Article Text

Abstract
Background Childhood-onset pulmonary arterial hypertension (PAH) is rare and differs from adult-onset disease in clinical presentation, with often unexplained mental retardation and dysmorphic features (MR/DF). Mutations in the major PAH gene, BMPR2, were reported to cause PAH in only 10–16% of childhood-onset patients. We aimed to identify more genes associated with childhood-onset PAH.
Methods We studied 20 consecutive cases with idiopathic or heritable PAH. In patients with accompanying MR/DF (n=6) array-comparative genomic hybridisation analysis was performed, with the aim of finding common deletion regions containing candidate genes for PAH. Three patients had overlapping deletions of 17q23.2. TBX2 and TBX4 were selected from this area as candidate genes and sequenced in all 20 children. After identifying TBX4 mutations in these children, we subsequently sequenced TBX4 in a cohort of 49 adults with PAH. Because TBX4 mutations are known to cause small patella syndrome (SPS), all patients with newly detected TBX4 mutations were screened for features of SPS. We also screened a third cohort of 23 patients with SPS for PAH.
Results TBX4 mutations (n=3) or TBX4-containing deletions (n=3) were detected in 6 out of 20 children with PAH (30%). All living patients and two parents with TBX4 mutations appeared to have previously unrecognised SPS. In the adult PAH-cohort, one TBX4 mutation (2%) was detected. Screening in the cohort of (predominantly adult) SPS patients revealed no PAH.
Conclusions These data indicate that TBX4 mutations are associated with childhood-onset PAH, but that the prevalence of PAH in adult TBX4 mutation carriers is low.
- Pulmonary hypertension
- Molecular genetics
- Copy-number
- Clinical genetics
- Developmental
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Pulmonary arterial hypertension (PAH) is an intrinsic vascular disease of the lungs, which leads to high pulmonary vascular resistance and increased pulmonary artery pressure. These result in right ventricular hypertrophy and, eventually, death from right ventricular failure. The histological findings in PAH include medial hypertrophy, intimal fibrosis and plexiform lesions. Thrombosis, inflammation and a disrupted balance between proliferation and apoptosis have been suggested to play a role in this typical vascular remodelling process.1
PAH is classified as group 1 in the five groups of the Updated Clinical Classification of Pulmonary Hypertension (PH) (Dana Point, 2008).2 PAH may be idiopathic (IPAH, subgroup 1.1) or heritable (HPAH, mutations in BMPR2, ALK1, or ENG, subgroup 1.2), associated with drugs or toxins (subgroup 1.3), connective tissue disorders or several other diseases (subgroup 1.4), or a specific disease of the newborn (persistent PH, subgroup 1.5).2
This study focuses on children in subgroups 1.1 and 1.2. Our aim was to identify genes associated with PAH in children.
PAH is a genetically heterogeneous disease, but the major gene identified for PAH is BMPR2, a gene of the TGF-ß family involved in BMP-SMAD signalling.3 Mutations in two other members of the TGF-ß super family (ALK1 and ENG) may also cause PAH, which in most patients is associated with hereditary haemorrhagic telangiectasia (HHT).4 ,5 Several infrequent mutations in genes of the SMAD-family are reported to be associated with PAH,6 ,7 and recently a CAV1 mutation was detected by exome sequencing in a three-generation family with PAH.8 Epigenetic factors may also contribute to the disease.9
Mutations in BMPR2 were reported to cause PAH in 10–16% of childhood-onset PAH patients,10–12 compared with 21–26% of adult-onset sporadic PAH patients and 69% of familial PAH patients,13 ,14 so the genetic aetiology of the disease may be different in childhood onset.14 ,15
Childhood-onset PAH is rare, with a prevalence estimated at 0.7 cases per million.16
There are, thus, no large cohorts available. It presents with more pulmonary vascular medial hypertrophy, and has a higher proportion of responders to acute pulmonary vascular dilatation tests than adult-onset PAH.17 ,18 PAH in children is frequently associated with mental retardation and/or dysmorphic features (MR/DF), without a syndrome diagnosis.12 This observation led to the strategy we chose for looking for PAH candidate genes. We performed array-comparative genomic hybridisation (aCGH) screening of all children with PAH and MR/DF, with the aim of detecting deletion regions harbouring candidate genes for PAH.
