Article Text

Abstract
Background Deadenylation regulates RNA function and fate. Poly(A)-specific ribonuclease (PARN) is a deadenylase that processes mRNAs and non-coding RNA. Little is known about the biological significance of germline mutations in PARN.
Methods We identified mutations in PARN in patients with haematological and neurological manifestations. Genomic, biochemical and knockdown experiments in human marrow cells and in zebrafish have been performed to clarify the role of PARN in the human disease.
Results We identified large monoallelic deletions in PARN in four patients with developmental delay or mental illness. One patient in particular had a severe neurological phenotype, central hypomyelination and bone marrow failure. This patient had an additional missense mutation on the non-deleted allele and severely reduced PARN protein and deadenylation activity. Cells from this patient had impaired oligoadenylation of specific H/ACA box small nucleolar RNAs. Importantly, PARN-deficient patient cells manifested short telomeres and an aberrant ribosome profile similar to those described in some variants of dyskeratosis congenita. Knocking down PARN in human marrow cells and zebrafish impaired haematopoiesis, providing further evidence for a causal link with the human disease.
Conclusions Large monoallelic mutations of PARN can cause developmental/mental illness. Biallelic PARN mutations cause severe bone marrow failure and central hypomyelination.
- Genetics
- Haematology (incl Blood transfusion)
- Copy-number
- Molecular genetics
- Neurology
Statistics from Altmetric.com
Introduction
Development of tissues during embryogenesis and their homeostasis after formation are highly regulated, and are delicately orchestrated by the expression of coding and non-coding RNAs at specific times and levels.1 Deadenylation is a major mechanism that regulates RNA function and fate by controlling the turnover and abundance of mRNAs and maturation of non-coding RNAs (eg, small nucleolar RNAs (snoRNAs)).2 ,3 Several deadenylases have been identified.4–7 Poly(A)-specific ribonuclease (PARN) is one of the major mammalian deadenylases2 that trims single-stranded poly(A) tails of mRNAs4 ,6 ,8–10 and oligoadenylated tails of H/ACA box snoRNAs3 and microRNAs.11 It plays a role in a variety of cellular processes,12 including stress response, cell migration and adhesion,13 as well as in Xenopus oocyte maturation and early development.6
To our knowledge, animal models of PARN loss have not been described. Two distantly related genes, parn-1 and parn-2 in Caenorhabditis elegans, have been studied.14 ,15 However, as discussed previously,2 parn-1 lacks the RRM motif, which provides the unique cap binding property of PARN, and the R3H domain in the nuclease domain of human PARN. parn-2 is actually the homologue of human TOE1.16
RNA biogenesis has emerged as a mechanism underlying several inherited diseases. These include defects in rRNA maturation as seen in Diamond blackfan anaemia, Shwachman–Diamond syndrome and dyskeratosis congenita,17–20 as well as defects in snRNA maturation,21 snoRNA transcription22 and mRNA maturation.23 Interestingly, dysregulation of mRNA adenylation in mice24 and in zebrafish25 impaired haematopoiesis. Complex sequential interplay between multiple genes is required to orchestrate haematopoiesis. Hence, it is possible that gene expression regulation by deadenylation/polyadenylation is important for promoting haematopoietic cell expansion and differentiation and preventing bone marrow failure.
Inherited bone marrow failure syndromes (IBMFSs) are rare, genetically heterogeneous disorders that manifest low blood counts due to impaired haematopoiesis and a high risk of cancer.26 They include disorders such as Fanconi anaemia and dyskeratosis congenita. IBMFSs frequently feature varying degrees of developmental delay, mental illnesses (eg, behavioural disorders and autism) and physical malformations. The genes mutated in IBMFSs function in fundamental processes such as DNA repair, telomere maintenance and ribosome biogenesis. Many patients do not have mutations in known IBMFS genes, and a large number of cases cannot be classified into known syndromes27 despite having typical features of an IBMFS (ie, have chronic bone marrow failure in addition to either physical malformations or family history of a similar inherited disorder or presentation in the first year of life).28
In the present work, we identified four patients with developmental delay or mental illness from three different families, who carried large monoallelic deletions in PARN. Importantly, one of the patients had severe bone marrow failure and neurological manifestations; this patient had biallelic disruption of PARN. We describe the results of genetic interrogation of these patients, the consequences of biallelic PARN mutations on RNA metabolic pathways, and demonstrate conserved function of PARN in haematopoiesis using the zebrafish. Taken together, these investigations establish a new IBMFS disease paradigm.
Methods
Genome-wide analysis of genetic alterations
Biological studies on patients and control subjects have been performed after written consents were obtained. Preparation of DNA, hybridisation to the Affymetrix Genome-Wide Human SNP 6.0 Array (Affymetrix, Santa Clara, California, USA) and analysis were performed as described.29 Minor allele frequency was referred from healthy controls from Database of Genomic Variants at The Center for Applied Genomics, Toronto (http://dgv.tcag.ca/dgv/app/home).
Comprehensive screening for mutations in IBMFS genes
Next-generation sequencing of a panel of 72 known IBMFS genes (see online supplementary table S1) is described in the online supplementary methods.
DNA quantitative real-time PCR
DNA quantitative real-time PCR (DNA-qPCR) was performed using Power SYBR Green (Applied Biosystems, Warrington, UK) as described,30 with primers designed within the PARN deletion regions, and FOXP2 as a reference gene (see online supplementary table S2).
Sanger sequencing
PCR amplifications and sequencing of genomic DNA and cDNA were performed as previously described.28 Amplification and sequencing primers are given in online supplementary tables S3 and S4.
Western blotting
Cell lysates were analysed as described previously.31 Polyclonal antibody against PARN dilution 1:1000 (Cell Signaling Technology, catalogue number 3894), custom-made antiserum32 and a monoclonal antibody against β actin (1:10 000) (Sigma-Aldrich, St Louis, Missouri, USA) were used.
PARN activity assays
Recombinant human PARN or mutants thereof were generated, expressed in Escherichia coli and purified as detailed in online supplementary methods and supplementary table S5. PARN activity assays33 were performed as detailed in online supplementary methods.
RNA-qPCR and poly(A) tail length assay
RNA-qPCR assays and poly(A) tail length measurements were performed with the primers listed in online supplementary table S6 and as detailed in online supplementary methods.
To measure poly(A) tail length, total RNA underwent reverse transcription using random hexamer and/or oligo dT-adapter primers (see online supplementary table S6) with or without RNase treatment. Subsequent poly(A) length assays were according to Murray and Schoenberg34 as detailed in online supplementary methods.
Cloning and sequencing of snoRNAs and scaRNAs
Adapter oligonucleotide was ligation to total RNA. Ligated RNA underwent cDNA synthesis using reverse primers (see online supplementary tables S6 and S5). SNORA63, SCARNA8 and TERC cDNAs were amplified with gene-specific primers. PCR products were subsequently cloned and sequenced (see online supplementary methods).
