


Abstract
Poly(ADP-ribose) polymerase-1 (PARP-1) is a member of the PARP enzyme family consisting of PARP-1 and several recently identified novel poly(ADP-ribosylating) enzymes. PARP-1 is an abundant nuclear protein functioning as a DNA nick-sensor enzyme. Upon binding to DNA breaks, activated PARP cleaves NAD+ into nicotinamide and ADP-ribose and polymerizes the latter onto nuclear acceptor proteins including histones, transcription factors, and PARP itself. Poly(ADP-ribosylation) contributes to DNA repair and to the maintenance of genomic stability. On the other hand, oxidative stress-induced overactivation of PARP consumes NAD+ and consequently ATP, culminating in cell dysfunction or necrosis. This cellular suicide mechanism has been implicated in the pathomechanism of stroke, myocardial ischemia, diabetes, diabetes-associated cardiovascular dysfunction, shock, traumatic central nervous system injury, arthritis, colitis, allergic encephalomyelitis, and various other forms of inflammation. PARP has also been shown to associate with and regulate the function of several transcription factors. Of special interest is the enhancement by PARP of nuclear factor κB-mediated transcription, which plays a central role in the expression of inflammatory cytokines, chemokines, adhesion molecules, and inflammatory mediators. Herein we review the double-edged sword roles of PARP in DNA damage signaling and cell death and summarize the underlying mechanisms of the anti-inflammatory effects of PARP inhibitors. Moreover, we discuss the potential use of PARP inhibitors as anticancer agents, radiosensitizers, and antiviral agents.
I. Poly(ADP-Ribose) Metabolism
A. Structure and Function of PARP-1
Poly(ADP-ribose) polymerase-1 (PARP-11; EC2.4.2.30) [also known as poly(ADP-ribose) synthetase and poly(ADP-ribose) transferase] is a nuclear enzyme present in eukaryotes. PARP-1 is a 116-kDa protein consisting of three main domains: the N-terminal DNA-binding domain containing two zinc fingers, the automodification domain, and the C-terminal catalytic domain (Mazen et al., 1989; de Murcia and Menissier de Murcia, 1994; de Murcia et al., 1994; Schreiber et al., 1995; Szabo, 2000; Smith, 2001) (Fig. 1). The primary structure of the enzyme is highly conserved in eukaryotes (human and mouse enzyme have 92% homology at the level of amino acid sequence) with the catalytic domain showing the highest degree of homology between different species; the catalytic domain contains the so-called PARP signature sequence, a 50-amino acid block showing 100% homology between vertebrates.

Comparison of the domain structures of some PARP enzymes. PARP-1 consists of three main domains: the N-terminal DBD, the automodification domain (AMD), and the C-terminal catalytic domain. Within the DBD, two zinc fingers are responsible for DNA binding and some protein-protein interactions. The DBD also contains an NLS within which the caspase-cleavage site (DEVD) can be found. The automodification domain contains a BRCT motif, which is common in many DNA repair and cell-cycle proteins. PARP-1 participates in various protein-protein interactions through the BRCT motif. The active site of the enzyme found in the C-terminal part is highly conserved in eukaryotes, and this 50-amino acid sequence is also known as “the PARP signature”. PARP-2 differs from PARP-1 in the structure of its DBD. Vault PARP (vPARP) has no DBD, and the N terminus is occupied by a BRCT motif. The enzyme has an NLS residing in the C-terminal part. The N terminus of tankyrase-1 and -2 has a histidine-, proline-, and serine-rich (HPS) region or N-terminal domain (NTD), respectively. The remaining parts [ankyrin repeats, sterile alpha motives (SAM), and catalytic domains] are basically the same.
PARP-1 functions as a DNA damage sensor and signaling molecule binding to both single- and double-stranded DNA breaks. Upon binding to damaged DNA mainly through the second zinc-finger domain, PARP-1 forms homodimers and catalyzes the cleavage of NAD+into nicotinamide and ADP-ribose and then uses the latter to synthesize branched nucleic acid-like polymers poly(ADP-ribose) covalently attached to nuclear acceptor proteins (de Murcia et al., 1994; de Murcia and Menissier de Murcia, 1994; Lindahl et al., 1995; Schreiber et al., 1995; Burkle, 2001; Smith, 2001). The size of the branched polymer varies from a few to 200 ADP-ribose units. Because of its high negative charge, the covalently attached ADP-ribose polymer dramatically affects the function of target proteins. In vivo, the most abundantly poly(ADP-ribosylated) protein is PARP-1 itself, and auto-poly(ADP-ribosylation) represents a major regulatory mechanism for PARP-1 resulting in the down-regulation of the enzyme activity. In addition to PARP-1, histones are also considered to be major acceptors of poly(ADP-ribose) (Tanuma et al., 1985; Nagele, 1995). Poly(ADP-ribosylation) confers negative charge to histones, leading to electrostatic repulsion between DNA and histones. This process has been implicated in chromatin remodeling, DNA repair, and transcriptional regulation. Several transcription factors, DNA replication factors, and signaling molecules [NF-κB (Oliver et al., 1999), AP-2 (Kannan et al., 1999), Oct-1, YY1 (Oei and Shi, 2001a,b), B-MYB (Cervellera and Sala, 2000), DNA-dependent protein kinase (Ariumi et al., 1999), p53 (Wesierska-Gadek et al., 1996; Kumari et al., 1998; Malanga et al., 1998; Simbulan-Rosenthal et al., 1999b, 2001b; Mendoza-Alvarez and Alvarez-Gonzalez, 2001; Wesierska-Gadek and Schmid, 2001; Tong et al., 2001), topoisomerase I, lamin B, and B23] have been shown to become poly-ADP-ribosylated by PARP-1. The effect of PARP-1 on the function of these proteins is carried out by noncovalent protein-protein interactions and by covalent poly-ADP-ribosylation. Poly-ADP-ribosylation is a dynamic process, indicated by the short (<1 min) in vivo half-life of the polymer (Whitacre et al., 1995). Two enzymes—poly(ADP-ribose) glycohydrolase (PARG) and ADP-ribosyl protein lyase—are involved in the catabolism of poly(ADP-ribose), with PARG cleaving ribose-ribose bonds of both linear and branched portions of poly(ADP-ribose) and the lyase removing the protein proximal ADP-ribose monomer (Davidovic et al., 2001).
The regulation of PARP-1 activity is established through different mechanisms. The best characterized mechanism is the down-regulation of enzyme activity through auto-poly-ADP-ribosylation (Kawaichi et al., 1981). Furthermore, nicotinamide, the smaller cleavage product of NAD+, also exerts inhibitory effect on PARP-1, allowing negative feedback regulation. Recently, the purines hypoxanthine, inosine, and adenosine also were identified as another class of endogenous PARP inhibitors (Virag and Szabo, 2001). The regulation of PARP activity by purines is possibly relevant under pathophysiological conditions in which intracellular levels of these metabolites reach levels that are high enough to efficiently inhibit PARP. Phosphorylation of PARP by protein kinase C also results in enzyme inhibition (Tanaka et al., 1987; Bauer et al., 1992). The abundance of PARP may also change under certain conditions, suggesting a transcriptional or posttranscriptional regulation (Bergeron et al., 1997; Tramontano et al., 2000; Doucet-Chabeaud et al., 2001). It is not yet clear whether PARP induction significantly alters the poly-ADP-ribosylating capacity of the cells, because even in resting cells, PARP-1 is one of the most abundant nuclear proteins. The interconnection of PARP activation and signal transduction pathways is supported by a report describing DNA strand break-independent PARP activation via the phospholipase C-inositol-1,4,5,-trisphosphate-calcium route (Homburg et al., 2000). The physiological or pathophysiological relevance of this pathway is poorly understood at present.
The biological role of poly(ADP-ribose) is complex and involves nine main functions. First, PARP-1 has been implicated in DNA repair and maintenance of genomic integrity (de Murcia and Menissier de Murcia, 1994; de Murcia et al., 1994,1997; Schreiber et al., 1995; Chatterjee et al., 1999b; Shall and de Murcia, 2000). This “guardian angel” function is indicated by delayed DNA base-excision repair and by a high frequency of sister chromatid exchange in PARP-1-deficient cells exposed to ionizing radiation or treated with alkylating agents (de Murcia et al., 1997). High levels of ionizing radiation and alkylating agents elicit higher lethality in PARP-1-deficient mice when compared with wild-type ones (de Murcia et al., 1997).
Second, PARP-1 also regulates the expression of various proteins at the transcriptional level. Of special importance is the regulation by PARP-1 of the production of inflammatory mediators such as the inducible nitric-oxide synthase (iNOS) (Hauschildt et al., 1992; Le Page et al., 1998; Szabo et al., 1998c; Oliver et al., 1999), intercellular adhesion molecule 1 (ICAM-1) (Zingarelli et al., 1998;Szabo et al., 2001b), and major histocompatibility complex class II (Otsuka et al., 1991). NF-κB is a key transcription factor in the regulation of this set of proteins, and PARP has been shown to act as a coactivator in the NF-κB-mediated transcription (Oliver et al., 1999). There is currently no consensus in the literature regarding whether the modulation of NF-κB-mediated transcription by PARP is dependent on the catalytic activity of the enzyme or, alternatively, on its physical presence (Hassa and Hottiger, 1999; Kameoka et al., 2000;Chang and Alvarez-Gonzalez, 2001; Hassa et al., 2001). Poly(ADP-ribosylation) of histones may also contribute to the transcription-promoting effect of PARP-1, because poly(ADP-ribose) confers negative charge to histones, leading to electrostatic repulsion between histones and DNA. Thus, poly(ADP-ribosylation) can loosen the chromatin structure and can thereby make genes more accessible for the transcriptional machinery. Nuclear receptor-mediated transcription is regulated by PARP-1 in a different manner: PARP-1 seems to suppress nuclear receptor-mediated transcription (Miyamoto et al., 1999).
