Article Text

Abstract
Background: Mutations in the surfactant protein C gene (SFTPC) have been recently associated with the development of diffuse lung disease, particularly sporadic and familial interstitial lung disease (ILD).
Objective: We have investigated the prevalence and the spectrum of SFTPC mutations in a large cohort of infants and children with diffuse lung disease and suspected with surfactant dysfunction.
Method and results: 121 children were first screened for the common SFTPC mutation, p.Ile73Thr (I73T). Ten unrelated patients were shown to carry this mutation. The I73T mutation was inherited in six cases, and appeared de novo in four. The 111 patients without the I73T mutation were screened for the entire coding sequence of SFTPC. Of these, eight (seven unrelated) subjects were shown to carry a novel mutant allele of SFTPC. All these seven new mutations are located in the BRICHOS domain except the p.Val39Ala (V39A) mutation, which is in the surfactant protein C (SP-C) mature peptide.
Conclusions: Our results confirm that SFTPC mutations are a frequent cause of diffuse lung disease, and that I73T is the most frequent SFTPC mutation associated with diffuse lung disease.
Statistics from Altmetric.com
Diffuse lung disease in children, including interstitial lung disease (ILD) and chronic lung disease, is a rare heterogeneous group of chronic disorders characterised by impaired gas exchange.1 Recent studies suggest that surfactant deficiency plays a role in the pathogenesis.2 Pulmonary surfactant is a complex mixture of proteins and lipids that prevents collapse at end expiration by reducing surface tension at the air–water interface of the lung alveoli.
Surfactant protein C (SP-C), a hydrophobic protein critically involved in surfactant homeostasis,3 is expressed by alveolar type II epithelial cells and encoded by a single gene located on chromosome 8 (8p21.3) called SFTPC. The human SFTPC gene is organised into six exons (I through V coding, VI untranslated). SP-C is synthesised as a 197 amino acid proprotein (proSP-C) that undergoes multiple processing steps to the mature SP-C peptide of 35 amino acids, to be finally released in the alveoli associated with the other surfactant proteins and phospholipids.3 Nogee et al first described an SFTPC mutation in 2001.4 The mutation, c.435+1G>A, located at the first base of intron 4, alters the normal donor splice site and results in the skipping of exon 4 and the deletion of 37 amino acids in the carboxy terminal domain of proSP-C. This mutation was observed in one allele of a patient suffering from chronic interstitial pneumonitis and was inherited following a dominant pattern. To date, 13 SFTPC mutations have been identified.4–11 They always occur in one single allele and have been associated with diffuse lung disease in children and adults. The mutation I73T (c.218 T>C) is the more prevalent mutation; others have been described in only one family. Dominant transmission with variable penetrance has been confirmed in familial cases, but a number of sporadic cases have also been reported with a de novo mutation. The phenotype associated with SFTPC mutations appears to be very variable. Indeed, neonatal forms leading to death in the first years of life as well as childhood and adult forms with chronic respiratory disease have been reported. Single nucleotide polymorphisms (SNPs) have also been detected both in the coding and non-coding sequence, with unclear consequences on proSP-C processing and function.2
The variable phenotype with unspecific biological findings makes the genetic diagnosis of SP-C inherited disease essential. Thus, the present study aims to investigate SFTPC variations in a large population of 121 children (120 unrelated) with diffuse lung disease and suspected surfactant dysfunction.
PATIENTS AND METHODS
Patients
One hundred and twenty-one newborns/children (60 male, 61 female) diagnosed with diffuse lung disease were recruited over a 5 year period (2002–2007) through a national programme on rare lung disease by several university hospital based paediatric pulmonology practices (Hôpital Armand Trousseau, Paris; Hôpital Necker Enfants Malades, Paris; Groupement Hospitalier Est, Lyon, and Centre Hospitalier Universitaire CHU Arnaud de Villeneuve, Montpellier). Seventy-four patients originated from Europe, 24 from North Africa, 13 from Africa, nine from Reunion Island and one from French Antilles. Among the 121 patients with diffuse lung disease, 86 had respiratory distress, 59 presenting with neonatal onset, and 18 patients died. Clinical observations included family background, physical signs at presentation, chest radiography and computed tomography (CT), and histological lung analysis when available. The protocol was accepted by the committee for the protection of individuals in biochemical research as required by French legislation, and written informed consent was obtained from the patients or their next of kin included in this study.
