Article Text

Abstract
BACKGROUND Alpha-1 antitrypsin (α1-AT) is the most abundant proteinase inhibitor within the lung. We have recently reported the surprising observation that cystic fibrosis patients with mild to moderate deficiency of α1-antitrypsin have significantly better pulmonary function than non-deficient patients. This study may have been biased as it did not include the most severely affected patients who have died in childhood or those who have undergone orthotopic lung transplantation. The prevalence of α1-antitrypsin deficiency alleles in this most severely affected group of patients with cystic fibrosis was therefore assessed.
METHODS DNA was obtained from neonatal blood spots from children with cystic fibrosis who had died from pulmonary disease and from formalin fixed lung tissue from transplanted cystic fibrosis patients. The common S and Z deficiency alleles of α1-AT were sought by amplification mutagenesis of the appropriate region of the α1-AT gene followed by restriction enzyme digestion with Xmn I and Taq I, respectively.
RESULTS Seventy nine patients were identified (seven dead, 72 transplanted). Two patients (2.5%) were heterozygous for the Z allele of α1-AT and four (5.1%) were heterozygous for the S allele. This is not significantly different from the incidence in the normal population of 4% and 8% for the S and Z alleles, respectively.
CONCLUSIONS These data support previous findings that deficiency of α1-AT is not associated with more severe pulmonary disease in cystic fibrosis and may be associated with milder lung disease. Further work is needed to clarify the mechanisms underlying the progressive lung damage in cystic fibrosis.
- α1-antitrypsin
- cystic fibrosis
- α1-proteinase inhibitor
- serpins
- bronchiectasis
Statistics from Altmetric.com
One suggested mechanism for cystic fibrosis is the imbalance between neutrophil elastase and proteinase inhibitors, of which the most important is α1-antitrypsin (α1-AT).1 2 Alpha1-antitrypsin is a highly polymorphic acute phase glycoprotein secreted by hepatocytes and monocytes. The common S (264Glu→Val) and Z (342Glu→Lys) deficiency alleles are found in approximately 12% of the UK population3 and result in plasma α1-AT concentrations in the homozygote of 60% and 10%, respectively, compared with the normal M homozygote. Deficiency of α1-AT is associated with early onset panlobar emphysema,4 bronchiectasis,5asthma,6 and cryptogenic fibrosing alveolitis,7 although this last association was not confirmed in a subsequent study.8 Current thinking would predict that α1-AT deficiency in patients with cystic fibrosis would lead to a greater excess of elastase and hence more severe pulmonary disease. We have found that cystic fibrosis patients with mild to moderate α1-AT deficiency unexpectedly have significantly better lung function than non-deficient patients.9 Moreover, Döring et al 10 also found that α1-AT deficiency was not associated with more severe lung disease and that this occurred despite significantly earlier colonisation with Pseudomonas aeruginosa. Both of these studies may have been biased as they did not include the most severely affected cystic fibrosis individuals who had either died or had been transplanted and who may have had an excess of α1-AT deficiency alleles.
We report here the prevalence of the common α1-AT deficiency alleles in these most severely affected patients with cystic fibrosis.
Methods
Neonatal blood spots from individuals screened for cystic fibrosis in East Anglia since 1979 were kindly provided by Dr Anthony Heeley, Department of Biochemistry, Peterborough Hospital, UK. Patients on whom blood spots were available who had died from respiratory disease were identified from the UK Cystic Fibrosis Survey and those who had been transplanted were identified form the UK Transplant Support Service Authority. The certified causes of death were provided by the UK Cystic Fibrosis Survey. Formalin fixed lung tissue had been stored from patients with cystic fibrosis who had undergone lung transplantation at Papworth Hospital since 1985.
ASSESSMENT FOR THE S AND Z α1-αT DEFICIENCY ALLELES
DNA was extracted from blood spots by boiling in 0.05 M NaOH and from formalin fixed lung tissue by DNAzol reagent (Gibco BRL). The S and Z alleles of α1-AT were sought by polymerase chain reaction (PCR) mutagenesis of regions of exons III and V, respectively, followed by restriction enzyme digestion with Xmn I andTaq I (New England Biolabs), respectively, using the PCR primers and a modification of the method of Andresen et al.11 The restriction enzyme recognised the wild type allele but not the deficiency allele. PCR conditions consisted of 12.5 pmol primers, 1.5 mM Mg2+, and one unit ofTaq polymerase (Promega) in a reaction volume of 25 μl. The PCR fragments were generated by 40 cycles of 94°C for 20 seconds, 50°C for 20 seconds, and 74°C for 30 seconds. Restriction enzyme digestion of 12 μl PCR product in a total volume of 20 μl was performed at 37°C and 65°C for a minimum of eight hours forXmn I and Taq I, respectively. The digestion products were run with previously sequenced controls on a 9% w/v polyacrylamide gel and a 3% w/v agarose gel for the S and Z α1-AT alleles, respectively, followed by staining with ethidium bromide. An additional restriction site present in the oligonucleotide primers acted as an internal control of enzyme efficiency. The presence of the S and Z alleles was confirmed by cycle sequencing with Thermosequenase and 33P labelled terminators (Amersham Life Science).