Third, PARP-1 regulates replication and differentiation. The involvement of PARP-1 in the regulation of replication is supported by observations that poly(ADP-ribose) metabolism is accelerated in the nuclei of proliferating cells (Tanuma et al., 1978; Kanai et al., 1981;Leduc et al., 1988; Bakondi et al., 2002a). Furthermore, PARP-1 is part of the multiprotein replication complex (MRC) (Simbulan-Rosenthal et al., 1996), indicated by copurification of PARP-1 with key components of MRC (Simbulan-Rosenthal et al., 1996; Dantzer et al., 1998). Moreover, several replication factors and centromere proteins have been shown to serve as substrates for PARP (Simbulan-Rosenthal et al., 1996;Saxena et al., 2002). Another mechanism by which PARP may regulate nuclear processes is poly(ADP-ribosylation) of histones facilitating the assembly and deposition of histone complexes on DNA during replication (Boulikas, 1990).
Fourth, poly(ADP-ribosylation) has been implicated in the regulation of telomerase activity. The overexpression of tankyrase-1, a recently discovered PARP enzyme, in telomerase-positive human cells resulted in a gradual and progressive elongation of telomeres (Smith and de Lange, 2000). Besides tankyrase-1, PARP-1 has also been implicated in the maintenance of telomere length. Genetic ablation of PARP-1 has been shown to result in telomere shortening (d'Adda di Fagagna et al., 1999) but others found no difference in telomere length of PARP-proficient and -deficient cells (Samper et al., 2001).
Fifth, PARP-1 activation has been proposed to represent a cell-elimination pathway (Berger et al., 1983, 1986; Schraufstatter et al., 1986b; Sims and Benjamin, 1987; Schreiber et al., 1995;Kleczkowska and Althaus, 1996) through which severely damaged cells are removed from tissues. PARP-1-mediated cell death occurs in the form of necrosis (Schreiber et al., 1995; Virag et al., 1998a,b), which is probably the least desirable form of cell death. During necrotic cell death, the cellular content is released into the tissue, exposing neighboring cells to potentially harmful attacks by proteases and other released factors. This scenario (Fig. 2) is best exemplified by cells that have been exposed to DNA-damaging stimuli. Mild genotoxic noxa cause PARP activation that facilitates DNA repair and cell survival. Severe DNA damage, however, causes overactivation of PAR,P resulting in the depletion of NAD+ and ATP and consequently in necrotic cell death (Fig. 2).

DNA-damage-induced PARP activation: a kiss of life or a kiss of death? DNA-damaging stimuli cause PARP activation. Activated PARP cleaves NAD+ into nicotinamide and ADP-ribose and polymerizes the latter on nuclear-acceptor proteins. Poly(ADP-ribosylation) facilitates DNA repair and thus permits cell survival. Severe DNA damage, however, leads to overactivation of PARP, resulting in NAD+ and ATP depletion and necrotic cell death.
Sixth, poly(ADP-ribose) polymer has been identified recently as an emergency source of energy used by the base-excision machinery to synthesize ATP (Maruta et al., 1997; Oei and Ziegler, 2000).
Seventh, similarly to ubiquitination, poly(ADP-ribose) may also serve as a signal for protein degradation in oxidatively injured cells (Ciftci et al., 2001; Ullrich and Grune, 2001; Ullrich et al., 2001a). Hydrogen-peroxide treatment of K562 cells caused a PARP-1-dependent up-regulation of 20S proteosome activity. During this process, the proteosome becomes poly(ADP-ribosylated), resulting in the enhanced degradation of poly(ADP-ribosylated) histones (Ullrich and Grune, 2001). Immunoprecipitation experiments demonstrated a protein-protein interaction of the functionally active PARP with the proteasome in correlation with the proteasome activity (Ullrich et al., 2001a).
Eighth, in addition to PARP-catalyzed covalent poly(ADP-ribosylation), poly(ADP-ribose) polymers can noncovalently bind to specific (ADP-ribose)n binding motifs in proteins, such as histones, XRCC1, p53, and DNA polymerase ε, and thereby modify their function (Althaus et al., 1993; Pleschke et al., 2000). Such (ADP-ribose) polymers can be formed during the catabolism of poly(ADP-ribose) by poly(ADP-ribose) glycohydrolase (Davidovic et al., 2001).
In the ninth and final function, poly(ADP-ribosylation) may also be involved in the regulation of cytoskeletal organization. A recent study reported morphological alterations in Drosophilaoverexpressing PARP-1 (Uchida et al., 2001). The overexpression of PARP-1 disrupted the organization of cytoskeletal F-actin, resulting in aberrant cell and tissue morphology. Furthermore, heat-induced PARP expression disrupts the organization of cytoskeletal F-actin in embryos and tissue polarity in adult flies. Whether these morphological alterations are indeed related to PARP-1 function or, alternatively, whether PARP-1 overexpression interferes with the function of cytoplasmic PARP enzymes remains to be seen.
B. PARP Homologs
Until recently, PARP activity was believed to result from the function of a single enzyme. After the observation that PARP-1-deficient cells have some residual PARP activity (Shieh et al., 1998), intensive research began to identify enzymes responsible for this activity. In the last 2 years, several other enzymes possessing poly(ADP-ribosylation) activity have been described (Smith, 2001) with the founding member of the PARP enzyme family now designated as PARP-1. Although research on the biological role of these novel PARP enzymes is in the embryonic stage, interesting differences in domain structure (Fig. 1), subcellular localization, tissue distribution, and ability to bind to DNA have already been established.
PARP-2 is a 62-kDa protein of unknown function (Ame et al., 1999). Human and mouse PARP-2 genes were mapped to 14q11.2 and 14C1, respectively. These loci are different from PARP-1 loci. The automodification domain is missing from PARP-2, and the DNA binding domain (DBD) is very different from that of PARP-1 (Ame et al., 1999). Somewhat surprisingly, even though the putative DBD of PARP-2 is devoid of any known DNA binding motifs, DNase I-treated DNA induced PARP-2 activation. DNA binding of PARP is facilitated by the high ratio of basic amino acids in the PARP-2 DBD. Moreover, PARP-2 is capable of auto-poly(ADP-ribosylation); however, it could not poly(ADP-ribosylate) histones, which are prototypical PARP-1 substrates. The enzyme localizes to nuclei and becomes activated in cells upon methylmethanesulfonate-induced DNA damage. PARP-2 has been shown recently to be cleaved by cysteinyl aspartate-specific protease (caspase)-8 (Benchoua et al., 2002).
Vault PARP has been found in vaults (Kickhoefer et al., 1999). Vaults are barrel-shaped ribonucleoprotein particles of arched morphology reminiscent of the vaulted ceilings of cathedrals (Kickhoefer et al., 1996). Their biological role is unknown at present. Vaults were proposed to be part of the nuclear pore complex and have also been implicated in multidrug resistance (Kickhoefer et al., 1996). Vault PARP has been found to associate with and poly(ADP-ribosylate) the major vault protein. The functional significance of this cytoplasmic PARP is as elusive as the biological role of vaults.
Tankyrase-1.
The chromosomal end-replication problem has been fascinating researchers for some years. The observation that tumor cells but not untransformed cells can prevent the shortening of their chromosomes by using a ribonucleoprotein enzyme named telomerase opened a new target area for anticancer drug development. Therefore, it is not surprising that among the novel PARPs, tankyrase, a telomere-associated enzyme (Smith et al., 1998), has attracted much attention. Tankyrase-1, a protein containing 24 ankyrin repeats, binds to and poly(ADP-ribosylates) telomere repeat-binding factor 1 (TRF1), a negative regulator of telomerase (Smith et al., 1998). Tankyrase-1 was also found to auto-poly(ADP-ribosylate) itself. Poly(ADP-ribosylation) probably releases TRF from telomeres and inhibits TRF function because overexpression of tankyrase-1 increased telomere length. Like vault PARP, tankyrase does not require DNA for activity. Overexpression of tankyrase-1 in the nucleus diminished the level of unmodified TRF1 in immunoblots and led to reduced immunofluorescence of TRF1 at interphase telomeres (Smith and de Lange, 2000). Long-term overexpression of tankyrase-1 in telomerase-positive human cells resulted in a gradual and progressive elongation of telomeres (Smith and de Lange, 2000). A PARP-deficient form of tankyrase-1 failed to affect TRF1 and did not alter telomere length dynamics, which is consistent with ADP-ribosylation of TRF1 being the main cause of altered telomere homeostasis. Recently, a new tankyrase-1 binding protein has been identified (Seimiya and Smith, 2002). TAB182, a 182-kDa protein has been shown to coimmunoprecipitate with tankyrase-1 from human cells and to serve as an acceptor of poly(ADP-ribosyl)ation by tankyrase-1 in vitro. Like TRF1, TAB182 binds to the ankyrin domain of tankyrase-1 (Seimiya and Smith, 2002).
Tankyrase-2, an extranuclear PARP, was originally described as a Golgi-associated protein also referred to as TNKL (Chi and Lodish, 2000; Lyons et al., 2001). Tankyrase-2 is ubiquitously expressed in all tested tissues. The enzyme has a unique N-terminal domain, but the rest of the enzyme (ankyrin repeat, sterile alpha motif, catalytic domain) shows 85% homology to tankyrase-1 at the amino acid level (Kaminker et al., 2001). During subcellular fractionation, tankyrase-2 associates with low-density microsome fraction (Lyons et al., 2001). Similarly to tankyrase-1, tankyrase-2 localizes predominantly to the perinuclear region in a pattern consistent with localization to the Golgi apparatus. Currently available antibodies, however, cannot differentiate between the two tankyrase enzymes. Therefore, upon the availability of specific tankyrase-1- and -2-specific antibodies, intracellular localization may need to be reevaluated. Both tankyrases interact with TRF1, as suggested by two-hybrid analysis and coimmunoprecipitation. In contrast to tankyrase-1, overexpression of tankyrase-2 triggered a 3-aminobenzamide-inhibitable necrotic-type cell death (Kaminker et al., 2001).
Tankyrase-3 has recently been identified in Gilbert de Murcia's laboratory (G. de Murcia, personal communication).