Patients and methods
Patients
Initial study population
Childhood-onset PAH patients
In the National Referral Center for Pediatric PH in The Netherlands, DNA was collected from 20 consecutive patients (10 male, 10 female), diagnosed with childhood-onset PAH (IPAH/HPAH, subgroups 1.1 and 1.2 in the 2008 Dana Point Classification)2 and enrolled at the referral centre between 2003 and 2010. Patient characteristics are shown in table 1.
Clinical characteristics and results of molecular analyses of 20 childhood-onset PAH patients
None of the patients had affected relatives, and six patients had unexplained MR/DF (patients 1, 2, 3, 15, 19, 20). Three patients had previously identified BMPR2 mutations (patients 7, 8 and 9; table 1), but were included because BMPR2 mutations have a low penetrance for PAH and the modifiers might be genetic.
No haemodynamic data were available for three patients because they died before cardiac catheterisation could be performed. Echocardiography and autopsy were performed in all three, and advanced pulmonary vascular lesions were present, including intima fibrosis and plexiform lesions. In one patient, histopathological features of pulmonary veno-occlusive disease (PVOD) were present, leading to the diagnosis being revised from PAH to PVOD postmortem.
Additional patient cohorts
After identifying TBX4 deletions and mutations, two additional cohorts were screened:
Adult-onset PAH patients
An additional cohort of 49 adult-onset PAH patients was tested for TBX4 mutations. The cohort included 40 IPAH and 9 HPAH patients with BMPR2 mutations; all diagnoses were confirmed with right heart catheterisation (RHC).
Small patella syndrome patients
Because TBX4 mutations are known to cause small patella syndrome (SPS), a highly variable lower limb malformation syndrome, a group of 23 patients with SPS was screened for clinical and echocardiographical signs of PAH; RHC was performed if indicated by the criteria proposed in the European guidelines for PH.19 This cohort included the two carrier parents and came from 11 families. They were aged 11–67 years (one patient was 11 years old, all the others were older than 20 years). They all carried disease-causing mutations in TBX4 but did not have known symptoms of PAH. Three of the families were described in the original paper on TBX4 mutations causing SPS.20
Genetic analyses
Technical details of the analyses are provided in the online supplementary data appendix S1.
aCGH analysis: screening for deletions
Of the 20 patients with childhood-onset PAH, six also had unexplained MR/DF. We performed aCGH analysis in these six patients (patients 1, 2, 3, 15, 19, 20), which is a standard diagnostic procedure in patients with MR/DF in our hospital. If a deletion or duplication was detected, their parents were also tested.
Mutation and deletion screening of TBX2 and TBX4 by direct sequencing and multiplex ligation-dependent probe amplification
From the six known genes detected by aCGH and located in overlapping 17q23.2 deletion regions, we selected TBX2 and TBX4 as candidate genes for PAH, based on their function and data in the literature. T-box genes are transcription factors known to be involved in several developmental and cardiovascular diseases and are connected with the BMP/SMAD pathway.21–23 All 20 childhood-onset PAH patients, regardless of whether we had previously identified a deletion or mutation in them, were then screened for mutations in TBX2 and TBX4. For patients in whom we found a mutation in one of these genes, we also screened the parents for the same mutation. Intragenic deletion detection for the TBX2 and TBX4 genes was performed by multiplex ligation-dependent probe amplification (MLPA).
Clinical evaluation of TBX4 mutation carriers
Patients with a TBX4 mutation, or with deletions including TBX4, were physically examined by a clinical geneticist and radiographs of the pelvis, knees and feet were made to study pelvic and lower limb malformations associated with SPS.