Results
Formalin fixed lung tissue was available from 72 patients with cystic fibrosis who had undergone heart-lung or double lung transplantation at Papworth Hospital and blood spots were present for seven cystic fibrosis patients who had died in childhood from pulmonary disease. The mean (SD) age of those who had been transplanted was 25.4 (6.1) years and of those who had died in childhood was 3.6 (3.4) years.
α1-AT PHENOTYPES
Z α1 - AT
PCR amplification of exon V generated a 97 bp fragment (fig 1, lane 2) that was fully digested in non-Z individuals by theTaq I restriction enzyme to a 64 bp fragment (lanes 4–7). The Z point mutation rendered the PCR product resistant to digestion and was seen as an 86 bp band (lane 3), having been shortened from the PCR product by digestion at the internal control site. The individual in lane 3 was heterozygous for the Z allele as both 86 and 64 bp fragments are generated.

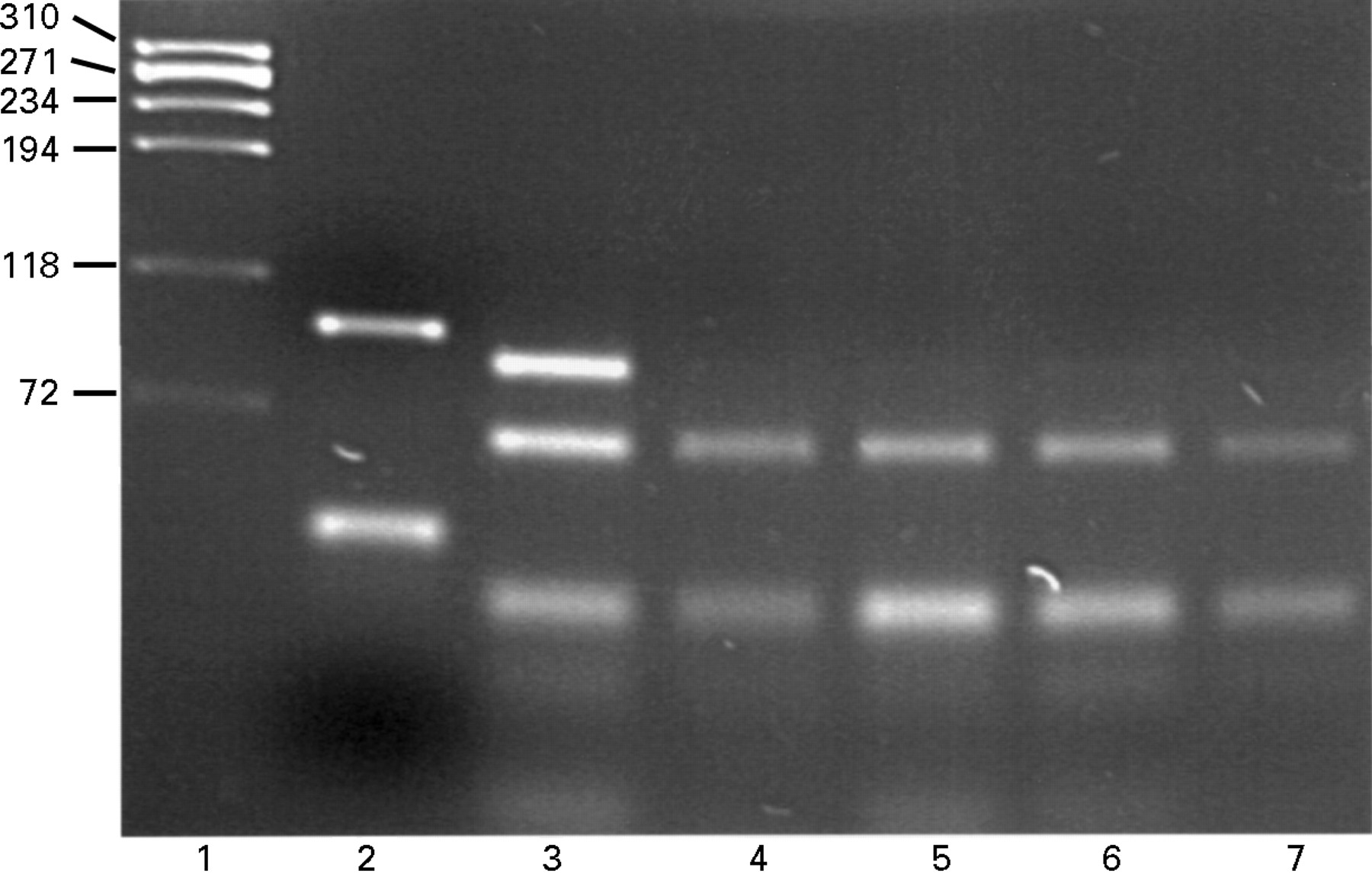{kind=link}
3% w/v agarose/ethidium bromide gel showing the results of restriction enzyme digestion with Taq I. Lane 1, DNA size markers (kb); lane 2, undigested product and primer dimers (lower band); lane 3, partial digestion suggesting the presence of one (undigested) Z allele; lanes 4–7, complete digestion indicating that no Z alleles were present. The difference in size between the upper bands of lanes 2 and 3 is due to a restriction site being present in the 3′ primer which acted as a control of enzyme efficiency.