Using differential display, a novel inducible PARP enzyme termed TcPARP has been identified in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated cells (Ma et al., 2001). Expressed 2,3,7,8-tetrachlorodibenzo-p-dioxin-inducible PARP, a 75-kDa protein exhibited PARP activity toward histones and was found to be homologs to RM1 and TIL, which are proteins induced during long-term potentiation (memory formation) and in tumor-infiltrating lymphocytes, respectively.
Poirier's group has isolated a cDNA from PARP-1-deficient fibroblasts that encodes a 55-kDa protein identical with the catalytic domain of PARP-1 (Sallmann et al., 2000). Based on currently available data, the possibility that short PARP represents a splice variant of PARP-1 cannot be excluded.
This list of novel PARP enzymes is far from complete. Gilbert de Murcia's laboratory has recently cloned a total of 16 novel PARP cDNAs (G. de Murcia, personal communication), indicating that PARP research soon faces a stimulating challenge to determine the likely distinct or sometimes overlapping biological roles of these new PARP homologs.
C. Poly(ADP-Ribose) Catabolism: Poly(ADP-Ribose) Glycohydrolase
Poly(ADP-ribosylation) is a dynamic process because poly(ADP-ribose) polymer is rapidly degraded by poly(ADP-ribose) glycohydrolase and ADP ribosyl protein lyase. The half-life of the polymer is estimated to be less than 1 min, indicating a concerted activation of poly(ADP-ribose)-synthesizing and -degrading enzymes. Since its discovery by Miwa and Sugimura in 1971 (Miwa and Sugimura, 1971; Miwa et al., 1974), PARG has not been investigated nearly as intensively as PARP-1. This is mostly because of difficulties in obtaining pure PARG enzyme. The difficulty lies in the low cellular abundance of the enzyme and its sensitivity to proteolytic degradation during purification (Davidovic et al., 2001). PARG is capable of hydrolyzing both terminal ADP-ribose units from poly(ADP-ribose) polymers via exoglycosidic activity and of removing larger oligo(ADP-ribose) fragments via endoglycosidic cleavage (Brochu et al., 1994; Davidovic et al., 2001). Because theK m value of PARG is much lower for larger (ADP-ribose)n polymers than for smaller ones (Hatekayama et al., 1986), the enzyme probably removes and catabolizes bigger fragments first. PARG then switches to exoglycosidic mode and removes ADP-ribose units one by one. The proximal ADP-ribose moiety is removed from the acceptor proteins by ADP-ribosyl protein lyase (Oka et al., 1984). The high specific activity of PARG compensates for the low abundance of the enzyme. Nearly 90% inhibition of PARG activity is required (by heat shock) for cellular poly(ADP-ribose) accumulation (Jonsson et al., 1988a,b). The rat and bovine PARG cDNA-s cloned by Sugimura's (Shimokawa et al., 1999) and Jacobson's groups (Lin et al., 1997), respectively, encode 109- to 111-kDa proteins sharing no homology with other proteins other than a protein sequence from Caenorhabditis elegans that is likely to be the PARG enzyme of this organism. Expressed PARG formed stable dimers through leucin zipper-like dimerization domains even under SDS-polyacrylamide gel electrophoresis conditions. PARG contains both a nuclear localization signal (NLS) and a nuclear export signal, providing support for the idea that PARG may shuttle between the nucleus and the cytoplasm (Shimokawa et al., 1999). A PARG shuttle may serve regulatory functions and may also allow PARG to participate in the digestion of poly(ADP-ribose) synthesized by cytoplasmic PARP enzymes.
Contrary to the plethora of articles reporting cellular and in vivo effects of PARP inhibition, there are very few articles on the biological role of PARG. Tannin derivatives have been most frequently used to inhibit PARG in vitro. The heterogeneous composition and significant vendor-to-vendor and batch-to-batch variation of tannin represents a major obstacle in PARG pharmacology. Nonetheless, oenothein B, a macrocircular ellagitannin PARG inhibitor, has been shown to suppress mouse mammary tumor virus transcription and to activate tumor-suppressing macrophages (Aoki et al., 1995). Furthermore, gallotannin and nobotanin B have been shown by Swanson's group to protect murine astrocytes from oxidative injury (Ying and Swanson, 2000). Moreover, the same PARG inhibitors and the PARP inhibitor 3-aminobenzamide have been tested in parallel for their cytoprotective effect in hydrogen peroxide,N-methyl-d-aspartate (NMDA), and DNA alkylating agent-induced neuronal and astrocyte cell death model (Ying et al., 2001). Even though inhibition of PARG and PARP had opposing effects on poly(ADP-ribose) formation, both approaches provided remarkable protection to the cells (Ying et al., 2001). We have also observed the cytoprotective effect of gallotannin in oxidatively stressed A549 lung epithelial cells and HaCaT keratinocytes (L. Virag, manuscript in preparation). These findings may open new avenues for pharmacological interventions targeting poly(ADP-ribose) metabolism. However surprising it may sound, it now seems that inhibition of both the poly(ADP-ribose) synthesizing enzyme (PARP) and the catabolizing enzyme (PARG) has similar cytoprotective effect in oxidatively stressed cells. The likely solution for this paradox is that removal of inhibitory poly(ADP-ribose) residues by PARG from the automodification domain of PARP is required for PARP to maintain its active state (Fig.3.). The inhibition of PARG results in hyper-auto-poly(ADP-ribosylation) of PARP and inhibition of the enzyme. In addition, PARG activity seems necessary for the high poly(ADP-ribose) turnover resulting in NAD+depletion and cell death triggered by DNA-damaging stimuli. These intriguing new data raise several questions: Is PARG inhibition a viable strategy for the treatment of diseases (reperfusion injury, inflammation, shock) in which PARP inhibitors proved useful? How does PARG inhibition and nontransient poly(ADP-ribosylation) affect DNA repair? Are PARP-assisted transcription machineries differentially regulated by PARG and PARP? To address these issues, specific and potent PARG inhibitors as well as efficient molecular biology tools to overexpress or genetically delete PARG are required. Considering the intense effort in this field of research, it is likely that PARG-deficient mice will soon become available and will certainly accelerate PARG research.

The role of PARG in the reactivation of PARP. PARP activation leads to automodification of PARP, resulting in PARP inhibition. By removing poly(ADP-ribose) from PARP, PARG reactivates PARP and allows for continuous NAD turnover.
D. PARP in DNA Repair
The assumption that PARP-1 may be involved in DNA repair was born simultaneously with the identification of the enzyme as a DNA binding protein. Several studies using various pharmacological PARP inhibitors have concluded that PARP-1 plays a role in DNA repair (Burkle, 2001;Ziegler and Oei, 2001). It has been shown, for example, that the PARP inhibitor 3-aminobenzamide retarded the rejoining of DNA strand breaks and enhanced the frequencies of unscheduled DNA synthesis and sister chromatide exchanges in MNNG-treated Chinese hamster ovary and HeLa S3 cells (Park et al., 1983). Furthermore, PARP inhibition rendered cells more sensitive to cytotoxicity induced by DNA-damaging stimuli (Szabo, 2000). Later, using random mutagenesis, Berger's group generated cell lines having low PARP activity and showed that these mutants were hypersensitive to ionizing and UV irradiation, topoisomerase I inhibitors, and a series of different alkylating agents, including alkylsufonates, alkylnitrosoureas, and nitrosoguanidine (Chatterjee et al., 1989, 1990a,b). When molecular biological manipulation has allowed dominant negative inhibition, depletion, or genetic ablation of PARP-1 by overexpression of its DNA binding domain, by expression of antisense PARP-1 RNA, or by homologous recombination, respectively, a contribution of PARP-1 in DNA repair and maintenance of genomic integrity becomes apparent (Molinete et al., 1993; Stevnsner et al., 1994; Schreiber et al., 1995; Shall and de Murcia, 2000; Smulson et al., 2000). Many studies using pharmacological inhibitors of PARP have problems associated with limited selectivity or specificity of many of the compounds used. Thus, from the large number of publications, we restrict our current discussion to what we have learned from the PARP-1-deficient mice and cells derived from them. However, the results obtained from studies using PARP-deficient experimental systems usually do not distinguish between findings related to the physical absence of the enzyme (i.e., “scaffolding” functions) and the lack of PARP's catalytic activity (i.e., the “enzymatic” function).
PARP knockout mice generated in de Murcia's laboratory were highly sensitive to death induced by ionizing radiation or monofunctional alkylating agents (de Murcia et al., 1997). Furthermore, exposure to methylmethanesulfonate or N-methyl-N-nitrosourea of embryonic fibroblasts derived from PARP−/−mice but not from PARP+/+ mice exhibited a reduced rate of proliferation because of their cell-cycle block in G2/M (de Murcia et al., 1997; Trucco et al., 1998). PARP-deficient cells also exhibited genomic instability, as evidenced by an increased number of micronuclei (chromatin fragments indicating chromosomal damage) with or without methylmethanesulfonate treatment. Furthermore, PARP−/− fibroblasts exhibited slower rejoining of DNA breaks, as measured with use of the comet (single-cell gel electrophoresis) assay indicating deficient ligation. It has also been investigated whether the short-patch repair system responsible for the replacement of a single mutated nucleotide or the long-patch repair system capable of replacing 7 to 14 nucleotides is more affected by the absence of PARP-1. It was found that lysates from PARP-1-deficient fibroblasts had no long-patch repair activity, and their short-patch repair activity was also reduced by approximately 50%, as compared with PARP-1-proficient cell lysates (Dantzer et al., 2000). From these data, the conclusion can be drawn that PARP-1 contributes to the maintenance of genomic integrity and also enhances base-excision repair in irradiated or alkylating agent-treated cells. The involvement of PARP-1 in genomic surveillance is also indicated by the interaction of PARP-1 with other nick sensors such as DNA ligase III, adaptor factors such as XRCC1 (Masson et al., 1998), and DNA repair effectors such as DNA polymerase β and DNA ligase III, components of the base-excision repair complex. Through the zinc-finger domains or the breast cancer susceptibility protein C terminus (BRCT) motif of the automodification domain, PARP-1 physically associates with these proteins, as indicated by the two-hybrid system or coimmunoprecipitation. Regulation of the activity of these proteins by PARP-1 is carried out both via physical interaction and poly(ADP-ribosylation). The exact nature of the regulatory role of PARP-1 within the base-excision repair complex, however, requires further investigation.
