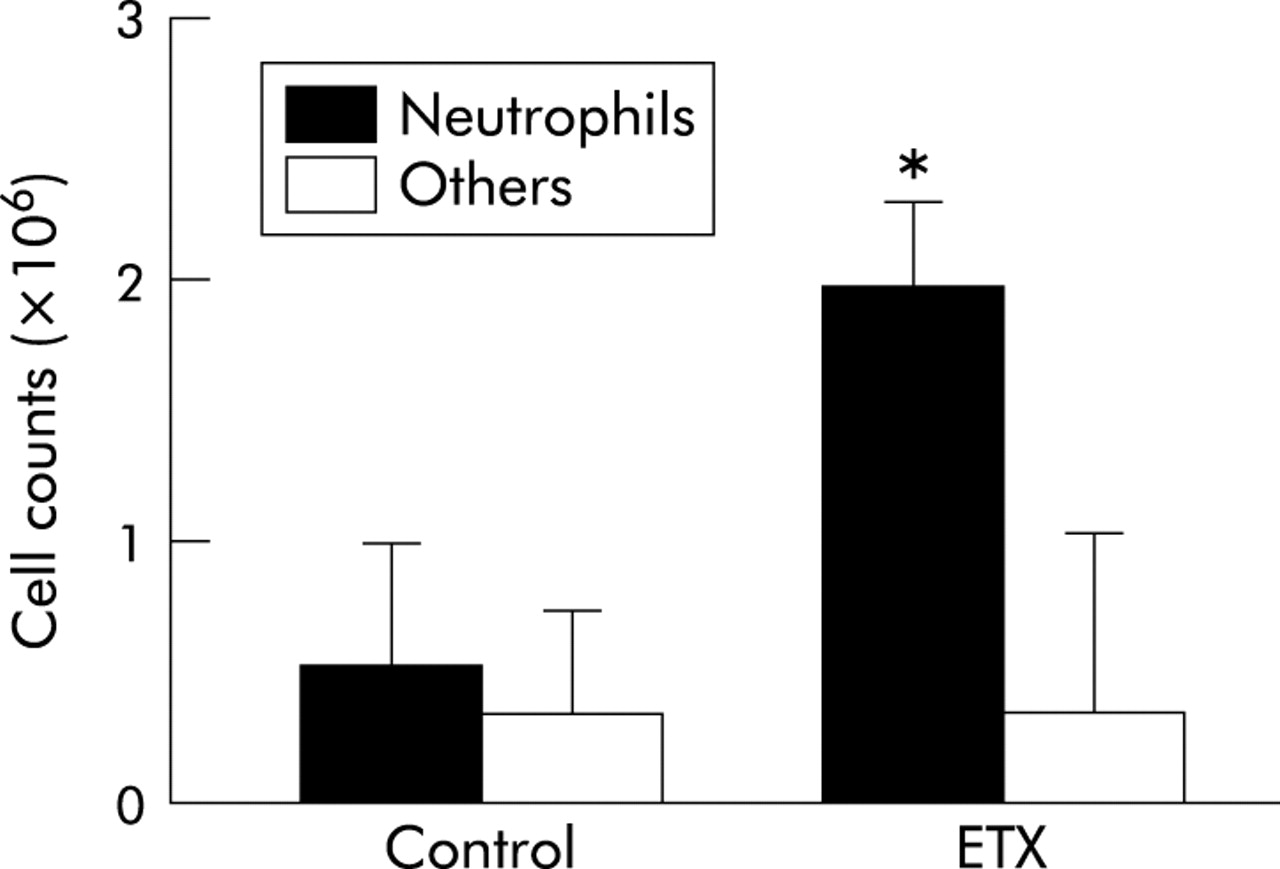
Flow cytometry of the lung vascular flush showed that endotoxin priming increased fluorescence intensity significantly (from 20.0 (2.9) to 60.6 (3.7), 95% CI of mean difference 24.6 to 56.6, p<0.05, fig 3A). Enhanced fluorescent intensity was inhibited to control levels—that is, 33% of endotoxin experiments—by pretreatment with cilostazol (22.0 (3.5), p<0.05 v endotoxin, fig 3B) or aspirin (18.7 (1.8), p<0.05 v endotoxin, fig 3C). Immunocytochemistry using FITC labelled antiplatelet antibody identified the existence of platelets with neutrophils in the lung vascular flush (fig 4). A smaller number of lymphocytes was found on the smear of the lung vascular flush but these lymphocytes were not associated with platelets. Endotoxin priming increased the neutrophil counts in the lung vascular flush significantly from 0.53 (0.50) to 2.00 (0.37) × 106 cells/lung (p<0.05), but there was no significant difference in the number of other cell types, mostly lymphocytes (fig 5).