Results
aCGH in six childhood-onset PAH patients with unexplained MR/DF
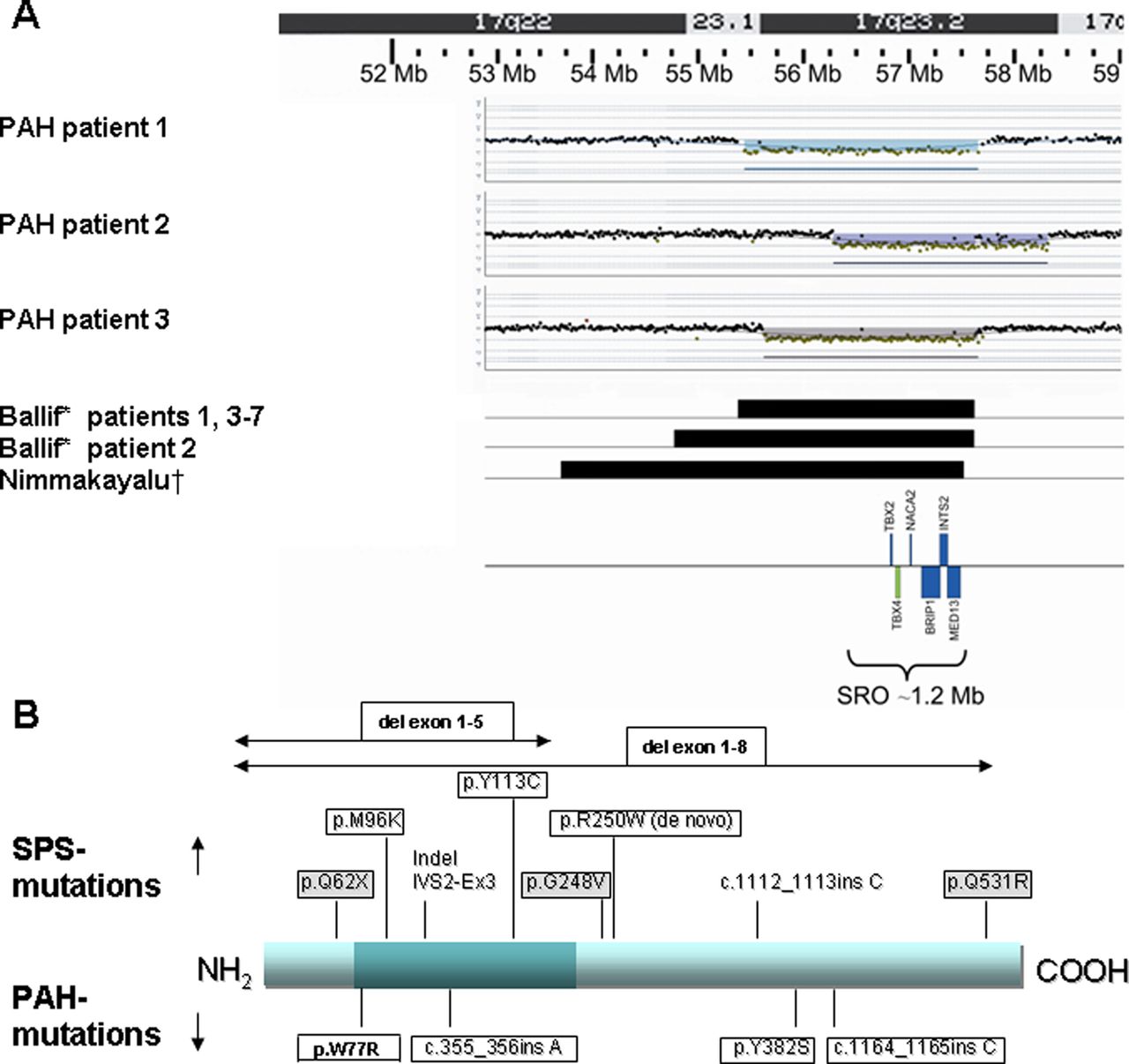
aCGH was performed in six childhood-onset PAH patients because of associated but unexplained MR/DF (table 1). In three of these (patients 1–3), we detected a microdeletion in 17q23.2 which was not present in either of their parents and was, therefore, considered to be causative. This deletion was not reported in the Database of Genomic Variants or in our departmental reference database of 800 healthy persons, but it was reported in the literature as associated with mental retardation and congenital malformations.24 ,25 The smallest region of overlap in these three patients was 1 Mb and contained six genes: TBX2, TBX4, NACA2, BRIP1, INTS2 and MED13 (figure 1A).