E. PARP-1 in Cell Death
In the last decade, PARP-1 has become widely known among cell biologists as the “death substrate” (Tewari et al., 1995). Indeed, PARP-1 was one of the first identified substrates of caspases, the main executioners of apoptosis (Kaufmann et al., 1993). Therefore, a role for PARP-1 in the regulation of apoptosis has been suggested. Even though there are data in the literature pointing toward a possible role of PARP-1 in apoptosis, more convincing evidence suggests the involvement of PARP-1 in necrosis. Here, we summarize the current knowledge on the role PARP-1 plays in the two main pathways of cell death: apoptosis and necrosis. Most of the studies related to PARP and cell death are likely to pertain to the major PARP isoform, PARP-1. Studies related to the potential role of the other PARP isoforms in cell death are scarce; in one such recent study, nevertheless, it was shown that overexpression of tankyrase-2 is able to induce cell death in fibroblasts (Kaminker et al., 2001).
1. Apoptosis.
During apoptosis, caspase-7 and caspase-3 cleave PARP-1 into two fragments: p89 and p24 (Tewari et al., 1995;Germain et al., 1999). These proteases recognize a DEVD motif in the nuclear localization signal of PARP-1 (Lazebnik et al., 1994), and cleavage at this site separates the DNA binding domain from the catalytic domain, resulting in the inactivation of the enzyme. Cleavage fragments contribute to the suppression of PARP activity because p89 and p24 inhibit homoassociation and DNA binding of intact PARP-1, respectively (Kim et al., 2000a,b; D'Amours et al., 2001). The existence of this positive feedback loop in caspase-mediated PARP-1 inactivation suggests that blocking PARP-1 activation is vital for the proper function of the apoptotic machinery. According to this concept PARP cleavage aims at preventing the activation of PARP by the ensuing DNA fragmentation and thereby aims at preserving cellular energy for certain ATP-sensitive steps of apoptosis. Experimental evidence supporting this hypothesis was provided by Herceg and Wang (1999), showing that the expression of a caspase-uncleavable, modified version of PARP in TNF-α-treated PARP-1 knockout fibroblasts leads to NAD+ depletion and necrosis. Inhibition of PARP activity by 3-aminobenzamide blocked both NAD+depletion and cell death. Recent work indicates that PARP-1 cleavage during apoptosis is not simply required to prevent excessive depletion of NAD and ATP, but it is also necessary to release the human Ca2+- and Mg2+-dependent endonuclease (DNAS1L3) from poly(ADP-ribosyl)ation-mediated inhibition (Boulares et al., 2001). Although caspase-mediated PARP-1 cleavage is well documented, little is known about the possible cleavage of novel PARP enzymes and PARG. Recently PARP-2 has been shown to be cleaved by caspase-8, a caspase which was considered to be an initiator caspase, the proform of which associated with cell-surface death receptors. In ischemia-induced neuronal apoptosis, caspase-8 trans locates into the nucleus and cleaves PARP-2 at a LQMD sequence. PARP-2 cleavage, similar to PARP-1 cleavage, separates the DNA binding end-catalytic domains and inactivates the enzyme (Benchoua et al., 2002). In addition to PARP-1 and PARP-2, PARG also becomes cleaved at a relatively early stage of apoptosis (Affar et al., 2001). However, the biological role of the cleavage of PARP-2 and PARG has not yet been investigated in detail.
The question arises whether poly(ADP-ribosylation) by PARP-1 affects the apoptotic process. Because the caspase-cleaved form of PARP-1 is catalytically inactive, such an apoptosis-modifying effect could only be exerted in the early phase of apoptosis, i.e., before caspase activation occurs. Data obtained with the use of pharmacological PARP inhibitors ranged from inhibition (Tanaka et al., 1995b; Kuo et al., 1996; Shiokawa et al., 1997; Guo et al., 1998; Richardson et al., 1999) to a lack of effect (Watson et al., 1995) and augmentation of apoptosis (Ray et al., 1992; Ghibelli et al., 1995; Payne et al., 1998; Tentori et al., 1999, 2001a,b; Berry et al., 2000) depending on cell type, culture condition, and apoptosis inducers used. A transient burst of poly(ADP-ribosylation) has been observed in various models of apoptosis, such as in camptothecin-treated HL-60 cells, anti-Fas plus cycloheximide-treated 3T3-L1 cells, and anti-Fas-treated Jurkat cells (Rosenthal et al., 1997; Simbulan-Rosenthal et al., 1998). Depletion of PARP-1 by antisense RNA expression or genetic ablation of PARP-1 gene in PARP-1−/− fibroblasts blocked poly(ADP-ribosylation) and also inhibited Fas-induced apoptosis (Simbulan-Rosenthal et al., 1999a). These data indicate that PARP-1-mediated poly(ADP-ribosylation) of nuclear proteins is required for apoptosis. However, several lines of evidence support the idea that PARP-1 is dispensable for apoptosis. In addition to pharmacological data, experiments on various PARP-1-deficient cells proved the proficiency of these cells to undergo apoptosis. Soon after PARP-1 knockout mice became available, a comprehensive series of experiments compared the sensitivity of PARP+/+ and PARP−/− hepatocytes, thymocytes, and primary neurons with Fas-, TNF-α−, etoposide-, dexamethasone-, and ceramide-induced apoptosis and found no difference between the knockout and the wild-type cells (Leist et al., 1997b). Wang et al. (1997) also found that cells lacking PARP underwent apoptosis normally in response to treatment with anti-Fas, TNF-α, γ-irradiation, and dexamethasone. Our group reported no difference in the apoptotic response of thymocytes in response to dexamethasone or anti-Fas treatment. Normal development of PARP knock out mice also argues against an essential role of PARP-1 in apoptosis (Wang et al., 1995). From these data, PARP seems to be dispensible for most forms of apoptosis; however, PARP cleavage is vital for the appropriate function of the apoptotic machinery.
Recent data indicate that PARP-1 also plays a central role in a caspase-independent apoptosis pathway mediated by apoptosis-inducing factor (AIF) (Yu et al., 2002). Translocation of AIF from the mitochondria to the nucleus is dependent on PARP activation in neurons and fibroblasts treated with various DNA-damaging stimuli such as MNNG, NMDA, or hydrogen peroxide (Yu et al., 2002).
2. Necrosis.
Various stimuli can trigger both apoptotic and necrotic cell death. It is important to note that necrosis is not simply another type of cell death; it represents a more severe form of cell demise compared with apoptosis. Viewing cell death from the point of view of the tissue or organ in which cell death takes place, such a distinction makes sense. In addition to numerous biochemical and morphological differences between apoptosis and necrosis, probably the most distinctive feature of necrosis is the disintegration of the plasma membrane, as opposed to the compaction of apoptotic cells. Leakage of cell content from necrotic cells into the surrounding tissue may contribute to organ injury, whereas apoptotic cells are rapidly cleared from the tissues by macrophages. Using NMDA- or peroxynitrite-treated neurons, Lipton's and Nicotera's groups elegantly demonstrated that apoptosis and necrosis are at two ends of a continuum in which apoptosis is caused by mild stimuli and necrosis is triggered by severe stimuli (Bonfoco et al., 1995; Nicotera et al., 1999). Furthermore, it has also been suggested that both ATP and NAD+ are important determinants of the mode of cell death, especially in oxidatively injured cells (Coppola et al., 1995; Klaidman et al., 1996; Leist et al., 1997a, 1999a; Mukherjee et al., 1997; Lelli et al., 1998; Lieberthal et al., 1998; Nicotera et al., 1998; Ran et al., 1999; Crowley et al., 2000). From these observations, it was plausible to hypothesize that PARP as a NAD+-catabolizing enzyme may serve as a molecular switch between apoptosis and necrosis. The initial studies on the role of PARP and cell death were performed using pharmacological inhibitors of PARP (most frequently, 3-aminobenzamide and nicotinamide) and have been previously reviewed (Szabo and Dawson, 1998). These agents can have additional actions, such as acting as free-radical scavengers. More recent studies using cells from PARP knockout animals confirmed the role of the PARP pathway in oxidant-mediated cell injury. In the first such study, Heller and coworkers (Heller et al., 1995; Wang et al., 1995) observed that islets of the PARP−/−mice are resistant to NO and oxidant-related injury when compared with the response in islets of the wild-type mice. Similarly, we observed that pulmonary fibroblasts from the PARP−/−mice are protected from peroxynitrite-induced cell injury when compared with the fibroblasts of the corresponding wild-type animals (Szabo et al., 1998c). Furthermore, Eliasson et al. (1997) demonstrated protection by PARP−/− phenotype in brain slices exposed to various oxidants. Thus, the more definitive studies using PARP knockout cells have now fully confirmed the conclusions of the earlier pharmacological studies. With respect to the mode of cell death, the earlier studies provided some clues that it is the necrotic type (e.g., in the Heller study, lactate dehydrogenase release was blocked in PARP-deficient islets), but direct investigations have not been conducted to characterize the mode of cell death. In hydrogen peroxide-treated HT-29 epithelial cells, the inhibition of PARP by 3-aminobenzamide inhibited necrosis but not apoptosis (Watson et al., 1995). One year later, Palomba et al. (1996) found that inhibition by 3-aminobenzamide of hydrogen peroxide-induced necrosis in U937 myeloma cells was associated by increased apoptotic DNA fragmentation and cell blebbing. Later, our group provided evidence for the possible role of PARP-1 in switching default apoptosis to necrosis in oxidatively injured cells. In our experiments, we used thymocytes from PARP-1+/+ and PARP-1−/−mice and compared their responses to peroxynitrite and hydrogen peroxide (DNA-damaging stimuli) as well as dexamethasone and anti-Fas treatment (non-DNA-damaging agents). While nongenotoxic stimuli (dexamethasone and anti-Fas) triggered equal apoptotic responses, as evidenced by phosphatidylserine exposure, caspase activation, and DNA fragmentation, in wild-type and PARP-1-deficient thymocytes, marked differences could be observed with DNA-damaging oxidative agents. Low concentrations of peroxynitrite and hydrogen peroxide induced apoptosis in both PARP-1+/+ and PARP-1−/− cells, with 3-aminobenzamide having no effect on the responses. At higher concentrations of the oxidants, necrotic cell death occurred as indicated by propidium iodide uptake, and necrosis was accompanied by decreased output of the apoptotic parameters caspase activation and especially DNA fragmentation. Necrosis and decrease of apoptosis could be prevented by 3-aminobenzamide, indicating that PARP activation was responsible for the apoptosis-to-necrosis switch in severely damaged cells. Furthermore, PARP-1 knockout cells responded to oxidative challenge with a concentration-dependent apoptosis and showed no switch to necrosis. Using pharmacological PARP inhibitors, we found similar biphasic responses in other cell types, including lymphoma cells, pancreatic acinar cells (L. Virág and C. Szabo, unpublished data), and HaCaT keratinocytes (Szabo et al., 2001), and other groups also reported protection by PARP inhibition from necrotic but not from apoptotic cell death (Ha and Snyder, 1999; Filipovic et al., 1999; Palomba et al., 1999; Tentori et al., 2001a). Comparison of PARP+/+ and PARP−/−fibroblasts provided further support for the existence of PARP-1-mediated apoptosis-to-necrosis switch in oxidatively challenged cells (Ha and Snyder, 1999). Recently, Moroni et al. (2001) published a series of investigations demonstrating that PARP activation may serve as a cell-death switch in vivo in oxygen-glucose deprivation-based models of cerebral ischemia. A role of PARP activation in necrosis is also consistent with the fact that the inhibition or absence of PARP provides the most remarkable protection in disease models such as stroke, myocardial infarction, or mesenteric ischemia-reperfusion injury, which are characterized predominantly by necrotic-type cell death (Miesel et al., 1995; Schreiber et al., 1995). It is noteworthy here that, in addition to the process of NAD+depletion and the induction of mitochondrial dysfunction, part of the PARP overactivation-induced cell necrosis may be related to intracellular acidification: when PARP catabolizes NAD+, the “by-product” of the reaction is H+, which directly induces intracellular acidification, having direct consequences for cell viability (Affar et al., 2002). It is still a widely held view that necrosis is a futile process that cannot be influenced by pharmacological means (although apoptosis is the sophisticated process which is under the control of a complex cellular machinery and is amenable to pharmacological intervention). The above-listed observations—demonstrating protection against cell necrosis by inhibition or inactivation of PARP-1—prove that the necrotic process, indeed, is amenable to pharmacological interventions. In fact, the massive protection seen in many inflammatory models and in models of reperfusion injury in the absence of functional PARP (see below) may indicate that necrosis and not apoptosis is the probable predominant form of cell death and organ dysfunction in many diseases.