Flow cytometry of the lung vascular flush. A representative histogram of three different measurements is shown in each group. Endotoxin priming increased fluorescence intensity (A). The increase in fluorescent intensity was inhibited by pretreatment with cilostazol (B) and aspirin (C). Vehicle controls for cilostazol and aspirin showed no inhibitory effects on fluorescence intensity (not shown). The cells in the lung vascular flush (105 cells/ml) were reacted with FITC labelled anti-rat platelet antibody and analysed by a flow cytometer. For negative controls the cells were reacted with FITC labelled normal rabbit IgG; no significant difference was seen in the very low levels of non-specific fluorescent intensity between experimental groups.
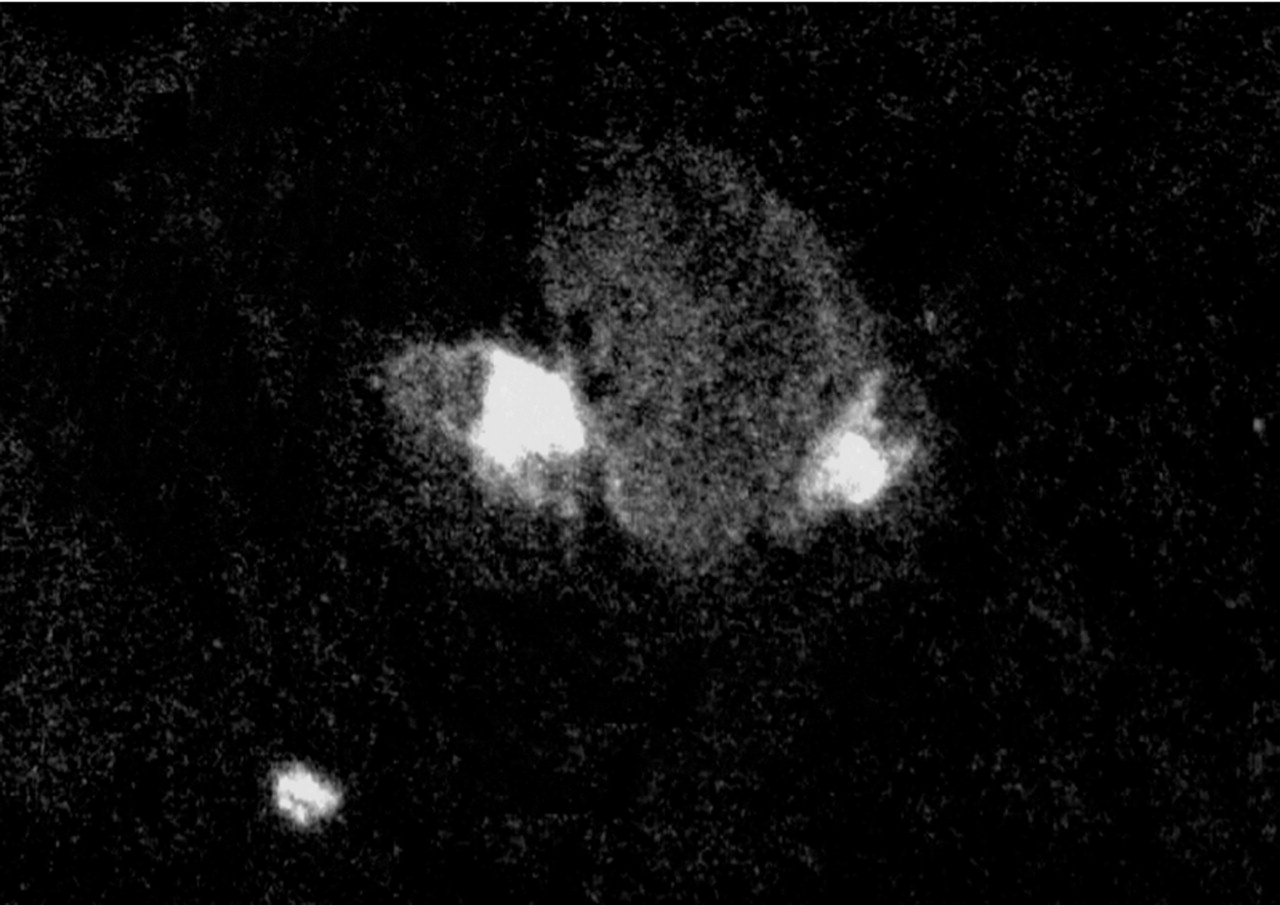
Immunocytochemistry of the lung vascular flush. FITC labelled anti-rat platelet antibody identified several platelets attached to neutrophils. Two platelets are shown in contact with a neutrophil. A number of