Ribosome profile analysis by sucrose gradient density ultracentrifugation
Skin fibroblast cells from patient and healthy controls were cultured and processed for ribosome profiling as we described previously.31
Characterisation of telomere length
Ficoll separated peripheral blood cells were analysed for telomere length by flow FISH in Repeat Diagnostic Laboratory (Vancouver, Canada) as previously described.35 The normal reference values of the laboratory are based on data from 391 healthy control persons ranging from birth to 100 years of age.
Lentivirus construction and cell culture
pGIPZ lentivector containing shRNA sequences (see online supplementary table S6) were obtained from the Signaling Identification Network facility at the Hospital for Sick Children, Toronto. Virus particles were generated after transfection of HEK293 cells as described.31
Marrow CD34 cells were separated by immunomagnetic beads as described,31 and were transduced with lentivectors, sorted and plated in methylcellulose cultures as described.36
The skin fibroblasts used herein had normal karyotype and were mycoplasma free. To evaluate cell growth, skin fibroblasts were seeded at 1.0×104 cells per 0.1 mL in 96-well plates. At various time points after plating, viable cell counts were determined using trypan blue dye exclusion using an automated cellometer.
Flow cytometry
Skin fibroblasts were serum starved for enrichment of cells at G0/G1. Exponentially growing cells were trypsinised, washed, replated at 30–40% confluency (105 cells/mL) in 5 mL Roswell Park Memorial Institute medium+0.5% fetal bovine serum and incubated 24–48 h at 37°C. Cell cycle analysis was done as described.36
Knockdown of parn in zebrafish
All zebrafish experiments were approved by the Dalhousie University Committee on Laboratory Animals, Protocol 13-007. Zebrafish husbandry is described in the online supplementary methods.
For gene knockdown, two morpholinos were used: translation-blocking morpholino (tparn) that targets the translation start-site, and splice-site blocking morpholino (sparn) that inhibits the parn exon 5/intron 5 splice junction. Details are given in online supplementary methods and supplementary table S7. The methods for injecting morpholinos and controls at the one-cell stage and confirmation of knockdown were as previously described37 (see online supplementary methods and supplementary table S7).
Dechorionated embryos were staged at 48 h post-fertilisation and fixed with 4% paraformaldehyde or stained with o-dianisidine peroxidise substrate (Sigma-Aldrich). Whole-mount in situ hybridisation assays for mpx and pu.1 were performed as published37 at 48 h post-fertilisation and is further explained in the online supplementary methods. Analyses of morphant and control embryos following whole-mount in situ hybridisation were blinded. Approximately 20–30 embryos were selected per replicate and three replicates were completed to ensure sufficient statistical power. Live embryos with normal gross development that reached the indicated predetermined time point post-fertilisation were used in experiments.
Rescue experiments were done by co-injecting mock vector or human PARN RNA (or mock) together with standard morpholino or tparn/sparn morpholino, respectively. Further details are given in the online supplementary methods.
Statistics
Student's t test was used to study statistical differences between groups. Data are presented as means±SE of the mean or mean±SD. Differences were considered statistically significant at p<0.05.
Results
Clinical phenotype and genomic characterisation
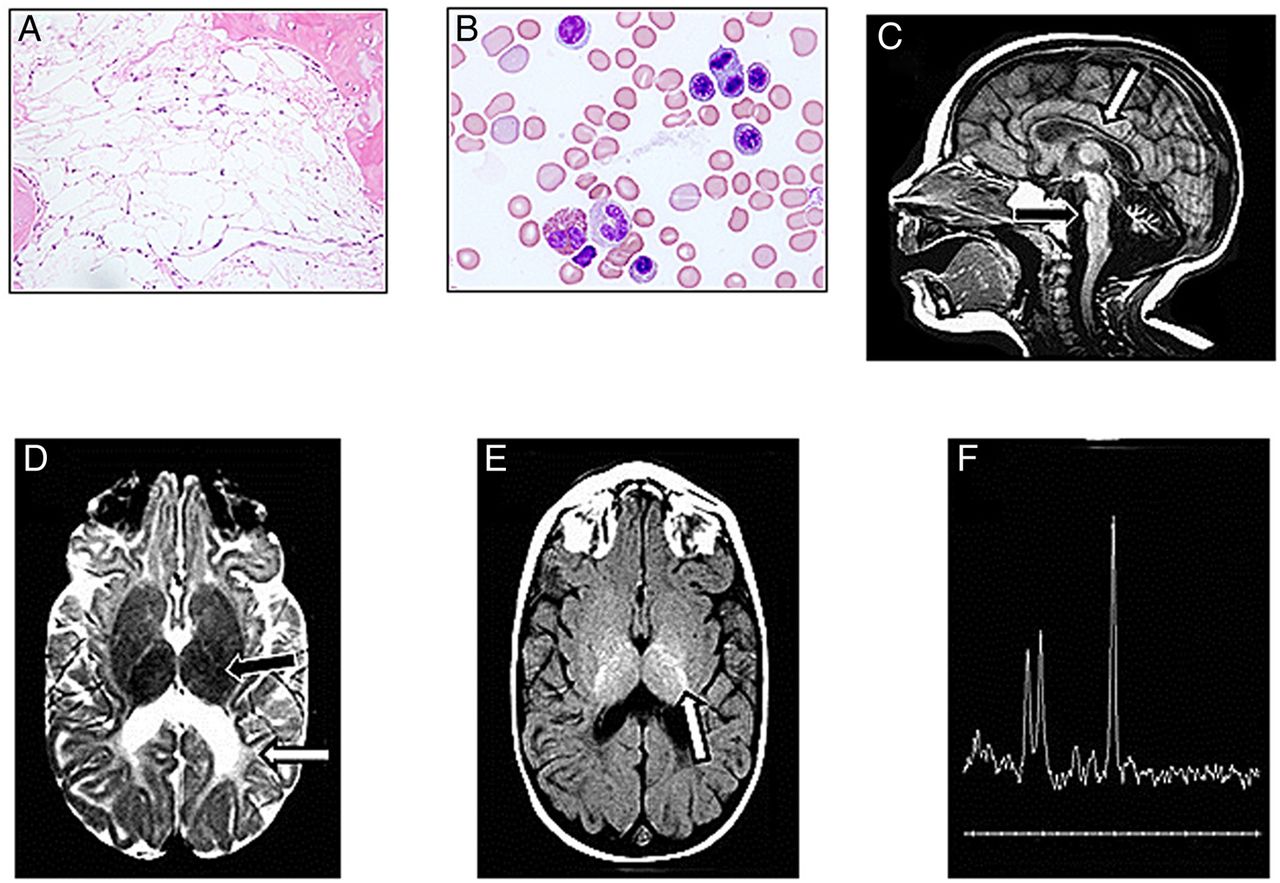
We identified four patients with deletions confined to the PARN gene (table 1). Patient 1 was identified as part of an effort to unravel causal alleles in cases with IBMFSs from our Canadian Inherited Marrow Failure Registry. This female patient had severe bone marrow failure and severe neurological defects. Bone marrow biopsy and aspirates showed cellularity of <5% (normal for this age is >70%) (figure 1A, B). MRI of the head showed very small volumes of all parts of the brain (figure 1C) and severe hypomyelination (figure 1D, E). MRI at 12 years of age showed no significant changes (see online supplementary figure S1A–C), suggesting no degenerative process. The metabolic spectra of the brain showed normal patterns (figure 1F), suggesting no metabolic or oxidative aetiology. Extensive investigation (table 1) did not reveal metabolic and infectious causes, and screening for mutations in 72 known IBMFS genes by next-generation sequencing (see online supplementary table S1) did not reveal a causal genotype.