Moderate PARP activation may decrease cellular NAD+ content without being fatal to the cells. Such moderately compromised cellular energetics may cause cell dysfunction. It seems that pharmacological inhibition of PARP, by improving cellular energetics, may rescue cells from this dysfunctional stage and thereby can restore cell function. Examples of such a restorative effect were found in ex vivo experiments in endothelial cells producing high levels of endogenous oxidants during diabetes (Soriano et al., 2001b) and in intestinal epithelial cells from colitic guts (Jijon et al., 2000). PARP-mediated dysfunction of intestinal epithelial cells manifesting in increased permeability has also been demonstrated in monolayers of peroxynitrite-treated CaCo-2BBe cells (Kennedy et al., 1998; Forsythe et al., 2002). Moreover, stress-induced immunosuppression could also be suppressed by genetic ablation of PARP-1, indicating the involvement of PARP-1 in stress-induced immune-cell dysfunction (Drazen et al., 2001).
3. Complex Role of PARP-1 in DNA Damage-Induced Cell Death.
After more than 30 years since the discovery of PARP-1, a considerable controversy still exists over the role the enzyme plays in DNA-damage signaling and especially DNA damage-induced cell death. The two sides of the coin are represented by groups claiming that PARP-1 is an indispensable cellular survival factor and by other scientists viewing PARP as a perpetrator of cell death, as described earlier in this article (Fig. 4). This controversy mainly results from differences in experimental approaches with special regard to the use of different DNA-damaging stimuli (alkylating agents, ionizing radiation, or free radicals and oxidants) (Fig. 4) and different cytotoxicity assays measuring either apoptosis or necrosis. This issue now seems to be resolvable in a unifying concept (Fig.5). According to this concept (Fig. 5), cells exposed to DNA-damaging agents can enter three pathways determined by the intensity of stimulus: 1) PARP-1 activated by mild genotoxic stimuli facilitates DNA repair by signaling cell-cycle arrest and by interacting with DNA repair enzymes such as XRCC1 and DNA-dependent protein kinase. As a result, DNA damage is repaired, and cells survive without the risk of passing on mutated genes. 2) More severe DNA damage induces apoptotic cell death during which caspases, the main executor enzymes of the apoptotic process, inactivate PARP-1 by cleaving it into two fragments (p89 and p24). This pathway allows cells with irreparable DNA damage to become eliminated in a safe way. Cleavage of PARP is believed to aim at preventing the activation of PARP by the ensuing DNA fragmentation and thereby preventing cells from the pathological sequelae of the third route in which cells die by necrosis, a less controlled mechanism posing danger for bystander cells. 3) The third route is induced by extensive DNA breakage that is usually triggered by a massive degree of oxidative or nitrosative stress (hydroxyl radical, peroxynitrite, nitroxyl anion). The overactivation of PARP depletes the cellular stores of its substrate NAD+ and consequently ATP. The severely compromised cellular energetic state inhibits the apoptotic cell death process to proceed, because many steps of apoptosis are known to depend on ATP (Kass et al., 1996; Richter et al., 1996; Stefanelli et al., 1997; Ferrari et al., 1998; Feldenberg et al., 1999; Chalmers-Redman et al., 1999; Leist et al., 1999b). PARP activation can quickly take predominance over caspase activation-mediated apoptosis because the very rapid activation of PARP that occurs within minutes after DNA damage, as opposed to the slower kinetics of caspase activation. Pharmacological PARP inhibition or the absence of PARP in PARP-deficient mice preserves cellular ATP and NAD+ pools in oxidatively stressed cells and thereby allows them to function normally or, if the apoptotic process has initiated, to use the apoptotic machinery and die by apoptosis instead of by necrosis. We and others have shown that inhibiting or deleting PARP—in parallel with decreased necrosis—results in a dramatic increase in the output of apoptotic parameters (caspase activity, DNA fragmentation, phosphatidylserine exposure), providing convincing support for this scenario (Palomba et al., 1996; Virag et al., 1998b).

The Yin-Yang of PARP activation. The death-promoting and the cytoprotective effects of poly(ADP-ribosylation) represent two seemingly opposing faces (the Yin and Yang) of PARP. Oxidative stress-induced DNA breakage causes a high level of PARP activation, leading to the depletion of NAD+and ATP and consequently to necrotic cell death. On the other hand, poly(ADP-ribosylation) facilitates DNA repair in cells subjected to treatment with alkylation agents or ionizing radiation.
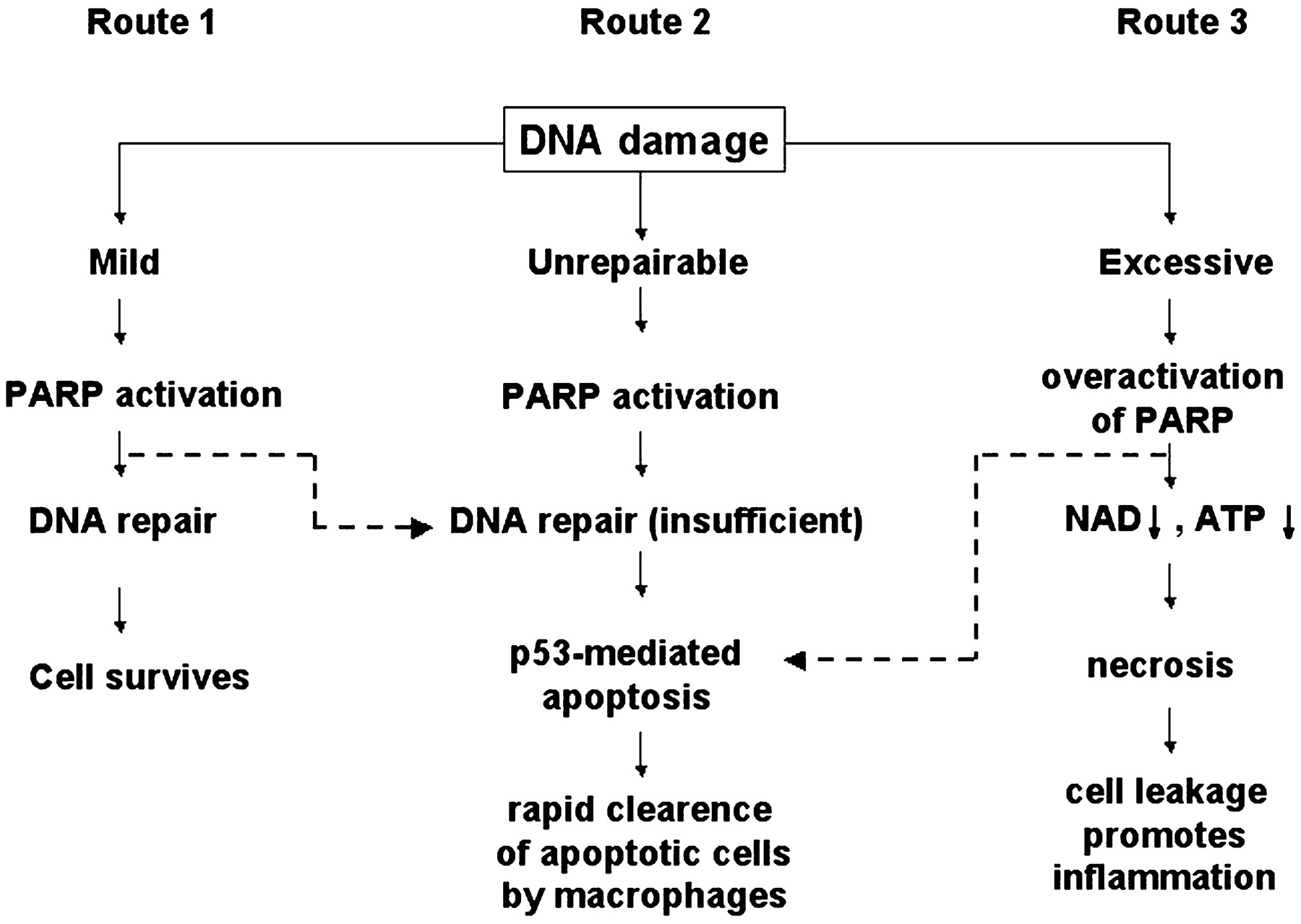
The intensity of DNA-damaging stimuli determines the fate of cells: survival, apoptosis, or necrosis. Depending on the intensity of the stimulus, genotoxic agents can trigger three different pathways. In the case of mild DNA damage, poly(ADP-ribosylation) facilitates DNA repair and thus survival (route 1). More severe genotoxic stimuli activate the p53-dependent (or possibly independent) apoptotic pathway (route 2). The most severe DNA damage may cause excessive PARP activation, depleting cellular NAD+/ATP stores. NAD+/ATP depletion blocks apoptosis and results in necrosis (route 3). The inhibition of PARP in cells entering route 1 inhibits repair and thus diverts cells to route 2 (broken arrow). The inhibition of PARP in cells entering route 3 preserves cellular energy stores and thus enables apoptotic machinery to operate (broken arrow).