neutrophils were found to exist with platelets in three different specimens of lung vascular flush of endotoxin primed rats, while very few neutrophils were associated with platelets in a smear obtained from control experiments (without endotoxin priming). A smear of the lung vascular flush was fixed with ethanol, reacted with FITC labelled polyclonal rabbit anti-rat platelet antibody and visualised via a fluorescence microscope at 450-490 nm of the emission wavelength. Original magnification ×600.
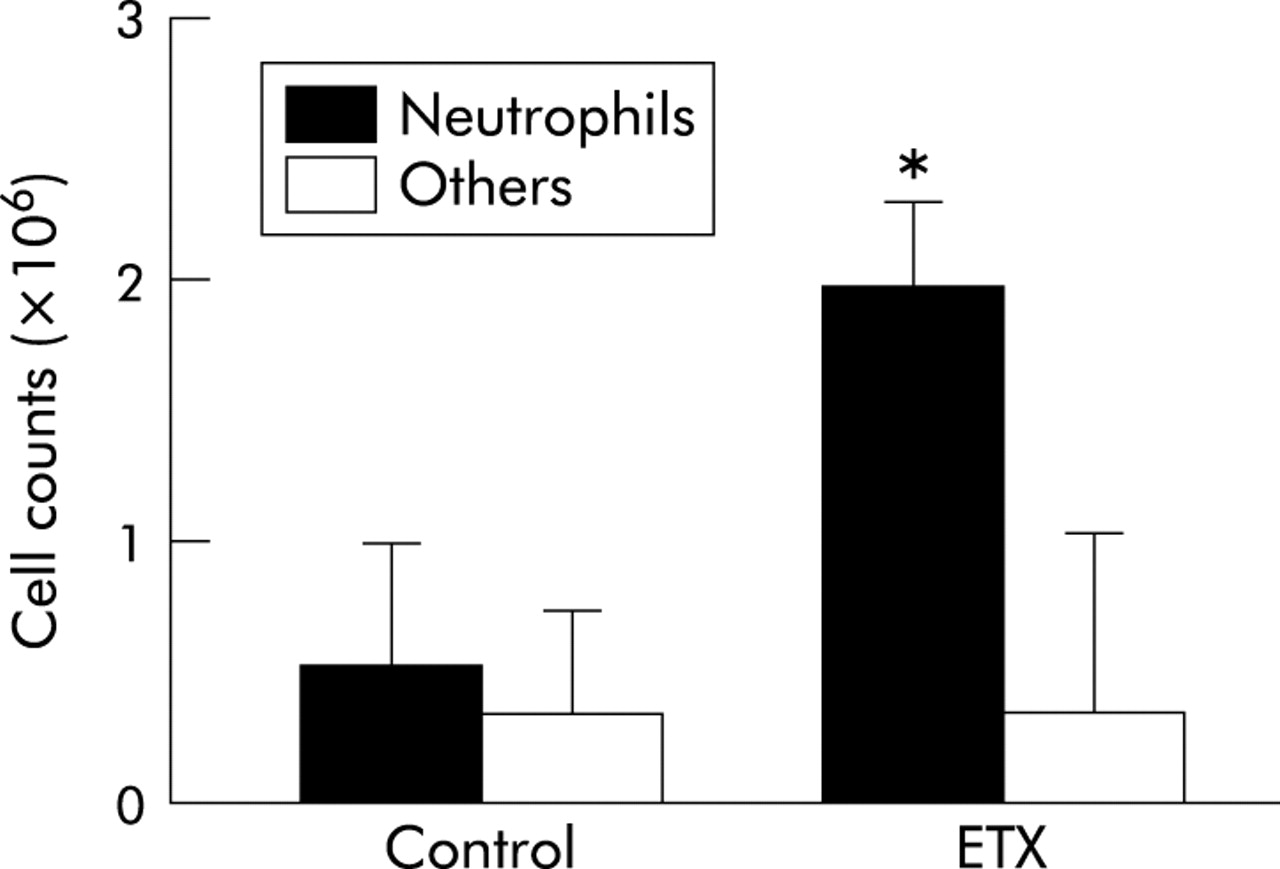
Neutrophil counts in the lung vascular flush. Endotoxin priming (ETX) increased the number of neutrophils in the lung vascular flush. There was no significant difference in the number of other cell types, mostly lymphocytes. Neutrophils were counted by staining lung vascular flush with a Giemsa solution. Data are shown as groups of mean (SE) (n = 5). *p<0.05 v control by ANOVA followed by a Bonferroni adjustment.