Summary of the clinical manifestations of patients with deletions in the PARN gene

The poly(A)-specific ribonuclease (PARN)-associated inherited bone marrow failure syndrome (patient 1) features severe bone marrow failure and severe hypomyelination. (A) Bone marrow biopsy at the age of 6 years shows haematopoietic cells that comprise <5% of the bone marrow spaces and almost complete replacement with fatty tissue (normal cellularity for children younger than 10 years is >60%). (B) Bone marrow aspirate at the age of 6 years shows morphologically normal and non-dysplastic precursors. (C) MRI of the patient brain at the age of 12 months. T1W sagittal image demonstrates a thin brainstem (black arrow), thin corpus callosum (white arrow) and a small cerebellum. (D) Axial T2W image demonstrates delayed myelin maturation of the posterior limb of the internal capsule (black arrow). There is decreased volume and abnormal signal of the periventricular white matter (white arrow). (E) Axial T1W image reveals myelin maturation in the posterior limb of the internal capsule (white arrow), appropriate for an infant 1 month of age. (F) Magnetic resonance spectroscopy at 12 years of age using echo time (TE) 144 shows normal metabolic spectra from grey and white areas.
Using Affymetrix SNP 6.0 array, we identified a rare alteration that deleted ∼22 kb of chromosome 16p13.12, which was not seen in any of the 2800 healthy controls (figure 2A and online supplementary figure S2A). This monoallelic deletion started at a region with low probe density between introns 13 and 14 and ended at intron 18. The deletion was validated by DNA-qPCR (figure 2B). cDNA amplification using primers that flank the deletion area (nucleotide 919–1531) revealed two fragments (a wild type, 613 bp, and a smaller mutant band) in the patient, but only the wild-type fragment in controls (figure 2C). The size of the mutant (closer to 269 bp than to 313 bp) indicates that the alteration deletes exons 14–18. This was confirmed by Sanger sequencing of the mutant cDNA fragment that indicated c.919–1262del344 (figure 2C). The deletion causes a reading frame shift and early truncation of the protein p.Asp307ValfsTer22. The mutant polypeptide lacks essential catalytic parts of the nuclease domain38 and the RNA recognition motif (figure 2D).
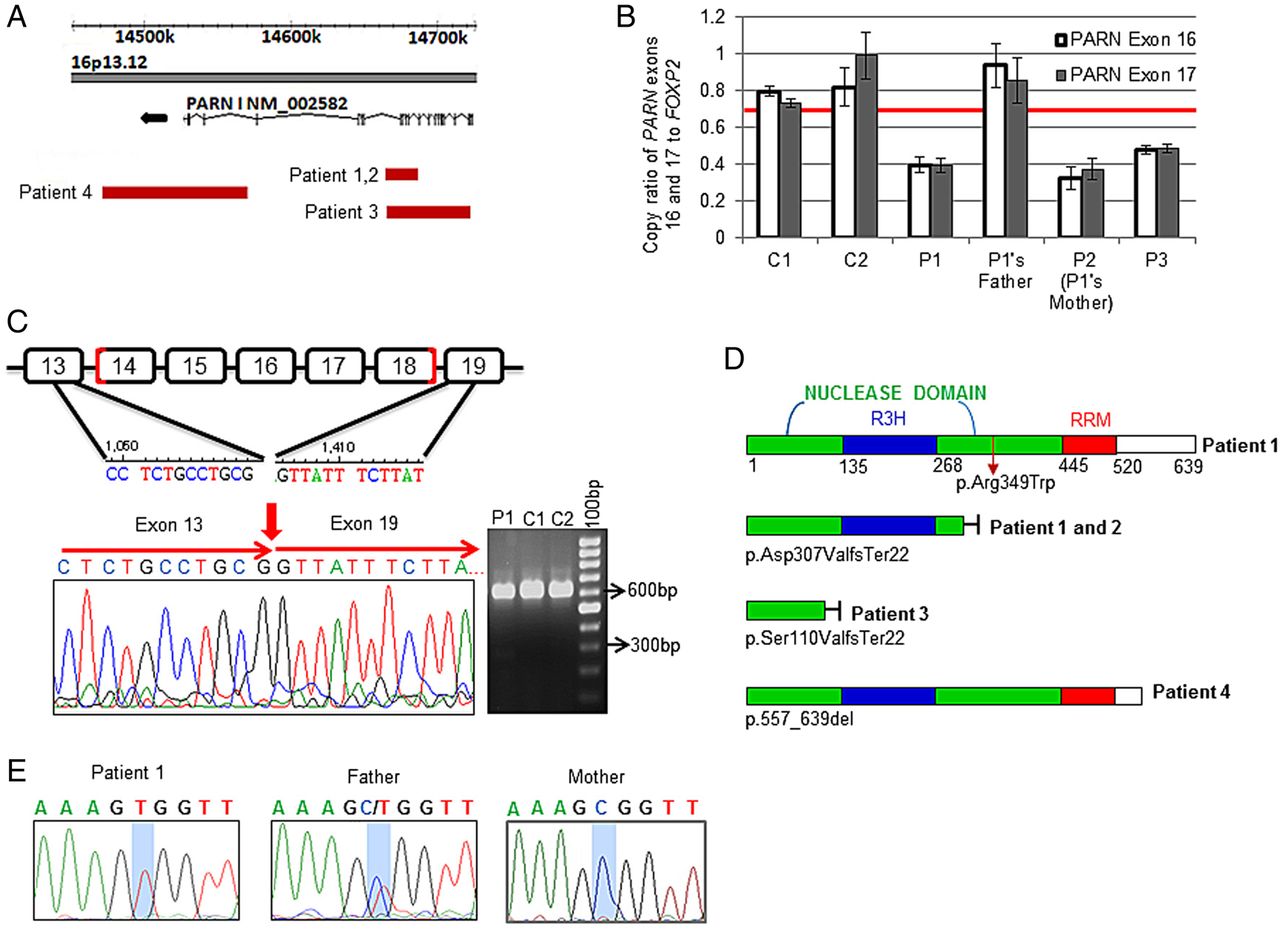
Genomic analysis of four patients with neurological disorders. (A) Patient 1 was tested by Affymetrix SNP 6.0 array. One copy deletion was found that started at a region with a low number of probes between introns 13 and 14 of PARN and ends at intron 18 (NM_002582.3, chr16:14658272–14679880). The deletion was inherited from the mother (patient 2). Analysis of patient 3's peripheral blood DNA by Agilent 180K oligonucleotide array showed a deletion of about 56 kb on chr16:14659344–14715111 that resulted in one copy loss between introns 5 and 18. Analysis of patient 4's peripheral blood DNA by 44K array-comparative genomic hybridisation revealed a de novo deletion of about 100 kb from intron 22 to downstream of exon 24 (chr16:14464480–14564129). (B) DNA qPCR of PARN compared with FOXP2 as an internal control was done using two sets of primers in the deletion area that include exon 16 or exon 17, respectively. The red line shows threshold for one copy number call. Average values from three replicates are shown. (C) PCR of a cDNA fragment between nucleotides 919 and 1531 shows the wild type (WT, 613 bp) and mutant (257 bp) PARN in patient 1. (D) Schematic drawing of the full-length PARN polypeptide in patient 1 with the missense mutation (upper part) and the truncated mutant in patients 1 and 2 (lower part). Patient 3 and 4 truncated mutant proteins were predicted based on the DNA deletion region. (E) Sanger sequencing of genomic DNA showed hemizygous, heterozygous and WT status for the c.1045C>T missense mutation in patient 1, father and mother, respectively.