According to the above scheme, cells with mild repairable DNA damage as well as cells with severely damaged cells are diverted by PARP inhibition to a common pathway (route 2), which means the elimination of cells by apoptosis (Fig. 5.). When repair of minor DNA damage is halted by PARP inhibition, DNA damage will trigger the apoptotic machinery via p53. On the other hand, inhibition of PARP in severely injured cells (which would normally undergo necrosis) preserves ATP for the apoptotic system, and cell death occurs via apoptosis instead of necrosis. Besides the severity of DNA damage, the type of DNA injury may also be relevant, because we have never observed sensitization by PARP inhibition to low-level (subapoptotic degree) oxidative challenge. Moreover, various free radicals and oxidants, but not alkylating agents and other DNA-damaging stimuli, target mitochondrial enzymes and the electron transport chain, exerting a direct inhibitory effect on central elements of cellular energy homeostasis (Radi et al., 1994;Lizasoain et al., 1996; Stachowiak et al., 1998; Richter et al., 1999;Boczkowski et al., 2001). Therefore, the ability of oxidatively injured cells to compensate for PARP-mediated energetic crisis is also compromised (Leist et al., 1999a). Moreover, PARP activation, probably via NAD+ and ATP depletion, also contributes to mitochondrial dysfunction, leading to the rapid deterioration of mitochondrial integrity (Virag et al., 1998a; Chong et al., 2002; Yu et al., 2002). Comprehending this complex role of PARP in the cell death process is important to understanding how PARP inhibition can protect from or enhance cytotoxicity depending on the nature and severity of DNA damage. Nevertheless, the exact reason of why PARP activation promotes cell death in certain systems or protects from cytotoxicity requires further investigation. The nature of genotoxic stimuli (oxidative stress, alkylating agents, ionizing radiation, etc.) and cellular metabolism are usually considered to be key factors in determining the role of poly(ADP-ribosylation) in the cell death process. Our unpublished observation that in thymocytes and A549 lung epithelial cells,N-methyl-N′nitro-N-nitrosoguanidine-induced cytotoxicity and oxidative stress-induced cytotoxicity are both mediated by PARP activation supports the idea that regardless of the nature of DNA-damaging noxa, PARP overactivation leads to necrotic cell death. Moreover, cell type-specific differences in cellular metabolism may be the key determinants of sensitivity to genotoxic stimuli.
F. PARP-1 in the Regulation of Cell Proliferation and Differentiation
To fulfill specific tasks and to function as specialized building blocks of tissues, cells need to undergo a series of proliferative steps during which they gain new functions and lose others. This strictly controlled process requires concerted gene activation and repression and results in differentiation into specialized cells functioning as hepatocytes, neurons, renal tubular cells, and so forth. Furthermore, many fully differentiated cell types such as lymphocytes, fibroblasts, and hepatocytes retain the ability to proliferate, such as in the course of immune response, wound healing, or liver regeneration, respectively. Moreover, after DNA damage, it is of primary importance to stop replications at certain check points to allow for the repair of DNA damage. From our current knowledge of PARP function, it is now widely accepted that PARP-1 is involved in the regulation of DNA replication, differentiation, and gene expression.
Involvement of PARP-1 in the regulation of replication is supported by observations that poly(ADP-ribose) metabolism is accelerated in the nuclei of proliferating cells (Tanuma et al., 1978; Kanai et al., 1981;Leduc et al., 1988; Bakondi et al., 2002a). Several lines of evidence suggest that PARP-1 is part of the MRC (Simbulan-Rosenthal et al., 1996). PARP-1 copurifies with DNA polymerase α and δ, DNA primase, DNA helicase, DNA ligase, topoisomerases I and II, and key components of MRC (Simbulan-Rosenthal et al., 1996; Dantzer et al., 1998; Bauer et al., 2001). Furthermore, several centromere proteins (Saxena et al., 2002) and replication factors such as DNA polymerase α, topoisomerase I and II, and proliferating cell nuclear antigen have been shown to be poly(ADP-ribosylated) (Simbulan-Rosenthal et al., 1996). Moreover, poly(ADP-ribosylation) of histones was also proposed to facilitate the assembly and deposition of histone complexes on DNA during replication (Boulikas, 1990). Nonetheless, the exact role of PARP-1 in the regulation of replication is still controversial. Inhibition of PARP by pharmacological or molecular biological means (anti-PARP-1 antisense, knockout cells, dominant-negative PARP inhibition by overexpression of the DNA binding domain of PARP) has been shown to inhibit replication, cell proliferation, and differentiation in various experimental models (D'Amours et al., 1999). However, PARP-1 has also been proposed to be a negative regulator for the initiation of DNA replication (Eki, 1994).
Given that replication and differentiation are closely coupled processes, the above-mentioned experimental data may provide rationale for a differentiation-modifying effect of PARP. Indeed, inhibition of PARP has been shown to interfere with differentiation in various cellular models. Some myeloid leukemia cell lines can be induced to undergo differentiation toward the monocyte/macrophage or neutrophil granulocyte linage. In NB4 acute promyelocytic leukemia and HL-60 acute myelocytic leukemia cells, PARP levels were dramatically modulated during monocyte/macrophage and neutrophilic differentiation (Bhatia et al., 1995). PARP inhibitors (5-methylnicotinamide, 3-methoxybenzamide, and 3-aminobenzamide) were found to inhibit differentiation of human granulocyte-macrophage progenitor cells to the macrophage lineage (Francis et al., 1983). Differentiation to the neutrophil-granulocyte lineage was much less affected (Francis et al., 1983). In other studies, overexpression of PARP arrested NB4 cells and blocked alltrans-retinoic acid-induced terminal neutrophilic differentiation (Bhatia et al., 1996). Furthermore, plasmacytic differentiation of Daudi lymphoma cells was impaired in the presence of PARP inhibitors (Exley et al., 1987). Importance of cell type-specific differences is also underlined by observations that benzamide PARP inhibitors induced melanogenesis and differentiation of melanoma cells (Durkacz et al., 1992). Poly(ADP-ribosylation) has also been implicated in erythroid differentiation (Rastl and Swetly, 1978; Morioka et al., 1979; Terada et al., 1979; Sugiura et al., 1984), chicken limb bud mesenchymal cell differentiation (Nishio et al., 1983; Cherney et al., 1985), and trophoblastic cell differentiation during tumorigenesis (Masutani et al., 2001).
G. PARP in the Regulation of Gene Expression
A possible role of poly(ADP-ribosylation) in the regulation of transcription has been indicated by findings reporting frequent association of PARP with transcriptionally active regions of chromatin (de Murcia et al., 1986; Lindahl et al., 1995). Furthermore, suppression of inducible-protein synthesis by PARP inhibitors has also been reported. For example, Yamada et al. (1990a) found that in pancreatic islet cells, nicotinamide and 3-aminobenzamide attenuated IFN-γ- and TNF-α-induced expression of class II but not class I major histocompatibility molecules. Similar results have also been reported in human thyroid cells and human astrocytes (Hiromatsu et al., 1992; Taniguchi et al., 1993; Qu et al., 1994). Moreover, inhibition of PARP by nicotinamide, 3-methoxybenzamide, and 3-aminobenzamide or 5-iodo-6-amino-1,2-benzopyrone (INH2BP) has been shown to inhibit cytokine-induced iNOS expression in various cell types (Hauschildt et al., 1991, 1992; Pellat-Deceunynck et al., 1994; Szabo et al., 1998). Furthermore, treatment of interleukin-1 β-stimulated rabbit synovial fibroblasts with 3-aminobenzamide resulted in reduced collagenase synthesis, indicating the involvement of PARP in the regulation of collagenase production (Ehrlich et al., 1995). Later, the role of PARP as transcriptional regulator was confirmed with PARP-deficient cells. Our group showed defective iNOS expression both at the protein and at the mRNA level in bacterial lipopolysaccharide (LPS)- and IFN-γ -stimulated PARP-1−/−fibroblasts compared with wild-type cells (Szabo et al., 1998). Furthermore, PARP inhibition or inactivation reduces the expression of ICAM-1, P-selectin, and E-selectin and of mucosal addressin cell adhesion molecule-1 in cytokine-stimulated human umbilical vein endothelial cells (Zingarelli et al., 1998; Oshima et al., 2001; Sharp et al., 2001). Moreover, decreased expression of these adhesion molecules has also been found after reperfusion injury in the hearts of PARP-1-deficient mice compared with their wild-type counterparts (Zingarelli et al., 1998).