Position of deletions in 17q23.2 and mutations in TBX4. (A) Position of breakpoints in 17q23.2 deletions in three childhood-onset PAH patients compared with eight patients with 17q23.2 deletions reported in the literature. Childhood-onset PAH patient 1: 55.4–57.6 Mb; childhood-onset PAH patient 2: 56.7–58.7 Mb; childhood-onset PAH patient 3:55.6–57.6 Mb; Ballif24: patients 1, 3–7: 55.4–57.6 Mb; Ballif24: patient 2: 54.8–57.6 Mb; Nimmakayalu25: one patient:53.7–57.5 Mb; SRO, smallest region of overlap. (B) The TBX4 protein, represented by a blue bar, with the dark part representing the T-box. The positions of TBX4 mutations in three childhood-onset PAH patients (mutations in boxes) and one adult-onset patient (p.W77R) are depicted on the lower side of the protein bar. The positions of TBX4 mutations in SPS patients from our SPS cohort (mutations in boxes, grey background if reported before in the literature), and from patients reported in the literature (no boxes) are depicted on the upper side of the protein bar.20 The mutation R250W was detected de novo in two unrelated SPS patients.
Direct sequencing and MLPA of TBX2 and TBX4 in childhood-onset PAH patients
No mutations were detected in TBX2. Three TBX4 mutations (figure 1B) were identified in three unrelated patients with normal intelligence and without previously detected SPS features (patients 4–6; table 1). Two mutations were insertions causing a frameshift, leading to a premature stop codon, which are predicted to lead to a truncated protein, and one was a non-synonymous mutation (Tyr382Ser), which is predicted to affect the protein function (see online supplementary data appendix S2). These mutations were not present in 1000 control chromosomes in our departmental database.
The parents of two of the three patients with a TBX4 mutation were available for testing: the mutation in patient 4 was maternally derived, while that in patient 5 was paternally derived. None of the patients with a TBX4 deletion/mutation also had a BMPR2 mutation.
Results of aCGH and sequencing are summarised in table 1.