Patient 1 was a single child of non-consanguineous parents of Greek and German descent. The mother (patient 2) had mental illness requiring hospitalisation. She carried the same deletion as her daughter (figure 2A). The father was reported as healthy and did not carry the deletion. The parents were not reported to have any blood disorders.
Two additional patients with deletions restricted to PARN were identified through collaboration with the Cytogenetics Laboratory, The Hospital for Sick Children, Toronto, and by searching the DECIPHER database (decipher.sanger.ac.uk). Patient 3 had global developmental delay, but less severe than patient 1 (table 1). She had no apparent haematological problems by the age of 5 years. Genomic microarray analysis revealed a de novo deletion of about 56 kb that resulted in one copy loss between intron 5 and the end of intron 18 of PARN (figure 2A and online supplementary figure S2B). The patient was enrolled on the Canadian Inherited Marrow Failure Registry and the deletion was validated by DNA-qPCR (figure 2B).
Patient 4 was enrolled on the DECIPHER study in Italy. The child had developmental delay (table 1) but normal blood counts. Genomic analysis revealed a deletion of about 100 kb, which resulted in one copy loss that begins at intron 22 of PARN and ends downstream of the gene (exon 24) (figure 2A and online supplementary figure S2C). The deletion was de novo and did not appear in the parents. The deletion was validated by a second independent array-comparative genomic hybridisation (data not shown). The patient had an additional small deletion on 4p32.1, which was inherited from his healthy father. This deletion disrupts the MAP9 gene. However, deletions in this gene have been previously found in healthy individuals.
The clinical phenotype of severe aplastic anaemia and marked hypomyelination was associated with biallelic mutations in PARN
The data above demonstrate four patients with deletions in the PARN gene and associated developmental delay or mental illness. Patient 1 had much more severe neurological disease than the other three patients, as well as bone marrow failure. To determine whether the identified phenotypes of the patients varied because of a different allelic dose, we sequenced the patient's genomic DNA. Indeed, patient 1 had a point mutation in exon 16 of the non-deleted allele, c.1045C>T (figure 2E), which leads to an amino acid change, p.R349W, in the nuclease domain (figure 3A). Analysis of next-generation sequencing data on >2000 healthy individuals and Sanger sequencing of DNA from 87 healthy individuals did not reveal individuals with this mutation. Recently, the p.R349W mutation was reported in a heterozygous state in one subject in Exome Aggregation Consortium (http://exac.broadinstitute.org) at an allele frequency of 8.32×10−6. Importantly, patients with one copy loss of PARN had a milder phenotype than patient 1 (ie, patients 2–4) and did not carry a point mutation on the second allele. The father of patient 1 was heterozygous for the point mutation (figure 2E). We screened 94 patients with IBMFSs, who had been enrolled in our registry for mutations in PARN. None of these patients was found to have mutations. Of the 94 patients, 31 had neurological phenotype in addition to bone marrow failure.

Consequences of biallelic poly(A)-specific ribonuclease (PARN) mutations on the encoded protein. (A) The location of arginine at position 349 in the nuclease domain (green) relative to RRM (red) in dimeric PARN are shown. (B) PARN protein expression in T cells from patient 1, her parents and healthy control subjects was analysed by western blotting using a commercial antibody against the N-terminal domain of the protein (Cell Signaling Technology). Forty micrograms of protein was loaded. (C) The disease-associated recombinant mutant, R349W, impairs the degradation of oligo(A20). 10 nM of oligo(A20) was incubated under conditions for in vitro deadenylation at 30°C for 10 min, in the absence or presence of mutant PARN (R349W) or wild-type PARN polypeptides as indicated. The concentrations of the mutant R349W PARN (range 4–256 nM) and the wild-type PARN (range 2–64 nM) that were used in the reactions are indicated. The reacted 5′-end-labelled RNA was fractionated by electrophoresis in 25% polyacrylamide gels (polyacrylamide/bisacrylamide 19:1). 5′-end-labelled marker oligonucleotides were separated in lane M and the positions and sizes of the fractionated markers are indicated. The positions of deadenylated reaction intermediates (A4 and A2) and product (A1) are as indicated.
Biallelic mutations in PARN resulted in severely reduced protein level
The expression of wild-type PARN mRNA in patient 1 was measured by qPCR (see online supplementary table S10) and by RNA sequencing, and was found to be twofold to threefold lower than normal controls. No reads for the mRNA allele with the deletion was detected, suggesting nonsense-mediated decay (complete transcriptome analysis of patient 1 will be published elsewhere).
The p.R349W mutation was predicted to damage the protein by SIFT and Polyphen software programs with high specificity scores of 0.915 and 0.999, respectively, at >95% confidence. Using a protein structure prediction software (http://molbiol-tools.ca/Protein_secondary_structure.htm), the mutation was predicted to change the α-helix motif that includes R349 to a β-sheet (figure 3A and online supplementary figure S3). Importantly, using both a commercial antibody against the N-terminus and antiserum that we had raised in rabbit against the whole protein,32 we found severely reduced protein expression in patient cells (figure 3B), consistent with autosomal-recessive inheritance. A small level of protein was detected only when a high quantity of protein was assayed (see online supplementary figure S3B). PARN protein level in the mother, who carried the deletion, was about 40% of that of the control's average; the protein level in the father, who carried the missense mutation, was about 62% of that of the control's average. Samples for protein analysis from patients 3 and 4 were not available.