S α1-AT
The amplification of exon III generated a 149 bp product which was digested by Xmn I to a 111 bp fragment in non-S individuals. The S allele was resistant to digestion producing a fragment of 133 bp that had been shortened at the primer control digestion site (data not shown).
Four patients with cystic fibrosis (5%; three transplanted, one died in childhood) were heterozygotes for the S allele of α1-AT and two (2.5%; one died in childhood, one transplanted) were heterozygotes for the Z allele. No Z or S α1-AT homozygotes were detected. The expected normal population frequency of the S and Z allele are 8% and 4%, respectively; 95% confidence intervals based on these figures are 1 to 13% and 0 to 9% for the S and Z allele, respectively, indicating that there was no significant increase in the S and Z allele frequencies found in our severely affected group of cystic fibrosis patients.
Discussion
The underlying mechanism by which a defect in the CFTR gene predisposes to the pulmonary disease in cystic fibrosis is not known, although a marked proteinase-antiproteinase imbalance in favour of neutrophil elastase within the lungs of patients with cystic fibrosis has been well documented.1 2 Proteinases have been shown to have many potentially deleterious characteristics such as the destruction of lung elastin,12 reduction of ciliary beat frequency,13 the stimulation of mucus pro duction,14 and the cleavage of immuno globulins15 and fibronectin.16 The association of severe α1-AT deficiency with emphysema4 gave rise to the concept of a proteinase-antiproteinase balance within the lung. This theory proposes that under normal conditions there is an equal balance between proteinases and antiproteinases, but in the presence of severe α1-AT deficiency this is disturbed in favour of the proteinases resulting in lung injury. Although mild to moderate deficiency of α1-AT is not associated with pulmonary disease in normal individuals, one might expect that such a deficiency would exacerbate lung damage in individuals with cystic fibrosis who have high levels of neutrophil elastase in their lungs. However, the common MS, S, and MZ α1-AT deficiency phenotypes, which result in mild to moderate deficiency of the protein, are not associated with worse lung disease10 but, paradoxically, predict those patients with cystic fibrosis with significantly better lung function.9 It is possible that these studies were biased by exclusion of the most severely affected patients who had died or had been transplanted. However, we show here that patients with mild to moderate deficiency of α1-AT do not have such severe lung disease that leads to early death or transplantation and that the results of these previous studies were unprejudiced by this factor.
It is unclear why mild to moderate deficiency phenotypes of α1-AT should be protective against cystic fibrosis lung disease but it is possible that they may be linked with other protective mutations. Indeed, a heterozygote advantage protecting against pulmonary tuberculosis has previously been proposed to account for the high incidence of the S and Z α1-AT alleles in the European population.17 A second possibility is that the effect of the mutation is through perturbation of the proteinase-antiproteinase balance within the lung. Recently it has been suggested that neutrophil elastase and other proteinases are not only destructive, but may also have beneficial effects in that they are important in bacterial killing18 and can downregulate inflammation19 by inhibiting neutrophil activation by cleavage of immunoglobulins, immune complexes, neutrophil receptors, and complement20 and by inducing apoptosis.21In the presence of α1-AT deficiency one could speculate that the regulatory effects of neutrophil elastase would occur earlier in the inflammatory process, thereby limiting its own damaging effect. Thirdly, it is possible that the role of neutrophil elastase within such a complex dynamic process as cystic fibrosis lung disease has been overemphasised and that other factors are of equal or greater importance, making mild to moderate deficiency of α1-AT unimportant. However, this would not explain the better lung function in our group of patients.
Larger epidemiological studies are required to confirm our results and further clinical studies may help to explain these findings, including a detailed assessment of the proteinase-antiproteinase balance within the lung of cystic fibrosis patients with normal and variant α1-AT alleles. This is an important area of research in view of ongoing studies designed to test the efficacy of nebulised α1-AT in the treatment of cystic fibrosis.
Acknowledgments
We are grateful to Dr Anthony Heeley, Department of Clinical Biochemistry, Peterborough Hospital for providing blood spots, the UK Transplant Support Service Authority and the UK Cystic Fibrosis Survey for allowing the identification of patients with cystic fibrosis. This work was supported by the East Anglian Locally Organised Research Scheme, the Cystic Fibrosis Trust (UK), and Papworth NHS Trust. RM is an Anglia and Oxford Research Fellow.