The 121 patients where the common 121ins2 SFTPB mutation was initially ruled out were analysed for the common I73T mutation. Then, the entire SFTPC coding sequence was performed in patients without the I73T mutation.
Mutation analysis
Genomic DNAs were extracted from patients’ blood using an automated BioRobot EZ1 workstation (Qiagen, Hilden, Germany). The common mutation I73T was screened using an allele specific amplification as previously published12 and confirmed by the direct sequencing of exon 3. The five coding exons of SFTPC and the exon–intron boundaries were analysed by direct DNA sequencing of polymerase chain reaction (PCR) products using the following primers: exon 1 sense 5′-ACCCAGGTTTGCTCTTGCT-3′, exon 1 antisense 5′-TGAATGGATCTGGATAAGGAAA-3′, exon 2 sense 5′-TGTTAGAATCCAGGCCACCT-3′, exon 2 antisense 5′-CGTGCCTCTTTCCTTCTAGC-3′, exon 3 sense 5′- CTCTTGGGAAAGAGGGAAGC-3′, exon 3 antisense 5-′GGGAGAGATGGATGTGGATG-3′, exon 4 sense 5′-CTAGTATGACTCCCGTGCCC-3′, exon 4 antisense 5′-TGAGGAACAGTGCTTTACAGG-3′, exon 5 sense 5′-TCAGCTGAGTCCACTCACTACC-3′, exon 5 antisense 5′-GTACCGGTCTGTGAGCTTCC-3′. Sequencing reactions were performed with Big Dye Terminator cycle sequencing kits from Applied Biosytems (Foster City, California, USA) and run on an ABI Prism 3130 Genetic Analyzer. Sequences were analysed using SeqScape software and compared with the reference sequence NM_003018.3. Mutations are named following the international nomenclature, starting at nucleotide 1 of the first codon (Human Genome Variation Society: Nomenclature for the description of sequences variation, http://www.hgvs.org/mutnomen/).
Multiple protein alignments were performed using the software SIFT (http://blocks.fhcrc.org/sift/SIFT.html) and the search for polymorphisms was carried out using the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/). The effect of substitution on the splicing site was studied using the software NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) and GeneSplicer Web Interface (http://www.tigr.org/tdb/GeneSplicer/gene_spl.html).
The control population consisted of 50 individuals of European descent, 50 individuals of North African descent, 50 individuals from African descent and 50 individuals of Reunion Island descent, all of whom have no report of pulmonary disease.
RESULTS
We screened a cohort of 121 patients with diffuse lung disease for the mutation c.218T>C (p.Ile73Thr, I73T). SFTPC mutation is present in 18 (17 unrelated) patients and seven novel mutations are described.
The I73T mutation was found in 10 patients without neonatal onset, four presenting with respiratory viral infection. I73T was inherited in six cases. In four cases, I73T was sporadic and thus absent in both parents. The 111 patients without I73T mutation were screened for all the coding sequence and the exon–intron boundaries of SFTPC. Seven novel SFTPC variations were identified (table 1).