The R349W mutation disrupts the deadenylation activity of PARN
In addition to the effect of the mutations on protein levels, the location of the R349W mutation in the nuclease domain raises the hypothesis that it also affects the catalytic properties of the enzyme. To test this hypothesis, we generated recombinant wild-type and R349W-mutated polypeptides. We incubated the polypeptides with oligo(A20) substrates under conditions for in vitro deadenylation. The mutant polypeptide had markedly reduced deadenylation activity (figure 3C). The mutant's kinetic activity was approximately 50-fold reduced compared with wild-type PARN (see online supplementary table S9). The deficiency was primarily caused by a decrease in Vmax and only partially by a change of Km. Importantly, using a standard procedure for generating control point mutations by introducing alanine at position 349 of the PARN polypeptide (R349A) did not reveal any major deficiency in its overall efficiency. This suggests a specific damaging effect by tryptophan at this residue.
PARN-deficient patient cells manifest PARN-associated defects in snoRNA and scaRNA trimming and aberrant ribosome profile
PARN trims oligoadenylated tails of H/ACA box snoRNAs and scaRNAs.3 Processed H/ACA box snoRNAs are critical for rRNA maturation by pseudouridylation of nascent rRNA before its cleavage.39 H/ACA box scaRNAs are localised in Cajal bodies and are involved in snoRNA and snRNA biogenesis.40 To ask whether PARN-deficient patient cells manifest defects in snoRNA/scaRNA processing similar to those described in PARN-knockdown cells,3 we analysed the oligoadenylated status of H/ACA and CD box snoRNAs/scaRNAs studied by Berndt and colleagues,3 in skin fibroblasts from patient 1 by q-PCR. In keeping with the results of that study, we found that several H/ACA box snoRNAs/scaRNAs were markedly enriched in their oligoadenylated status compared with control cells, while their general abundance was only slightly affected (figure 4A, online supplementary figures S4A, B and S5A, B, and supplementary table S10). For example, we found that the relative oligoadenylated level of SNORA63 and SCARNA8 increased by >200-fold and 30–100-fold, respectively.

Oligoadenylation status of small nucleolar RNAs (snoRNAs) and scaRNAs in poly(A)-specific ribonuclease (PARN)-deficient patient cells. (A) Patient 1 and control skin fibroblast RNA was analysed by qPCR. Results are representative of two independent experiments in triplicates. Random hexamer (RH/grey bars) or oligo(dT)-primed (dT/black bars) cDNAs were used together with specific primers. RNA levels (mean±SD) were cross-normalised to GAPDH and are plotted as results in patient versus control cells. Online supplementary figures S4, S5 and supplementary table S10 depict the complete set of quantified data and comparison to two additional controls. PCR amplification of oligoadenylated SNORA63 (B), SCARNA8 (C) and TERC (E) show a clear ‘oligo(dT)adaptor’ primed PCR product in the patient (lane 4 in each panel), which is absent in normal fibroblasts (lanes 2 and 3 in each panel) and disappears if the RNA is treated with oligodT/RNaseH (lane 7 in each panel). SNORA63, SCARNA8 and TERC are present in all samples investigated, if primers specific for the ‘coding’ part are used (lanes 8–10 in each panel). (D) Skin fibroblasts were lysed with buffer containing cycloheximide. Equal amounts of RNA were layered on 5–45% sucrose gradients. Absorbance of each fraction at A254 was recorded by the Brandel Density Gradient Fractionator. Ribosomal 40S, 60S, 80S peaks are indicated. As characteristic to skin fibroblasts, polysomes are scarce. A representative diagram of three independent experiments is shown. (F) Telomere length in peripheral blood lymphocytes was measured by flow FISH (Repeat Diagnostic Laboratory). The dots represent subject results. The lines represent percentiles among 391 healthy control persons aged 0–100 years. *Non-oligoadenylated snoRNAs in patient 1 (P1); **oligoadenylated snoRNAs in patient 1 (P1); ***oligoadenylated TERC in normal controls (N1 and N2).
Next, we measured the poly(A) tail length34 of SNORA63 and SCARNA8. In keeping with the qPCR results, we detected oligoadenylated species in patient 1's fibroblast RNA that were not abundant in control cells (figure 4B, C, lanes 2–4) or in RNase H control (figure 4B, C, lanes 5–7 compared with 2–4). The oligoadenylated species of SNORA63 and SCARNA8 contained up to ∼40 and ∼20 additional nucleotide residues, respectively, compared with the corresponding non-adenylated species. Sequencing of the cDNA clones of SNORA63 and SCARNA8 revealed large fractions of oligoadenylated 3′-ends in the patient cells (see online supplementary figure S6). The oligoadenylated tails of SNORA63 and SCARNA8 were frequently added to a nucleotide located just downstream of the mature 3′-end. In some cases, the position of the oligoadenylated tail overlapped with an adenosine residues in the genomic sequence, making it impossible to unambiguously determine the site of oligoadenylation. The analysis of SCARNA8 revealed 3′-end truncated versions of the molecule in both normal and patient cells, which may represent yet unknown 3′-end variants of SCARNA8 or degradation/turnover intermediates.
Taken together, PARN-deficient patient cells manifested PARN-associated deficiencies in snoRNA/scaRNA trimming. Given that snoRNAs41 and scaRNAs42 are essential for rRNA maturation and since rRNA maturation defects underlie several IBMFSs,17 ,18 ,20 we hypothesised that ribosome assembly might be altered in the PARN-associated disease. Characteristic defects in patients with ribosomopathies include reduced levels of one or more of the ribosome subunits 40S,43 60S44 or the assembled 80S28 ,45 ribosome. Patient 1's fibroblasts manifested an abnormal ribosome profile; in particular, there was marked reduction of the mature 80S ribosome subunit (figure 4D).
PARN-deficient cells exhibit deficiencies in telomerase RNA component (TERC) trimming
The H/ACA box is also present at the 3′-end of TERC/SCARNA19 and is essential for its stability and assembly into the telomerase pre-ribonucleoprotein complex.46 Since TERC mutations have been shown to cause bone marrow failure, we asked whether deficiency of PARN affects TERC abundance, TERC oligoadenylated state and telomere maintenance. The abundance of oligoadenylated TERC in PARN-deficient patient cells was significantly increased although its total levels were slightly reduced (figure 4A, online supplementary figure S4A, B, and supplementary table S10). Poly(A) tail studies (figure 4E) showed that TERC oligoadenylated tails included up to 30 added adenosine residues. Moreover, cloning and sequencing of TERC showed a large repertoire of oligoadenylated species in patient 1's cells compared with controls. Remarkably, the oligoadenylated pattern of TERC in patient cells was almost identical to what was observed for SNORA63 and SCARNA8. This suggests that biogenesis of TERC is dependent on proper PARN activity.