The question arises as to how PARP regulates transcription. One component of the transcription-regulating activity of PARP may be the regulation of chromatin structure and function. Poly(ADP-ribosylation) confers negative charge to histones resulting in electrostatic repulsion between DNA and histones. Loosening histone-DNA interactions may render DNA regions more accessible to the transcriptional machinery and thus may enhance transcription. Indeed, it was reported that basal PARP activity regulates histone shuttling and nucleosomal unfolding (Althaus et al., 1994).
Meisterernst et al. (1997) provided further molecular details to our understanding of how PARP regulates transcription. Their work identified PARP-1 as a functional component of the positive cofactor-1 activity. PARP enhanced transcription by acting during preinitiation complex formation, but it did so at a step after the binding of transcription factor IID. This transcriptional activation was independent of DNA damage and required the amino-terminal DNA binding domain but not the carboxyl-terminal catalytic region (Meisterernst et al., 1997). The coactivator function of PARP was suppressed by NAD+, probably as a result of auto-ADP-ribosylation. These results supported a model in which the binding of PARP-1 to DNA and members of the transcription complex facilitates transcription, whereas catalytic activity of PARP has a transcription-inhibitory effect.
Another important milestone in establishing PARP as a transcriptional regulator was a report from de Murcia's laboratory (Oliver et al., 1999). Given the known anti-inflammatory and transcription inhibitory effect of PARP inhibition, they hypothesized a possible interaction between PARP-1 and NF-κB, a key transcription factor regulating the expression of several elements of inflammation such as cytokines, chemokines, adhesion molecules, and inflammatory mediators (e.g., iNOS, and the inducible form of cyclooxygenase). They showed that PARP-1-deficient cells were defective in NF-κB-dependent transcription activation, but not in its nuclear translocation, in response to TNF-α (Oliver et al., 1999). Treating mice with LPS resulted in the rapid activation of NF-κB in macrophages from PARP-1+/+ but not from PARP-1−/− mice. PARP-1-deficient mice were extremely resistant to LPS-induced endotoxic shock (Oliver et al., 1999). The molecular basis for this resistance relied on an almost complete abrogation of NF-κB-dependent accumulation of TNF-α in the serum and a down-regulation of iNOS, leading to decreased NO synthesis.
Recent studies attempted to delineate the relative importance of PARP catalytic activity versus PARP as a structural protein in its stimulatory role on NF-κB activation, and they yielded contrasting results. For instance, Hassa et al. (2001) demonstrated that a PARP-1 mutant lacking enzymatic and DNA binding activity interacted comparably with the wild-type PARP-1 with p65 or p50, concluding that the enzymatic activity of the enzyme is not essential for its interaction with NF-κB. In contrast, Chang and Alvarez-Gonzalez (2001) concluded that NF-κB p50 DNA binding was dependent on the presence of NAD+; DNA binding by NF-κB p50 was not efficient in the absence of NAD+ and was blocked in the presence of 3-aminobenzamide, allowing for the conclusion that NF-κB p50 DNA binding is protein-poly(ADP-ribosyl)ation-dependent. It is possible that these interactions are dependent on the cell type, the model system, and the nature of the stimulus used. Using the hydrochloride salt ofN-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide (designated PJ34), a potent and specific PARP inhibitor, a suppression of NF-κB-mediated gene expression was found in immunostimulated macrophages (Jagtap et al., 2002), but no alterations in NF-κB activation were seen in endothelial cells stimulated in the presence of high extracellular glucose concentration (Soriano et al., 2001c).
The identification of genes, the expression of which is regulated by PARP, has also been carried out using DNA chip technology. In wild-type and PARP-deficient fibroblasts, 91 of 11,000 genes were found to be differentially expressed (Simbulan-Rosenthal et al., 2000), suggesting a role for PARP-1 as a basal transcriptional regulator. This technology will hopefully be used for the systematic delineation of genes affected by PARP in stimulated cells and also in vivo in various forms of inflammation.
II. Pharmacological Inhibition of PARP
The endogenous inhibitor of PARP, nicotinamide, and the compound 3-aminobenzamide have long served as “benchmark” inhibitors of PARP, i.e., experimental agents suitable for laboratory investigations. These compounds inhibit the enzyme with a low potency, have limited cell uptake and cellular residence time, and exert nonspecific effects, for example, antioxidants (Wilson et al., 1984; Cantoni et al., 1987;Farber et al., 1990; Szabo et al., 1998c). More recently, several other classes of more potent and selective PARP inhibitors have been synthesized. Most PARP inhibitor compounds fall into the categories of monoaryl amides and bi-, tri-, or tetracyclic lactams. A common structural feature for these inhibitors is a carboxamide attached to an aromatic ring or the carbamoyl group built in a polyaromatic heterocyclic skeleton to form a fused aromatic lactam or imide. Most PARP inhibitors act as competitive inhibitors of the enzyme, i.e., the inhibitors block NAD+ binding to the catalytic domain of the enzyme, although some benzamides have also been shown to exert additional effects, such as inhibition of the binding of PARP to DNA (McLick et al., 1987). With the exception of some preliminary studies comparing the inhibitory effect of phenanthridinones on PARP-1 versus PARP-2 (Perkins et al., 2001), the issue of isoform selectivity has not yet been explored in detail, although considering the highly conserved active center of PARPs, it is likely that potent competitive PARP inhibitors will inhibit the catalytic activity of all PARP isoforms.
In 1992, Banasik and colleagues at the Department of Clinical Science and Laboratory Medicine, Kyoto University Faculty of Medicine, Japan, conducted what was at the time considered a large-scale screening of known small molecules on the isolated PARP enzyme. This screening yielded many interesting lead structures that subsequently were the subjects of extensive structure-activity optimization (Banasik et al., 1992). Constraining the monoaryl amide compounds by the formation of lactam-generated bicyclic compounds, the two-ring PARP inhibitors were found superior in potency and specificity over the monoaryl amide series. Systemically designed constrained 3-aminobenzamide analogs have been developed by using nicotinamide or 3-aminobenzamide as a template (Griffin et al., 1995; Watson et al., 1998). The amide group of nicotinamide or 3-aminobenzamide is free to rotate relative to the plane of the aromatic ring. Only certain orientation of the amide group with respect to the nitrogen of the pyridine ring of nicotinamide or the substitution at the 3-position of benzamide might be accommodated for PARP inhibition. The compounds 3,4-dihydro-5-methyl-isoquinolin-1(2H)-one and benzoxazole-4-carboxamide are examples of this approach (Griffin et al., 1995, 1996, 1998). Dihydroisoquinolin-1(2H)-nones, 1,6-naphthyridine-5(6H)-ones, quinazolin-4(3H)-ones, thieno[3,4-c]pyridin-4(5H)ones and thieno[3,4-d]pyrimidin-4(3H)ones, 1,5-dihydroxyisoquinoline, and 2-methyl-quinazolin-4[3H]-one are also potent inhibitors of PARP (Yoshida et al., 1991; Watson et al., 1998; White et al., 2000).
Three or more ring structure PARP inhibitors have also been identified. 1,8-Napthalimide derivatives and (5H)-phenanthridin-6-ones are representative of the tricyclic family (Banasik et al., 1992; Watson et al., 1998), with recent modifications on the latter class yielding many potent compounds (Soriano et al., 2001c; Li et al., 2001; Jagtap et al., 2002), including PJ34, a potent, water-soluble, orally bioavailable compound with marked in vivo activities (see also below). An inherent disadvantage for these planar heteroaromatic compounds is the poor solubility in water and many organic solvents. Certain tetracyclic lactams have also been identified as potent PARP inhibitors. A member of this latter class of compounds, 1,11b-dihydro-[2H]benzopyrano [4,3,2-de]isoquinolin-3-one (GPI 6150), inhibits PARP in vitro with aK i of 60 nM and demonstrates efficacy in rodent models of focal cerebral ischemia, traumatic brain injury, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced damage to dopaminergic neurons, regional myocardial ischemia, streptozotocin-induced diabetes, septic shock, and arthritis (Zhang et al., 2000; Mazzon et al., 2001). According to Zhang and Li (2000), a common structural feature of several classes of PARP inhibitors is either the presence of a carboxamide or an imide group built in a polyaromatic heterocyclic skeleton or a carbamoyl group attached to an aromatic ring. The oxygen atom from this carbonyl group seems to serve as a hydrogen acceptor, and the hydrogen atom from the amide or imide group serves as a proton donor in the hydrogen-bond interaction with the enzyme (Zhang and Li, 2000). Consensus structural requirements for PARP inhibitors acting at this nicotinamide-binding site include the following: 1) amide or lactam functionality is essential for effective interaction with the binding pocket; 2) an NH proton of this amide or lactam functionality should be conserved for effective bonding; 3) an amide group attached to an aromatic ring or a lactam group fused to an aromatic ring has better inhibition than an amide group attached to a nonaromatic ring or a lactam group fused to a nonaromatic ring; 4) optimal cis-configuration of the amide in the aromatic plane is required for maximal inhibitory activity; and 5) constraining mono-aryl carboxamide into heteropolycyclic lactams usually increases potency (Zhang and Li, 2000). Recently, the structural basis responsible for PARP inhibition has been carried out in a computational study using a docking approach into the crystallographic structure of the catalytic domain of PARP via the AutoDock program (version 2.4; The Scripps Research Institute, La Jolla, CA) and using and comparing 46 inhibitors available through the literature. These and related data may become useful for the design of new selective and potent PARP inhibitors (Costantino et al., 2001).