Once it was established that neutrophils existed in contact with platelets in endotoxin primed lungs and that in vivo pretreatment with cilostazol and aspirin inhibited adherence of platelets and neutrophils, we then examined whether these antiplatelet agents inhibit fMLP induced thromboxane release. The thromboxane B2 level in the lung perfusate collected 2 hours after the fMLP challenge was very low in control experiments, but fMLP caused a significant increase in thromboxane B2 levels in the endotoxin primed lungs (59 pg/ml, 95% CI 34 to 94 pg/ml v 8696 pg/ml, 95% CI 7146 to 10418 pg/ml, p<0.05, fig 6). Pretreatment with cilostazol and aspirin completely inhibited the impact of endotoxin priming on the fMLP induced thromboxane release (368 pg/ml, 95% CI 92 to 771 pg/ml, p<0.05 v endotoxin, and 58 pg/ml, 95% CI 3 to 167 pg/ml, p<0.05 v endotoxin, respectively; fig 6).
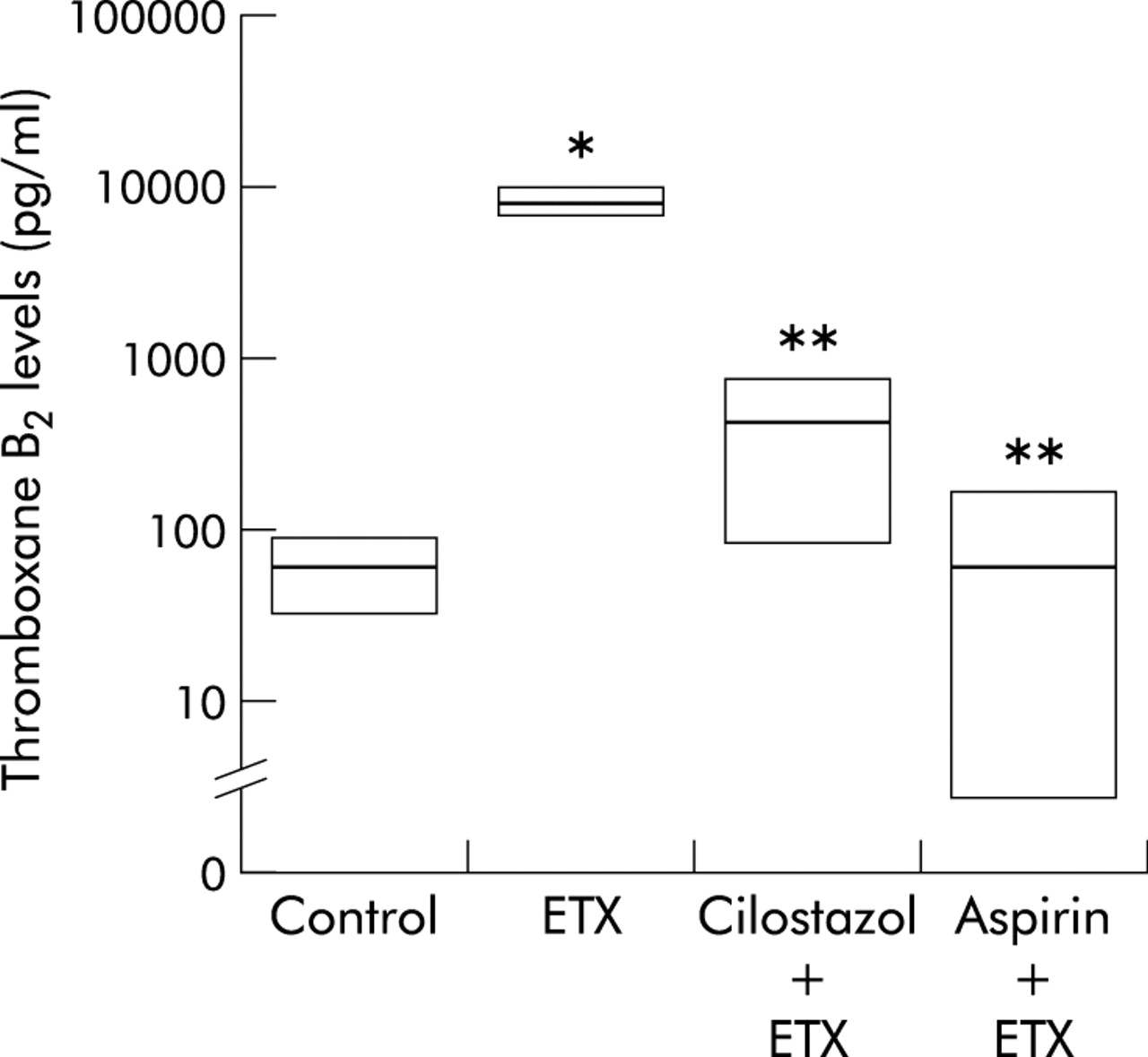