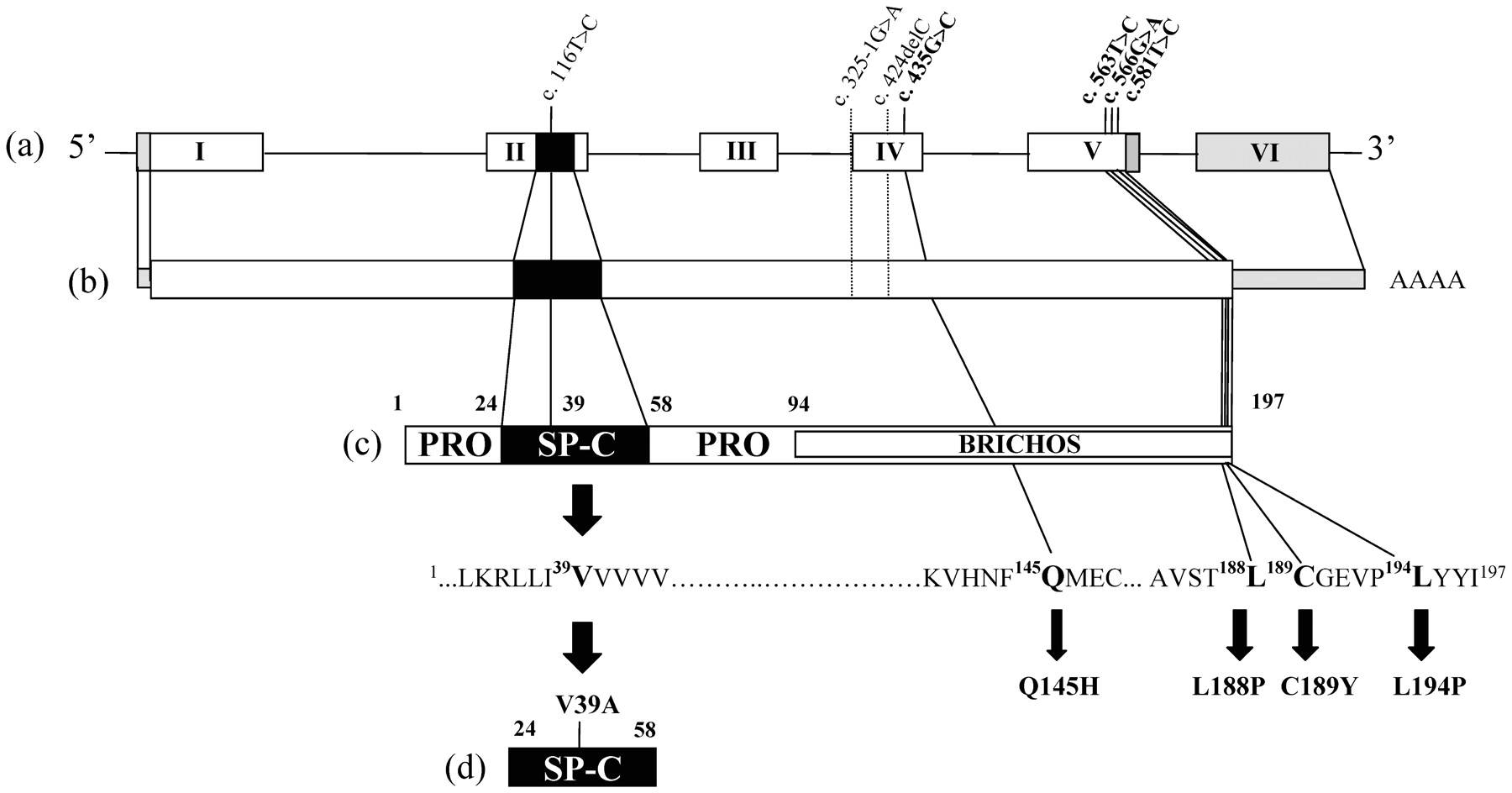
Two mutations, C189Y and L194P, were inherited (fig 1), whereas the mutations c.325-1G>A and c.424delC were sporadic and absent in the patient’s parents and probably appeared de novo. Samples of familial individuals of index cases with V39A, Q145H and L188P were unavailable. The novel variants segregated with the disease in the familial cases (C189Y and L194P) and have not been previously reported as polymorphisms, suggesting that they are pathogenic. In addition, each new variant was not detected in 100 control alleles of the same origin. The mutation c.325-1G>A located at the last base of intron 3 is presumed to abolish the normal acceptor splice site using the GeneSplicer prediction software, and to result in the skipping of exon 4 and the deletion of 37 amino acids in the carboxy terminal domain of proSP-C. The mutation c.424delC in exon 4 is a frameshift mutation that induces a premature stop codon at position 185. The variation c.435G>C resulting in the substitution of glutamine 145 by histidine, involved the last base of exon 4. This substitution is supposed to abolish the donor splicing site of exon 4 following the software of splice site prediction, and to result in the skipping of exon 4. The V39A mutation is located in mature SP-C. The six other novel variations have been detected in the BRICHOS domain (fig 2).