Clinical evaluation of TBX4 deletion and mutation carriers
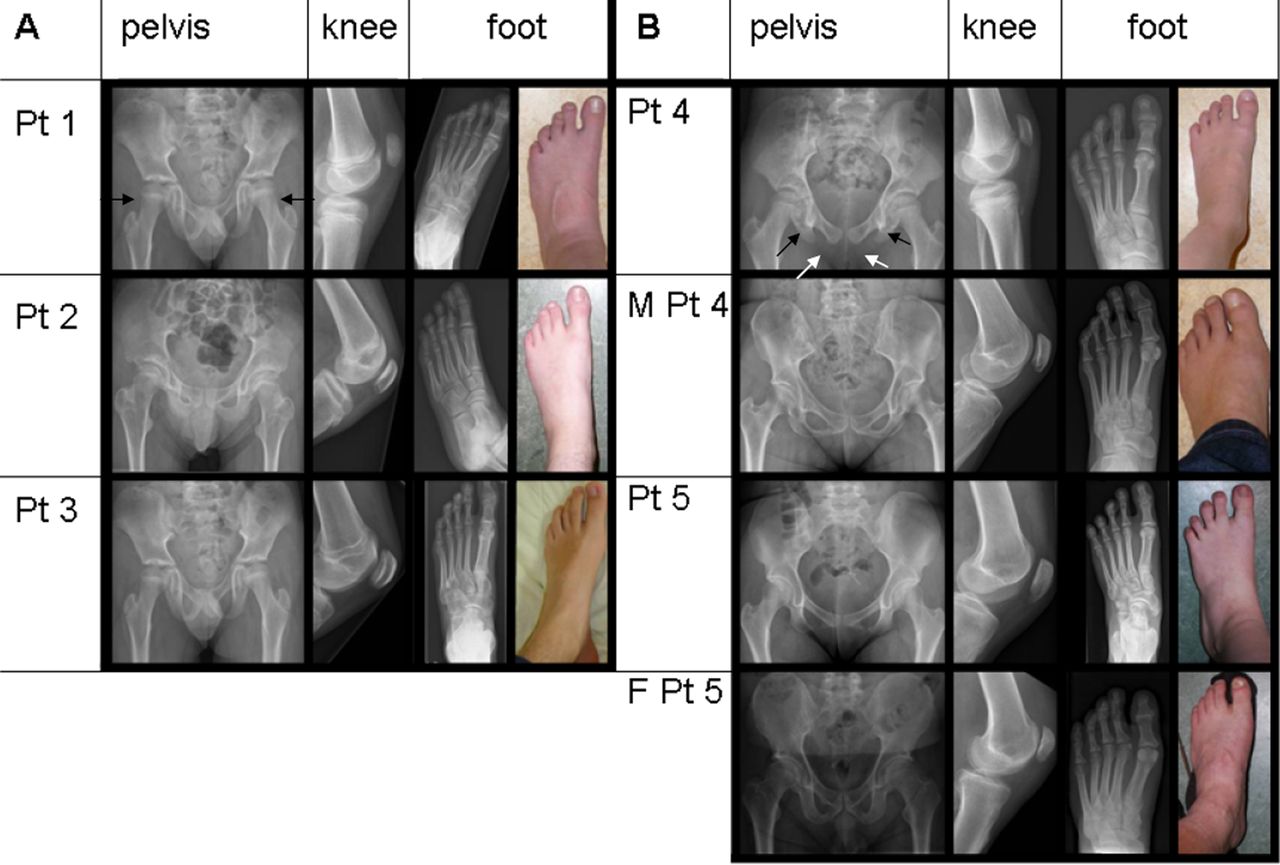
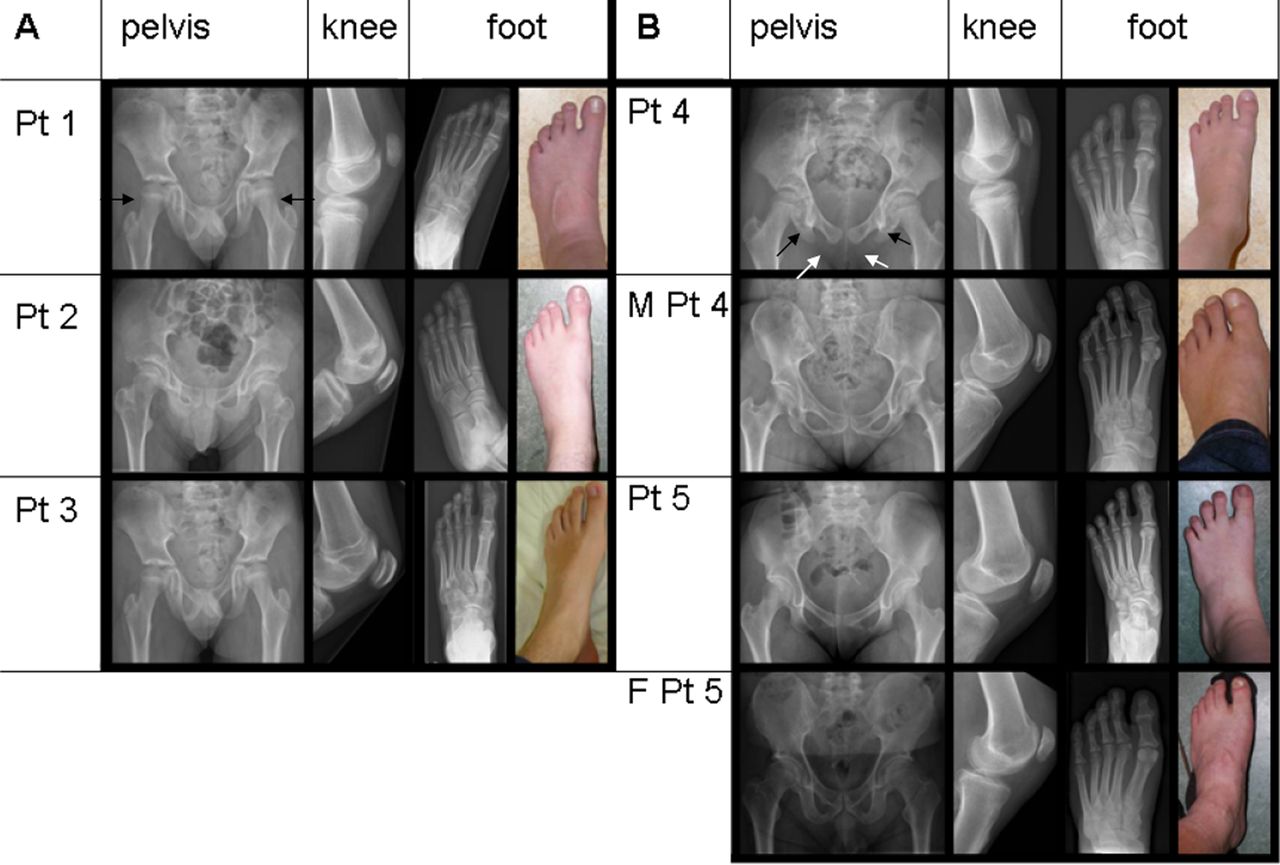
Since TBX4 mutations are known to cause SPS, a clinical geneticist (WSK-F) physically examined the childhood-onset PAH patients with a TBX4 mutation or deletion and their carrier parents for clinical and radiological features of SPS. In all the five patients still alive, and in the two carrier parents, skeletal malformations were found characteristic for SPS, including a history of patella luxation, a hypoplastic or absent patellae, a large gap between the first and second toes, and relatively long second and third rays of the feet. There were also absent or irregular ossification of the ischiopubic junction of the pelvis, axe-cut notches in the acetabulum, and a broad neck of the talus (figure 2A,B). The SPS features had not been recognised earlier because this syndrome is rare, is not associated with mental retardation, and the skeletal findings are only recognised after a thorough examination and x-rays of the pelvis and lower limbs.
{kind=link}
{kind=link}
Pelvic and lower limb malformations characteristic for small patella syndrome. Radiographs and photographs showing variable expression of pelvic and lower limb malformations associated with TBX4 deletions (A) and mutations (B) in five childhood-onset PAH patients (patients 1–5) and in two mutation carrier parents (M, mother, F, father). Radiographs of the pelvis showing long femoral necks, especially in patient 1 (black arrows), axe-cut notches (black arrows in patient 4) and lack of or abnormal ossification of the ischio-pubic junction (unblackened arrows in patient 4) in all patients. x-rays of left knees showing small patellae, and x-rays and photographs of left feet, showing long toes in patient 3, and a large gap between digits I and II and relatively long second and third rays in all patients.