Next, we asked whether the increased level of oligoadenylated or polyadenylated TERC is associated with defects in telomeres as seen in patients with mutations in telomere pathway genes.47 Patient 1's peripheral blood lymphocyte telomeres were very short and much lower than the first percentile for age-matched healthy controls (figure 4F). Similar results were obtained when the various T, B and natural killer lymphocyte subsets and granulocytes were analysed (data not shown). Blood cell telomere length of the mother (patient 2) and the father were higher than the first percentile.
Differentiation of haematopoietic stem cells/early progenitors requires PARN
The above results demonstrate defects in snoRNAs/TERC, ribosome profile and telomere length in cells with biallelic PARN mutations. Since defects in ribosome biogenesis and telomere maintenance underlie several IBMFSs, we aimed to consolidate the relationship between PARN and marrow failure by determining whether it is critical for normal haematopoiesis. The PARN-associated disease was characterised by a profound reduction of marrow haematopoietic cells (figure 1A, B). Importantly, purification of marrow CD34+ haematopoietic stem cells/progenitors from patient 1 resulted in only 1.3% positive cells (average percentage obtained from healthy controls in our lab is 2.53%, range 1.43–3.8%). In vitro clonogenic assays of CD34+ cells revealed markedly reduced potential to form colonies; one granulocytic–monocytic colony/3000 plated cells (see online supplementary figure S7A, B) compared with an average of 23 granulocytic–monocytic colonies (range 8–37), 46 erythroid-burst forming units colonies (range 9–83) and 4 mixed colonies (range 1–14) from 9 healthy control subjects.
In order to determine whether PARN plays a role in blood cell formation, we knocked down PARN in marrow CD34+ cells from healthy individuals using a lentivirus expressing shRNA against PARN (figure 5A). PARN-knockdown resulted in smaller colonies (figure 5B) and in marked reduction in haematopoietic colony formation compared with controls (figure 5C). Knocking down PARN by a second shRNA showed similar results (see online supplementary figure S7C).

Poly(A)-specific ribonuclease (PARN) is critical for normal haematopoiesis from human CD34+ haematopoietic stem cells/early progenitors. (A) PARN was knocked down in normal human marrow CD34+ cells using a pGIPZ lentivirus expressing either specific PARN shRNAs or scrambled shRNA control. Knockdown efficiency was confirmed by qPCR. (Low number of viable cells remained after transduction, which did not allow complementation of mRNA measurement by protein measurement.) (B) Clonogenic assays of PARN-knockdown and control CD34+ cells. Representative colonies are shown. (C) Colony production from CD34+ cells expressing either PARN shRNA or scrambled RNA control. The left panel shows results of four separate experiments containing 2–3 replicates of shRNA1 (PARN KD1) and 2–3 replicates of the control. Error bars represent range. Statistical significance was determined with Student's t test (*p<0.01; ** p<0.001). (D) Skin fibroblasts from patient 1 and control subjects were cultured and counted daily by an automatic cellometer. The results represent two independent experiments with four replicates each. (E) 24 h after synchronisation by low serum conditions, cells were cultured for 30 h and analysed by flow cytometry (N=3). The ratio of cells in S/G2/M phases to cells in G1/G0 was analysed (N=3). BFU-E colonies, erythroid burst-forming units; GEMM, mixed colonies producing granulocytes, erythrocytes, megakaryocytes and macrophages; GM, granulocytic–monocytic colonies; total, sum of all colonies; KD, knockdown; STD, standard or mock injection.