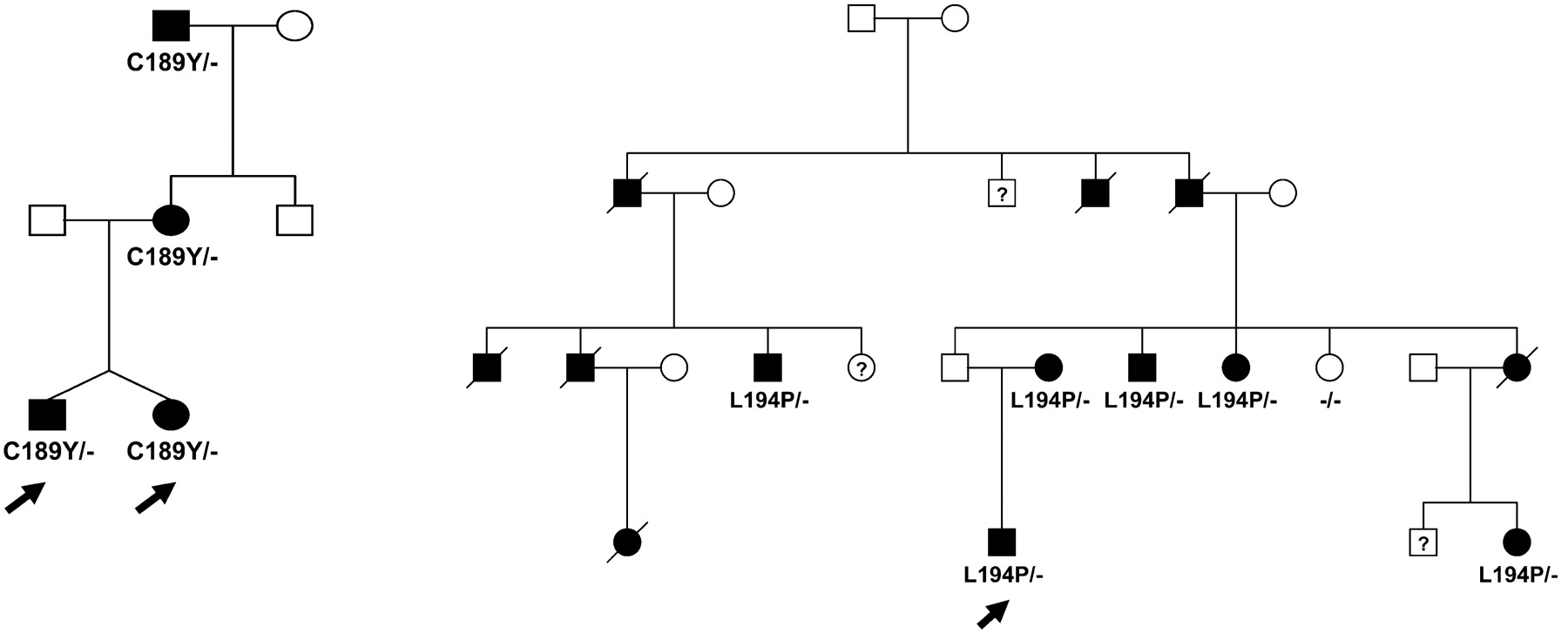
Pedigree of families with the new SFTPC mutations C189Y and L194P.

{kind=link}
{kind=link}
Localisation of new SFTPC mutations. Schematic representation of SFTPC gene. (a) (exon I to VI), corresponding mRNA (b), pro-SP-C (c) and SP-C (d) protein sequence with annotated newly identified mutations.
Interestingly, the age of onset was significantly lower in patients with new mutations compared with those carrying the I73T mutation (table 2). No other significant differences between the two groups of patients were observed.
DISCUSSION
The identification of SP-C alterations has led to significant advances in the diagnosis of diffuse lung disease. Our study on a large cohort of patients confirms that the common mutation I73T in the SFTPC gene is a frequent cause of diffuse lung disease. In addition, we described seven new pathogenic SFTPC variants associated with respiratory diseases occurring during the first months of life.