As for all 20 patients, these six SPS-PAH patients fulfilled the Dana Point criteria for PAH groups 1.1 (idiopathic) or 1.2 (heritable). In one of our PAH patients with a TBX4 mutation, postmortem examination of lung tissue revealed features of PVOD. This observation is comparable with what is seen in patients with BMPR2 mutations who have also been reported with PVOD, and it confirms the similarities between the genetic backgrounds of PAH and PVOD suggested previously.26 ,27
TBX4 mutation screening of 49 adult-onset PAH patients
In our cohort of adult-onset PAH patients, we detected one probably disease-causing mutation (c.229T>C, p.W77R), and one variant of uncertain significance (c.104C>T, p.A35V) in TBX4 (see online supplementary data). Both patients are dead, and no symptoms of SPS had been recognised. Although both patients had PAH diagnosed in adulthood, an undetected, mildly progressive childhood onset of the disease in these patients cannot be ruled out retrospectively.
Echocardiographic screening of 23 SPS patients
Echocardiographic screening of 23 SPS patients (including the two carrier parents of patients 4 and 5) from 11 families, without clinical symptoms of PAH, revealed a tricuspid regurgitation velocity of 3.3 m/s (‘PH possible’) in a 57-year-old patient (not a parent of a childhood-onset PAH patient), that could not be explained by associated conditions. RHC revealed that she did not fulfil the criteria for PAH. In two patients, both older than 60 years of age, a tricuspid regurgitation velocity of 2.8 m/s and 3.1 m/s respectively (‘PH possible’) was attributed to chronic obstructive pulmonary disease (COPD) in one patient and left ventricular dysfunction in the other. Finally, in 20 patients, a tricuspid regurgitation velocity <2.8 m/s (‘PH is unlikely’) was measured.
Discussion
In our cohort of patients with childhood-onset PAH, we detected a deletion of 17q23.2 harbouring the TBX4 gene or a TBX4 point mutation in 30% of the patients (6/20). Mutations in TBX4 have been reported as the cause of SPS.20 We detected features of SPS in all the living TBX4 mutation carriers. As SPS is an autosomal dominant disease with an offspring recurrence risk of 50%, we consider that SPS families should be informed about this evident association with childhood-onset PAH.