To investigate whether the PARN-deficiency phenotype is also characterised by a defect in cell growth, we studied the ability of PARN-deficient skin fibroblasts from patient 1 to expand in vitro. Compared with controls fibroblasts, PARN-deficient cells featured 3.5-fold to 12.5-fold slower growth (figure 5D). Cell cycle analysis of the PARN-deficient skin fibroblasts showed a 7% increase in the number of cells that stayed in G1/G0 and a 7% decrease in the number of cells that progressed to S/G2/M compared with healthy controls (figure 5E). This suggests impairment in transition of cells from the G1 to S phase, similar to the defect reported in PARN-knockdown gastric cancer cells.48 Apoptosis was not statistically different from controls cells (see online supplementary figure S8).
PARN is required for normal haematopoiesis in zebrafish
To strengthen the hypothesis that PARN is critical for normal haematopoiesis and study whether the role of PARN is conserved throughout evolution, we knocked down parn in zebrafish embryos. Zebrafish parn protein shares 64% sequence homology to its human counterpart.
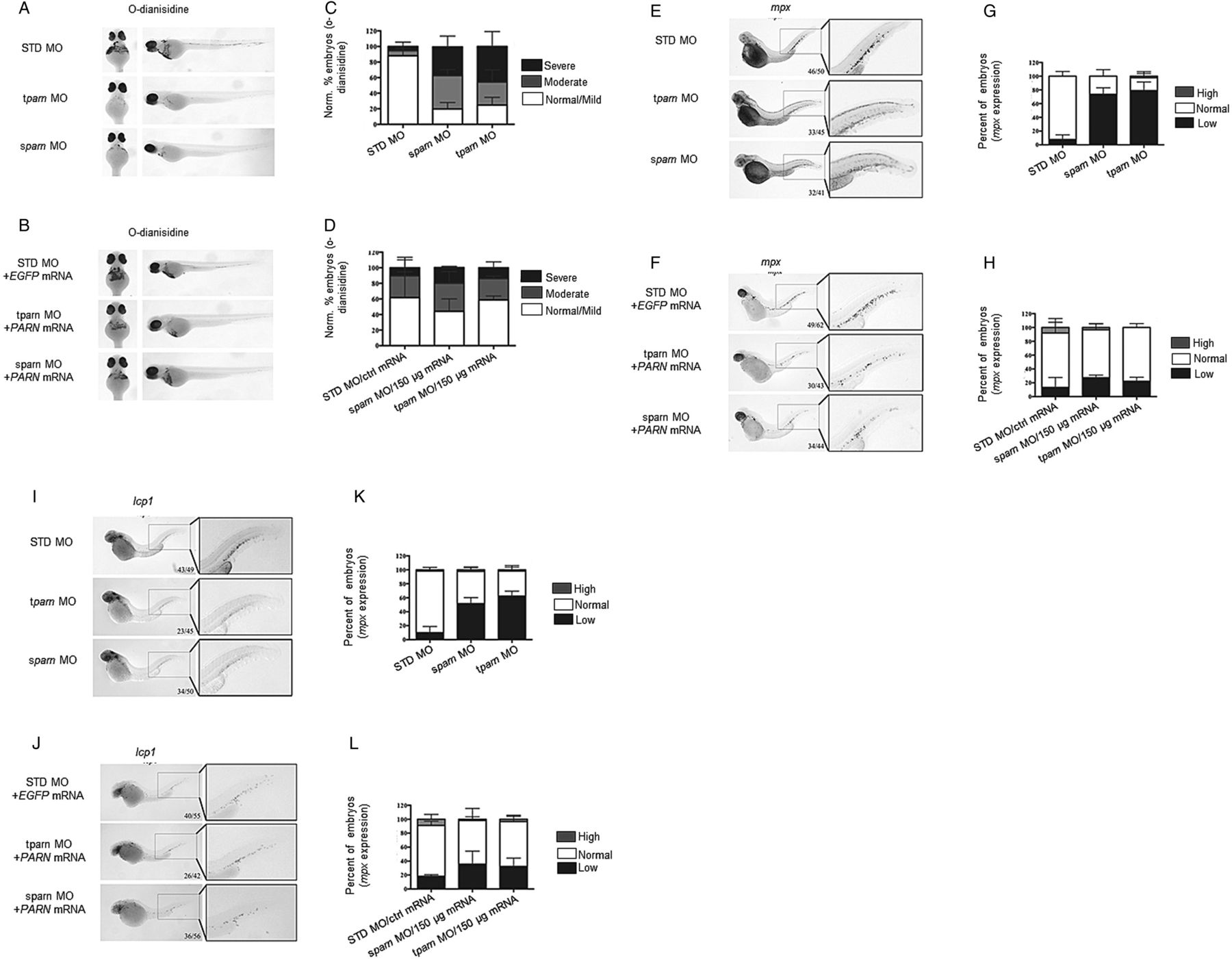
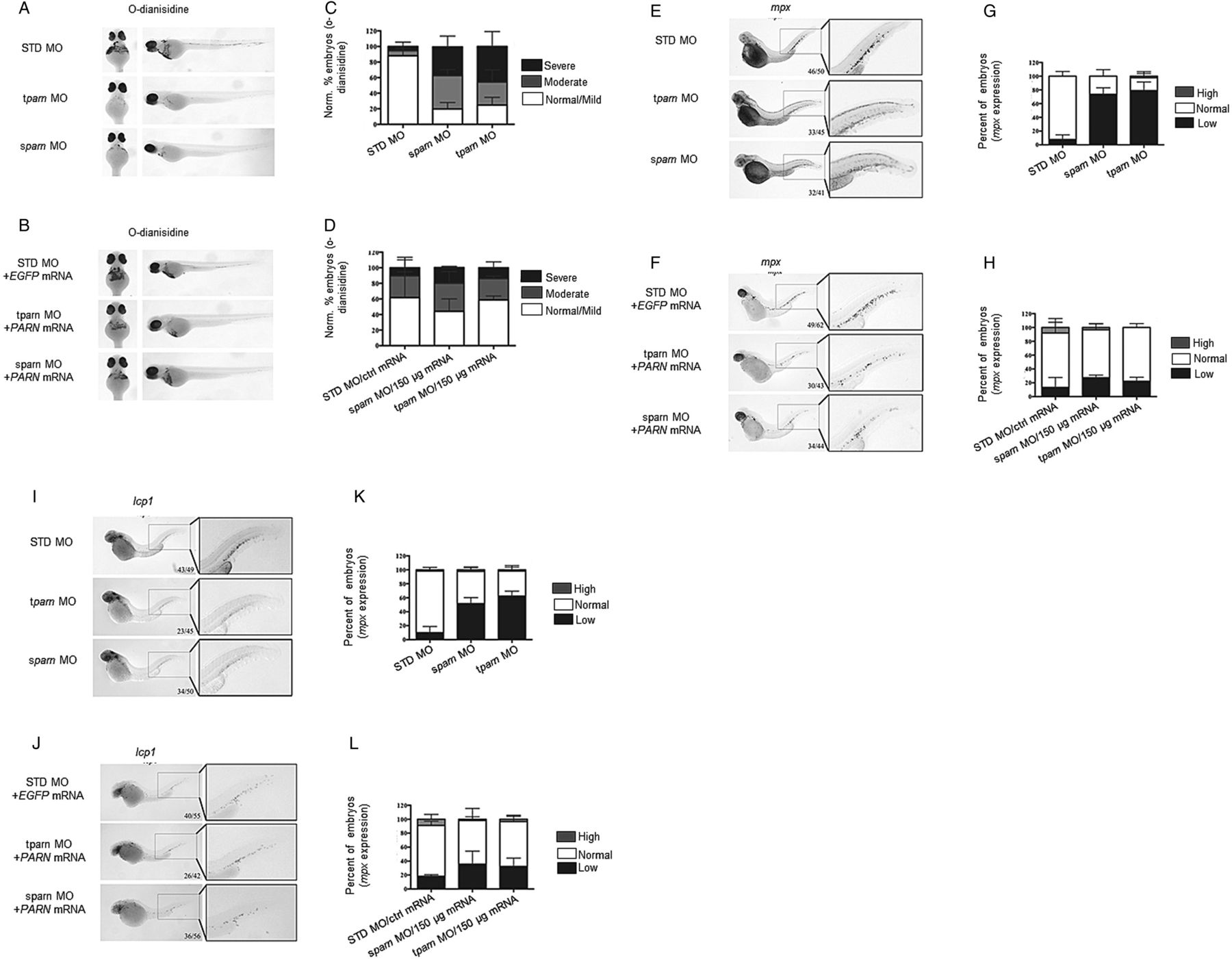
Embryos at the one-cell stage were injected with either translation start-site or splice-site blocking morpholino (tparn morpholino and sparn morpholino, respectively, see online supplementary figure S9A, B). At 48 h post-fertilisation, both morphants were anaemic as demonstrated by reduction in o-dianisidine staining (figure 6A, C) and leukopenic as evidenced by reduced expression of myeloid markers (figure 6E, G, I, K). Importantly, when human PARN mRNA was co-injected with tparn or sparn morpholino (see online supplementary figure S9C), these haematopoietic phenotypes were largely rescued, resulting in a return of normal haemoglobin production (figure 6B, D) and myeloid marker expression (figure 6F, H, J, L, respectively).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Poly(A)-specific ribonuclease (PARN) is critical for normal haematopoiesis in zebrafish. Wild-type (WT) or pigment-lacking casper zebrafish embryos were injected with a standard control (standard or mock injection (STD)) morpholino (MO) (0.625 mM), a splice-site blocking (sparn) or translation-blocking (tparn) antisense MO (0.625 mM) targeting parn at the one-cell stage. Rescue experiments include combined injection of STD MO (0.625 mM) with EGFP mRNA (150 ng/μL), sparn MO with human PARN mRNA or tparn with human PARN mRNA (0.625 mM respective parn MO and 150 ng/μL mRNA). (A–D) O-dianisidine staining for haemoglobin at 48 h post-fertilisation (hpf) in casper embryos. (A and C) Both tparn and sparn MO reduces O-d staining compared with controls. (B and D) Injection of human PARN restores this lack of haemoglobin. (E–L) Whole-mount in situ hybridisation for myeloperoxidase (mpx) and l-plastin (lcp1) at 48 hpf of STD, tparn and sparn morphants. (E–H) Knockdown of parn results in a decrease of mpx expression compared with STD MO controls, which may be rescued by injection of human PARN mRNA. (I–L) Similarly, the loss of parn results in the loss of lcp1 compared with STD MO controls, which can be rescued upon injection with human PARN mRNA.