Effect of cilostazol and aspirin on thoromboxane B2 levels in the lung perfusate. fMLP caused a significant increase in thromboxane B2 levels. Pretreatment with cilostazol and aspirin completely inhibited the fMLP induced thromboxane release. Vehicle controls for cilostazol and aspirin showed no inhibition (not shown). Thromboxane B2 levels were determined using a thromboxane B2 ELISA kit. Data are shown as groups of median with 95% confidence interval (CI) (n = 5). *p<0.05 v control; **p<0.05 v endotoxin experiment (ETX) (Kruskal-Wallis test followed by Steel test).
Endotoxin priming enhanced the decrease in lung weight induced by fMLP (from −4.9 (4.9) to −63.9 (16.0) mg, 95% CI of mean difference −99.5 to −10.5 mg, p<0.05, fig 7). The effect of endotoxin priming on the fMLP induced decrease in lung weight was completely inhibited by pretreatment with cilostazol (–6.0 (8.2) mg, p<0.05 v endotoxin) and aspirin (−4.3 (6.3) mg, p<0.05 v endotoxin, fig 7). The effect of endotoxin priming was also completely inhibited by mecrofenamate (−6.4 (6.3) mg, p<0.05 v endotoxin) and seratrodast (−2.2 (3.1) mg, p<0.05 v endotoxin, fig 7).