Although our study is descriptive, and we have no functional data, the finding of deletions and mutations in 30% of a childhood-onset PAH cohort indicates an association and is unlikely to be a coincidence, since these deletions and mutations are not detected in healthy controls. The small size of the cohort of patients with childhood-onset PAH is a major limitation of our study, but is a direct consequence of the rarity of this disease.
The three PAH patients with 17q23.2 deletions had mild mental retardation, all in addition to PAH and SPS, possibly caused by haploinsufficiency of other genes in the deletion region. The three patients with point mutations in TBX4 had normal intelligence. In this small patient series, we could not determine significant differences between PAH patients with or without TBX4 mutations, with regard to age at PAH diagnosis (2.2–15.4 years vs 0.3–15.8 years), or pulmonary vascular disease characteristics. Mortality (1/6 vs 6/14) appears lower in the TBX4-group, but the numbers are too small to draw any conclusions.
Microdeletions on chromosome 17q23.2, including the TBX4 gene, have been reported previously, with patients having pelvic and lower limb malformations and PH.24 ,25 However, there was no clear link between the loss of TBX4 and PH in these patients. Our identification of mutations in the TBX4 gene suggests that TBX4 is a PAH-associated gene.
PAH has not been reported in human SPS patients previously, but the total number of reported SPS patients is low. We demonstrated that none of our 23 predominantly adult SPS patients with heterozygous TBX4 mutations had PAH. These results indicate that the variable expressivity of SPS does not often include PAH in TBX4 mutation carriers, similar to the low frequency of PAH in families with HHT caused by ALK1 and ENG mutations. The low penetrance of PAH in BMPR2 mutation carriers (20%) also indicates that PAH is not a purely monogenic disease.28 ,29
In our relatively small paediatric cohort, TBX4 mutations were more frequent (6/20; 30%) than BMPR2 mutations (3/20; 15%). In adult patients, this frequency appeared to be lower: TBX4 mutations in 1/49 (2%) versus BMPR2 mutations in 9/49 (18%). From these data, it is clear that PAH is a heterogeneous disease. It is unclear, however, why TBX4 mutations are apparently less frequent in adult-onset PAH. One obvious reason could be that most TBX4 mutation carriers do not survive till adulthood. However, the family histories of the SPS patients did not indicate unexplained deaths in childhood (data not shown). Different pathogenic mechanisms may indeed underlie PAH in adults and in children, as suggested previously.15 A second potential explanation for the early onset of PAH in TBX4 mutation carriers could be that TBX4 is only expressed in embryonic and, maybe, childhood pulmonary vasculature, but not in aged pulmonary vasculature, but unfortunately, insufficient data on TBX4 expression are available to confirm or reject this hypothesis. A third potential explanation could be that in TBX4 mutation carriers, as in BMPR2, ALK1 and ENG mutation carriers, unknown modifying factors play a role which ultimately trigger (or silence) the disease, and that these factors are age-specific.