The mutation I73T has been first reported in one allele of an infant with severe respiratory insufficiency and a histological pattern of non-specific interstitial pneumonia (NSIP) and pulmonary alveolar proteinosis (PAP).7 9 Furthermore, in a cohort of 116 children with ILD, this mutation was present in seven cases and appeared to be common.12 The mutation was inherited or observed de novo. The phenotype associated with I73T was described as eminently variable, ranging from fatal cases to asymptomatic adult carriers.12 13 In our cohort of 121 patients with diffuse lung disease, we found 10 unrelated patients with diffuse lung disease harbouring the I73T mutation. This result confirms the high frequency of this mutation in diffuse lung disease.
Besides this frequent mutation, only 12 SFTPC mutations have been previously described. We report in this study seven new mutations. The splice site mutation c.325-1G>A is thought to affect the acceptor splice site of exon 4, and may result in the skipping of exon 4. Thus, this mutation probably has the same effect that the previously reported mutation c.435+1G>A affecting the donor site of exon 4.4 The deletion c.424delC induces the translation of an abnormal protein. The missense mutations V39A, Q145H, L188P, C189Y, and L194P are believed to be pathogenic because they have not been observed in control chromosomes of the same origin. Moreover, the amino acids involved are conserved in mammalian SP-C sequences, and the mutation segregates with the pulmonary disease in familial cases (C189Y and L194P). The phenotype associated with C189Y appeared to be variable, severe in the first months of life and asymptomatic in adults. In contrast, L194P is associated with pulmonary fibrosis in adults. The mutation V39A is the first valine of the polyvaline stretch of mature SP-C. The polyvaline stretch is believed to be a crucial actor of the secondary structure of SP-C. Indeed, studies of purified mature SP-C have shown that the polyvaline stretch is highly α-helical and capable of spanning phospholipid bilayers in the liquid crystalline phase.14
To date, most of the mutations previously identified in the SP-C gene are localised in the propeptide. In our study, with the exception of the V39A located in the mature peptide, the other new mutations are in the propeptide. Based on functional studies that ascertain the role of SFTPC mutations in the pathogenesis of diffuse lung disease, two mechanisms have been suggested.2 10 First, owing to a heterozygous mutation, abnormal proSP-C could disrupt the regular processing of proSP-C within the cell and consequently inhibit SP-C production in the alveoli. This is the case for the I73T and E66K mutations altering intracellular trafficking.7 10 Also, as shown in the Δexon4 and L188Q mutations, aberrant proSP-C forms aggregates that could be toxic for alveolar type II cells and induce an unresolved inflammatory response.15–17 Moreover, in two patients, diffuse lung disease have also been associated with SP-C deficiency without any detectable SFTPC mutation.9 Thus, alteration of SP-C metabolism or complete SP-C deficiency is critical in diffuse lung disease pathophysiology.
Interestingly, we found three novel variants, L188P, C189Y and L194P, located in the C-terminus of proSP-C BRICHOS domain (F94-I197)—a highly conserved domain. The BRICHOS domain is composed of about 100 amino acids in the proprotein. It is found in several proteins associated with degenerative and proliferative diseases.18 The BRICHOS domain is thought to be involved in protein processing and is assumed to have a chaperone-like function. It plays a critical role in proSP-C targeting and processing.16 19 20 Indeed, in previous studies, two patients have been found to carry a mutation encoding substitution (L188R and L188Q) of the same conserved leucine residue altered in our cohort (L188P). The L188R mutation has been detected in one infant with ILD.5 On the other hand, the L188Q mutation is detected in large familial pulmonary fibrosis kindred, either in children or adults with cellular interstitial pneumonitis or usual interstitial pneumonitis.6 We have observed L188P in an 18-year-old patient who had diffuse lung disease during the first year of life. Unfortunately, familial samples were not available.
Descriptions of the reported new mutations will be the subject of further functional studies, which will improve knowledge on the pathogenesis of associated surfactant diffuse lung disease in children.
Acknowledgments
We thank the patients and their families for their cooperation in this study. We also thank Catherine Meunier, France Michel, Corinne Chauve, Isabelle Sargis, Magali Niasme, and Cristina Das Neves for their expert technical assistance.
REFERENCES
Supplementary materials
Footnotes
LG and RE contributed equally to this work
Funding: This work was supported by the Département de la Recherche Clinique et du Développement (PHRC 2007, “Surfactant disorders associated with chronic lung disease in children”).
Competing interests: None declared.
Patient consent: Obtained.
Linked Articles
- Correction