Effect of cilostazol and aspirin on the weight of perfused lungs. Endotoxin priming enhanced the decrease in lung weight induced by fMLP (95% CI of mean difference −99.5 to −10.5 mg). The decrease in lung weight was completely inhibited by pretreatment with cilostazol and aspirin, as well as mecrofenamate (mecro) and seratrodast (serat). Cilostazol, aspirin, mecrofenamate, or seratrodast had no significant influence on lung weight in rats without endotoxin priming (not shown). Vehicle controls for cilostazol and aspirin had no inhibition (not shown). The lungs were perfused with Krebs-Henseleit solution at a constant pressure difference which made lung weight stable during perfusion. fMLP (10−6 M) caused a transient decrease in lung weight which lasted for up to 15 minutes. The decrease in lung weight induced by fMLP was therefore considered to be representative of a decrease in lung vascular volume due to vasoconstriction. Data are shown as mean (SE) groups of 5. *p<0.05 v control; **p<0.05 v endotoxin experiment (ETX) (ANOVA followed by Bonferroni adjustment).
Endotoxin priming also enhanced the increase in lung filtration coefficient induced by fMLP (from 0.63 (0.32) to 2.40 (0.22) mg/min/cmH2 O/g, 95% CI of mean difference 1.17 to 2.37 mg/min/cm H2O/g, p<0.05, fig 8). Again, the effect of endotoxin priming on the fMLP induced increase in lung filtration coefficient was completely inhibited by cilostazol (0.22 (0.13) mg/min/cm H2O/g, p<0.05 v endotoxin) and aspirin (0.15 (0.33) mg/min/cm H2O/g, p<0.05 v endotoxin), as well as mecrofenamate (0.18 (0.18) mg/min/cm H2O/g, p<0.05 v endotoxin) and seratrodast (0.22 (0.21) mg/min/cm H2O/g, p<0.05 v endotoxin; fig 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of cilostazol and aspirin on lung filtration coefficient. Endotoxin priming also enhanced the increase in lung filtration coefficient induced by fMLP (95% CI of mean difference 1.17 to 2.37 mg/min/cm H2O/g). The increase in lung filtration coefficient was completely inhibited by cilostazol and aspirin, as well as by mecrofenamate (mecro) and seratrodast (serat). Cilostazol, aspirin, mecrofenamate, or seratrodast had no significant influence on lung filtration coefficient in rats without endotoxin priming (not shown). Vehicle controls for cilostazol and aspirin had no inhibition (not shown). The lung filtration coefficient was determined before and after fMLP challenge by the isogravimetric method15 with modification. Data are shown as mean (SE) groups of 5. *p<0.05 v control; **p<0.05 v endotoxin experiment (ETX) (ANOVA followed by Bonferroni adjustment).
DISCUSSION
Our first question was whether endotoxin priming enhances adherence of neutrophils and platelets in the lung capillaries. A marked increase in lung MPO activity confirmed that endotoxin causes recruitment of neutrophils into the lung capillaries. In addition, we found accumulation of a number of platelets in the lungs of endotoxin primed rats. Some of these platelets were found to adhere to neutrophils since flow cytometry of the lung vascular flush showed that endotoxin priming increased fluorescence intensity of platelets. Although we did not separate neutrophils from other cell types such as lymphocytes in the lung vascular flush, the cells attached by platelets were mostly neutrophils. Fluorescent immunocytochemistry identified several platelets on the polymorphonuclear cells in the smear of the lung vascular flush. In addition, the population of neutrophils in the lung vascular flush was >80% in endotoxin primed rats. Our results are consistent with a previous report that examined cell sedimentation in endotoxin primed rats by electron microscopy and found that it was composed not only of neutrophils but also platelets, although it did not examine the effect of antiplatelet agents.6
Once it was clear that endotoxin priming enhanced the adherence of neutrophils and platelets in the lung capillaries, we then determined whether pretreatment with antiplatelet agents would interfere with the neutrophil–platelet interaction. Two different antiplatelet agents—cilostazol and aspirin—were examined. Cilostazol is a selective inhibitor of the type III phosphodiestalase14 which displays powerful inhibition of platelet aggregation17 and has been used in the treatment of various peripheral vascular diseases.18 Aspirin, a classic non-selective cyclooxygenase inhibitor, has also been widely used to prevent platelet aggregation in the lung. Interestingly, both cilostazol and aspirin completely inhibited the accumulation of platelets in the lung but neither had any effect on the increased lung MPO activity. This suggests that antiplatelet agents inhibit the adherence of neutrophils and platelets in the lung capillaries, even though recruitment of endotoxin primed neutrophils occurs. Flow cytometry of the lung vascular flush showed that both cilostazol and aspirin completely inhibited endotoxin induced adherence of neutrophils and platelets.