TBX4 belongs to the T-box gene family. Mutations in T-box genes have been found to be associated with several developmental disorders.30–34 T-box genes are transcription factors involved in the establishment of cell-type-specific gene expression patterns, probably by epigenetic mechanisms through interaction with histone-modifying enzymes.35 Epigenetic mechanisms are also believed to play a role in PAH.9 ,36
The expression of Tbx4 is reported in zebrafish, chicken, mouse, dog and other animals.37–42 During early mouse development Tbx4 is expressed in the body wall, the genital papilla, the atrium of the heart, the limbs (predominantly the hindlimbs), in the mesenchym of the lung, mandible and trachea and in the tail mesoderm.37
Most animal studies focus on the role of Tbx4 in limb development. Tbx4 is reported to play a crucial role in lower limb development in both chickens and mice, and is reported to influence the activity of members of the FGF, BMP and Wnt pathways, which are known to be implicated in vertebrate limb outgrowth.37 Ectopic Tbx4 activation appears to initiate differentiation into legs and is, therefore, suggested to play a role in determining hindlimb identity.38 ,43 ,44 Furthermore, an association with clubfeet has been reported in humans with TBX4 containing microduplications.45 However, clubfeet are not a feature of SPS. The precise mechanism leading to skeletal malformations in SPS patients with TBX4 mutations has not yet been elucidated.20 ,46
In addition to expression in the limbs, Tbx4 is also reported to be expressed at the position where the lung buds form in chicken, and to be expressed at high levels in the lung mesenchyme during organogenesis in the mouse. 37 ,47 ,48 Mouse models studying the development of the respiratory system indicate an important role for Tbx4 and Tbx5 in lung branching through interaction with Fgf10. 49 ,50 Decreased Tbx4 activity leads to a decrease in foci expressing Fgf10, a known inducer of Bmp4. In normal lung development, Fgf10 and Bmp4 expression lead to activation of the BMP pathway.51
Although Tbx4 governs multiple processes during respiratory tract development, its role in the development of pulmonary vasculature is insufficiently known. Tbx4-null mice are embryonic lethal due to allantois failure, and since PAH is a postnatal diagnosis, these studies do not allow conclusions regarding associated PAH in these animals. However, Tbx4-mutant embryos are reported to show failure of endothelial cells to form vessels in the allantois.43
Since it now appears that mutations in BMPR2, ALK1, ENG and TBX4 are associated with PAH, there may be a common mechanism leading to PAH developing. BMPR2, ALK1 and ENG are involved in the BMP/TGF-β/SMAD signalling pathways. The balance between the BMP/TGF-β/SMAD signalling pathways probably influences whether a cell type is directed more towards a differentiating or a proliferating state. In particular, the BMP pathway may promote differentiation, whereas the TGF-β pathway may push the system towards a higher proliferation status. A disrupted balance between these two pathways, that is, towards a more proliferative state, may result in PAH.1 A decreased activation of the BMP pathway through FGF10 and BMP4, as reported in animal models focusing on lung and limb development, might link TBX4 mutations in humans to the BMP/TGF–β/SMAD signalling pathways involved in PAH.
More research is needed to elucidate modifying factors and downstream targets of TBX4, which could eventually lead to the development of novel therapeutic interventions.
Conclusions and recommendations
We conclude that SPS is associated with childhood-onset PAH, since we detected whole-gene deletions and point mutations of TBX4 in 6/20 patients (30%). In contrast with childhood-onset PAH, adult-onset patients showed a low frequency of TBX4 mutations (2%). Signs of SPS should be carefully looked for and asked about in patients with PAH, especially in the young, and TBX4 analysis and aCGH can be considered in patients with extrapulmonary features.
TBX4 mutations appear to have a variable expressivity, which seems age-dependent, because 23 SPS patients (22 adults, one child) with TBX4 mutations did not show PAH at screening. Nevertheless, in young patients with SPS, we feel screening for PAH should be considered.
TBX4 mutations might contribute to PAH through a decreased activation of the BMP pathway. Future studies on the expression of TBX4 and on the histopathology of lung vasculature in animal models or studies in inducible pluripotent stem cells may explain the mechanism underlying this association of TBX4 mutations and childhood-onset PAH.
Acknowledgments
We thank Jackie Senior, of the Department of Genetics, University of Groningen, for editing the manuscript, Rianne Kinds, Lennart Johansson and Eddy de Boer of the Department of Genetics, University of Groningen for performing the sequencing, aCGH and MLPA and Pieter Neerincx of the Department of Genetics, University of Groningen for evaluating the control samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online appendix 1
- Data supplement 2 - Online appendix 2
Footnotes
-
RMWH and RMFB contributed equally.
-
Contributors WSK-F, RMWH and RFMB conceived the study, WSK-F wrote the paper. WSK-F, EMHFB, MTRR, EML, JMD, AVD, AV-N and ESH phenotyped the patients, KKD-B, LHH, JJPG and BS-R performed and interpreted array CGH and sequencing of BMPR2, TBX2 and TBX4. All authors read, revised and approved the final version of the manuscript.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The clinical screening of 23 SPS patients for signs of PAH was approved for by the Regional Committee on Research Involving Human Subjects Nijmegen-Arnhem (0006-0119/2011/054). All further clinical data and DNA samples were obtained in the course of routine clinical care, no new patient samples were required.
-
Provenance and peer review Not commissioned; externally peer reviewed.