Discussion
In this study, we report for the first time four patients from three different families with developmental delay or mental illness, who carried large monoallelic PARN deletions that have not been reported in healthy controls. This clearly suggests that large PARN deletions in humans result in a neurological phenotype. Also, we identified a disease phenotype that is characterised by biallelic PARN mutations, severe bone marrow failure, severe hypomyelination, short telomeres and aberrant ribosome profile. This combination of clinical and laboratory abnormalities is consistent with a diagnosis of Hoyeraal-Hreidarsson syndrome, a severe form of dyskeratosis congenita.
Telomere-related disorders result in multiple different phenotypes. In contrast to patient 1 in our study, four patients with biallelic PARN mutations have very recently been reported49 and had other classical features of dyskeratosis congenita such as mucocutaneous changes. Other differences between patient 1 and the other four reported cases is the very severe aplastic anaemia that she manifested. Importantly, our patient responded to treatment with danazol. Interestingly, in contrast to the patient that was evaluated for protein expression by Tummala and colleagues, patient 1 in our series had markedly reduced protein levels and more severe haematological and neurological phenotype. Interestingly, monoallelic PARN mutations have recently been described in idiopathic pulmonary fibrosis;50 a phenotype seen in some patients with telomere-related disorders. Our patients with monoallelic deletions did not manifest this phenotype.
We demonstrated that severe deficiency of PARN in patient cells was associated with loss of PARN-associated specific H/ACA box RNAs, snoRNA/scaRNAs and TERC. This correlated with defects in two snoRNA-dependent and TERC-dependent pathways with essential roles in normal haematopoiesis, ribosome profile and telomere length. The critical role of PARN in normal haematopoiesis was supported by a clear deficiency of blood cells when the gene was knocked down in haematopoietic stem cells/progenitors and in zebrafish. Using this first in vivo parn-deficient animal model, we showed that introduction of human PARN in parn-knockdown zebrafish embryos rescued the phenotype. Unfortunately, introduction of virus particles containing mock or PARN sequences into patient fibroblasts resulted in marked cell death and severely impaired cell growth despite using three different lentivirus constructs; thus, we were not able to rescue human PARN-deficient fibroblasts.
The father of patient 1 who had monoallelic missense PARN mutation was not reported to have an apparent phenotype, while patients 2–4 who had monoallelic PARN deletions had only a neurological phenotype. This might be due to either an incomplete penetrance of monoallelic missense mutations or to a more damaging effect of large deletions than the missense mutation. Our data also suggest that biallelic mutations with one large deletion and one damaging missense mutation lead to a substantially more severe phenotype than monoallelic deletion.
The identified defects in ribosomes and telomeres unravel previously unknown functions of PARN and suggest a new disease mechanism in which PARN deficiency disrupts the polyadenylated state of H/ACA box RNA molecules that in turn influences ribosome profile and telomere length. The disorder described herein belongs to the spectrum of disorders associated with NOP10, NHP2 and DKC1 in which both telomere maintenance and ribosome assembly are affected. However, in contrast to the primary deadenylation defect in our case, in the above dyskeratosis congenita subtypes the encoded proteins directly bind to telomerase complex and nascent pre-rRNA complex. The severe telomere shortening in patient 1 is similar to that seen in dyskeratosis congenita.47 The ribosome defect is similar to that seen in Shwachman–Diamond syndrome.31 ,45 The mechanism of bone marrow failure and other phenotypes in IBMFSs with impaired telomeres and ribosome biogenesis is unclear. Future studies focused on the link between the snoRNAs/scaRNAs aberrations that we identified and telomere/ribosome homeostasis will likely help decipher the mechanism for the IBMFS phenotype. The consequences of impaired PARN-related mRNA deadenylation also require further investigation to elucidate how these defects result in severe blood phenotypes, providing additional insights into haematopoietic stem cell regulation through RNA processing.
Acknowledgments
The authors thank Dr Suzan Blazer for providing and interpreting the results of the MRI, and Dr Mohamed Abdelhaleem for providing and interpreting the results of the bone marrow test.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors SD designed and performed experiments, analysed data, and wrote the manuscript. SMRG designed and performed biochemical and qPCR experiments, analysed data and wrote the manuscript. APD designed and performed experiments to generate morpholino knockdown of zebrafish parn and whole mount in situ hybridisation studies and wrote and edited the manuscript. MN designed and performed biochemical and qPCR experiments, analysed data and wrote the manuscript. BB performed experiments and analyzed data. AJC and AJ designed and performed zebrafish experiments. AR, MM, AM, SB and LT contributed critical clinical information and biological samples. . . HL designed and performed experiments, analysed data and provided technical assistance. MSa performed experiment and analysed the data. MK contributed critical clinical information. MSh performed karyotyping studies, study design, data analysis, interpretation of results and wrote the manuscript. DP, conducted bioinformatics analysis and data analysis. JNB designed and supervised the zebrafish experiments and wrote the manuscript. SWS supervised bioinformatics analysis and data analysis, and provided control data set, genomic DNA from healthy controls. AV designed and supervised the biochemical and RNA-qPCR experiments and wrote the manuscript. YD conceived, designed and supervised research, analysed data and wrote the paper.
Funding The authors acknowledge the support to this work from the Canadian Institute for Health Research (funding reference 102528) and the Nicola's Kids Triathlon to YD, and financial support from the Swedish Research Council and the Linneus Support from the Swedish Research Council to the Uppsala RNA Research Centre to AV, Cancer Care Nova Scotia Peggy Davison Clinician Scientist Award for JB and the Beatrice Hunter/Dalhousie University Cancer Research Training Program for APD. The Italian Cell lines and DNA bank of Rett Syndrome, X-linked mental retardation and other genetic diseases, member of the Telethon Network of Genetic Biobanks (project no. GTB12001), funded by Telethon Italy, and of the EuroBioBank network, provided us with specimens. The authors also thank Dr. Suzan Blazer for providing and interpreting the results of the magnetic resonance imaging, and Dr. Mohamed Abdelhaleem for providing and interpreting the results of the bone marrow test. This study makes use of data generated by the DECIPHER Consortium. A full list of centres that contributed to the generation of the data is available from http://decipher.sanger.ac.uk.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Research Ethics Board, Hospital for Sick Children, Toronto, Canada.
Provenance and peer review Not commissioned; externally peer reviewed.