What is the physiological significance of the activity of antiplatelet agents on the interaction between neutrophils and platelets? We found that both cilostazol and aspirin completely inhibited fMLP induced thromboxane release, so we compared the pulmonary haemodynamic response to fMLP in the presence and absence of antiplatelet agents. Again, both cilostazol and aspirin completely inhibited the fMLP induced decrease in lung weight. The magnitude of this observed weight change is coincident with a decrease in the lung capillary volume caused by pulmonary vasoconstriction at precapillary sites rather than at post-capillary sites. The decrease in lung weight lasted for 15 minutes following the introduction of fMLP. The relatively short life time of fMLP activity on pulmonary blood vessels also suggests the involvement of thromboxane as the primary mediator. In addition, two different types of thromboxane inhibitors—a thromboxane A2 receptor antagonist seratrodast and a cyclooxigenase inhibitor mecrofenamate—completely inhibited the fMLP induced decrease in lung weight.
Our results show that fMLP, which stimulates neutrophils but not platelets,7,8 causes pulmonary vasoconstriction via thromboxane released from platelets. It is very likely therefore that the fMLP induced release of thromboxane did not occur in neutrophils alone but was the result of coordination with platelets. Through interactions with specific receptors on the surface of neutrophils, fMLP activates phospholipase A2 and metabolises phospholipids to arachidonic acids (AA). The attached platelets may share AA with neutrophils which can be further metabolised via cyclooxygenase and thromboxane synthase to thromboxane A2.10 In addition, neutrophils may induce another isozyme of cyclooxygenase, cyclooxygenase (COX)-2, by endotoxin priming19 and metabolise AA to prostaglandin H2 which can also be used as a substrate for thromboxane synthase in platelets.
Interestingly, we found that thromboxane not only caused pulmonary vasoconstriction but also an increase in permeability. Although thromboxane has been considered to mediate pulmonary vasoconstriction and not permeability, several lines of evidence have recently indicated that thromboxane is capable of increasing microvascular permeability in various tissues.20,21 Thromboxane activates phospholipase C and increases both inositol 1,4,5-triphosphate (IP3) production and intracellular free Ca2+ levels via thromboxane A2 receptor, thereby resulting in the contraction of targeted cells such as vascular smooth muscle cells.22 Thromboxane may also influence stress fibres in endothelial cells and increases fluid movement via intercellular junctions.23 We found that endotoxin priming enhanced the impact of fMLP on the lung filtration coefficient. This enhanced action of fMLP in the endotoxin primed lungs was again completely inhibited by pretreatment with cilostazol and aspirin, as well as seratrodast and mecrofenamate which were introduced into the lung perfusate. This indicates that an increase in lung vascular permeability after the fMLP challenge is a result of an enhanced release of thromboxane.
Several experimental studies suggest that aspirin may be beneficial in treating acute lung injury.24,25 In addition, the efficacy of thromboxane A2 receptor antagonist has been examined in endotoxin induced acute lung injury models.26 The results of this study appear to suggest that platelets in the lung capillaries do not function merely as bystanders but, more importantly, they interact with neutrophils in the production of thromboxane which plays an important role in the modulation of pulmonary haemodynamics under the condition of systemic inflammatory response. We found that the pharmacological interruption of the neutrophil–platelet interaction ameliorated fMLP induced pulmonary vasoconstriction and increased permeability by inhibiting thromboxane release in endotoxin primed rats. We therefore suggest that the use of antiplatelet agents may be potentially beneficial in the treatment of acute lung injury.