The most potent compounds from the recent bi- and tricyclic structures can be characterized by low-micromolar to mid-nanomolar inhibitory potencies in whole-cell-based assays and by effective inhibition of PARP and effective biological effects in the low milligram-per-kilogram dosing range. For example, PJ34 and related compounds inhibit PARP activation in whole-cell-based assays in the concentration range of 10 nM to 1 μM, with an EC50 in the 100- to 300-nM range, and they exert in vivo anti-inflammatory and anti-reperfusion actions in the dose range of 3 to 30 mg/kg (Mabley et al., 2001a;Soriano et al., 2001b,c; Jagtap et al., 2002). Because PARP-1 inhibition is an active and highly competitive area of investigation, it is likely that the most potent and effective compounds (i.e., the likely candidates for drug development) are not yet available in the scientific literature but rather may ultimately emerge in the various databases of published patents and pending patent applications. The published scientific and patent literature has recently been overviewed by Cosi (2002).
In addition to selective, potent enzymatic inhibition of PARP, several additional approaches have been described to inhibit the cellular activity of PARP in cells or in experimental animals. Somewhat surprisingly, the initial steps of oxidant-induced DNA single-strand breakage and PARP activation concern a step that involves the mobilization of intracellular calcium. Thus inhibition of intracellular calcium mobilization protects against oxidant-induced PARP activation, NAD+ depletion, and cell necrosis, as demonstrated in thymocytes (Virag et al., 1999) and in intestinal epithelial cells (Karczewski et al., 1999). Similar to calcium chelators, intracellular zinc chelators have been shown to protect against oxidant-mediated PARP activation and cell necrosis (Virag and Szabo, 1999). As mentioned earlier, intracellular purines (inosine, hypoxanthine), in addition to a variety of effects, also exert biological actions as inhibitors of PARP (Virag and Szabo, 2001). Calcium chelation, zinc chelation, and purines have been shown to exert a variety of cytoprotective and anti-inflammatory effects in experimental models in vitro and in vivo. It remains to be determined whether and to what extent PARP inhibition contributes to these beneficial effects.
III. Beneficial Effects of PARP Inhibition in Various Pathophysiological States
A. Activation of PARP in Pathophysiological Conditions
Multiple lines of evidence demonstrate that PARP becomes rapidly activated in various pathophysiological conditions, and its activation is prolonged and sustained. For example, direct detection of poly(ADP-ribose) polymer accumulation has demonstrated the activation of PARP in stroke induced by middle cerebral artery occlusion and reperfusion (Endres et al., 1998a) and in the heart after myocardial infarction and heart transplantation (Liaudet et al., 2001a; Faro et al., 2002; Fiorillo et al., 2002; Szabo et al., 2002). Similarly, PARP activation has been demonstrated in the gut, heart, and lung in hemorrhagic and septic shock (Liaudet et al., 2001a; Watts et al., 2001; Goldfarb et al., 2002; Jagtap et al., 2002; Soriano et al., 2002), in the lung of mice subjected to a model of acute respiratory distress syndrome (Liaudet et al., 2001a) as well as in the heart and blood vessels of diabetic animals (Soriano et al., 2001c; Pacher et al., 2002b). Although the trigger of PARP activation in vivo is difficult to delineate, from in vitro data, we can assume that the proximal initiator of PARP activation is DNA single-strand breakage, which can be induced by a variety of environmental stimuli and free radicals/oxidants, most notably hydroxyl radical, peroxynitrite, and nitroxyl anion (Table 1). In response to oxidative stress, DNA damage occurs; PARP becomes activated and, using NAD+ as a substrate, catalyzes the building of homopolymers of ADP ribose units, thereby triggering cells and organ dysfunction, which can culminate in full-fledged necrosis, as described above.
Pathophysiologically relevant sources of oxidant and free radical species capable of PARP activation
DNA single-strand breakage is an obligatory trigger for the activation of PARP. Peroxynitrite is a labile, toxic oxidant species produced from the reaction of superoxide and NO (Beckman et al., 1990; Szabo, 1996). This species, as well as the hydroxyl radical, are the key pathophysiologically relevant triggers of DNA single-strand breakage (Schraufstatter et al., 1986a, 1987). Moreover, nitroxyl anion, a reactive species derived from nitric oxide, is a potent activator of DNA single-strand breakage and PARP activation in vitro (Chazotte-Aubert et al., 1999; Bai et al., 2001) (Table 1).
Approximately 10 or more years ago, it was generally assumed that triggers of DNA single-strand breakage are restricted to severe environmental toxic agents (e.g., genotoxic or cytotoxic drugs) or various forms of ionizing radiation (Gu et al., 1995; Lazebnik et al., 1995). The research into the potential role of PARP in pathophysiological processes gained a new momentum in the mid-1990s by studies linking the formation of NO—an endogenously produced, reactive free-radical species produced from l-arginine by a family of enzymes termed NO synthases—to DNA single-strand breakage and PARP activation, with subsequent energetic changes in the cell (Radons et al., 1994; Zhang et al., 1994). Subsequent studies clarified that the actual trigger of DNA single-strand breakage is peroxynitrite, rather than NO (Szabo et al., 1996a): NO donors, in the absence of oxidative stress, are unable to induce DNA single-strand breakage, even at high concentrations. The identification of peroxynitrite as an important mediator of the cellular damage in various forms of inflammation stimulated significant interest into the role of the PARP-related suicide pathway in various pathophysiological conditions. Endogenous production of peroxynitrite and other oxidants has been shown to lead to DNA single-strand breakage and PARP activation. For example, in immunostimulated macrophages and smooth muscle cells, which simultaneously produce NO and superoxide and thus peroxynitrite from endogenous sources (Ischiropoulos et al., 1992; Tewari et al., 1995;Zingarelli et al., 1996a), DNA single-strand breakage has been demonstrated, and the time course of the strand breakage was shown to parallel the time course of NO and peroxynitrite production (Zingarelli et al., 1996a). Similarly, in brain slices, activation of NMDA receptors (a trigger for enhanced NO, superoxide, and peroxynitrite production) led to peroxynitrite-mediated DNA single-strand breakage and PARP-related cell injury (Zhang et al., 1994; Snyder, 1996). In a recent study using coculture of activated macrophages and hepatocytes, it was concluded that activated macrophage-derived NO and its oxidative metabolite, peroxynitrite, play key roles in hepatocyte injury during inflammation and cause subsequent DNA damage (including a significant degree of DNA single-strand breakage, but also other types of DNA base modifications) in surviving hepatocytes (Watanabe et al., 2001). Similarly, the ability of activated neutrophils to induce DNA single-strand breakage in neighboring cells is well documented (Shacter et al., 1988).
Recent work indicates that in intact mammalian cells, the process of DNA single-strand breakage by peroxynitrite may not be a direct result of peroxynitrite interacting with nuclear DNA, but it may also be related, at least in part, to a cascade involving the endogenous production of oxidants from the mitochondria and other cellular sources. For instance, in thymocytes exposed to peroxynitrite, there is a time-dependent gradual increase in mitochondria-derived reactive oxygen species generation (Virag et al., 1998a). In a study in human hepatocytes, there is a persistent and marked increase in DNA damage after treatment with NO or peroxynitrite generators that seems to come from the disruption of electron transport in the mitochondria (D'Ambrosio et al., 2001). Cantoni et al. (1987) proposed that peroxynitrite mediates the inhibition of mitochondrial complex III and, under these conditions, electrons are directly transferred from ubisemiquinone to molecular oxygen. Hydrogen peroxide is produced by the dismutation of superoxides, and this process was proposed to be the actual species mediating the peroxynitrite-dependent DNA cleavage (Guidarelli et al., 2000). As discussed above, calcium- and zinc-dependent (possibly mitochondrial) steps may also be important in the processes triggering peroxynitrite-induced DNA single-strand breakage (Karczewski et al., 1999; Virag et al., 1999).
B. PARP Activation and Cell Necrosis: Implications for Pathophysiology
Cochrane and colleagues have investigated in detail the time course of PARP activation and compared and contrasted its time course in relation to the time course of various other free radical-induced cytotoxic processes (Schraufstatter et al., 1986a, 1987, 1988). Various cell types were exposed to oxidants that are generated from stimulated leukocytes, including H2O2, superoxide, and hypochlorous acid (HOCl). The target cells used were P388D1 murine macrophage-like tumor cells, human peripheral lymphocytes, GM1380 human fibroblasts, and rabbit alveolar macrophages. In this experimental system, cell lysis could only be prevented when catalase was added within the first 30 to 40 min of H2O2 exposure, indicating that early metabolic changes determined the fate of the cell. Within seconds after the addition of H2O2 to the cells, activation of the hexose monophosphate shunt was observed, which is indicative of increased glutathione cycle activity. At the same time, DNA strand breaks (determined by an alkaline unwinding technique) were detected. The DNA breakage resulted in the rapid activation of PARP (within minutes after the addition of H2O2). At the same time, ATP and NAD+ concentrations decreased and nicotinamide accumulated extracellularly. Approximately 15 min after oxidant exposure, free intracellular Ca2+concentrations, as determined by Quin-2 fluorescence, started to increase because of the release of calcium from intracellular store. These findings collectively indicated the rapid activation and central role of PARP in the pathogenesis of oxidant-induced cell injury. The above-described changes eventually culminate in the stage of cell dysfunction and, ultimately, in necrosis. When this happens on the scale of an organ (e.g., during the ischemia and reperfusion of the brain or the heart), necrosis on a large scale leads to the loss of organ function and ultimately to death. Physicians have a long history of following the various plasma markers of cell necrosis using clinical tests. For instance, the measurement of troponin-C or creatine kinase (intracellular enzymes that spill into the extracellular space during necrosis) plasma levels in the blood of patients with myocardial infarction has long been used in the diagnosis and follow-up of myocardial infarction. Similarly, the measurement of plasma levels of the so-called “liver enzymes” (alanine aminotransferase and aspartate aminotransferase) in the plasma correlates with the degree of liver failure, i.e., the proportion of leaky and necrotic hepatocytes in the patient (Braunwals et al., 2001).
A good example of the transition from normal to dysfunctional and ultimately necrotic cell is the intestinal epithelial cell. Several studies have investigated the role of PARP in intestinal epithelial barrier function, an active process that is highly dependent on cellular ATP concentration. In vitro exposure of human CaCo-2BBe enterocyte cell monolayers to peroxynitrite rapidly induced DNA strand breaks and triggered an energy-consuming pathway catalyzed by PARP (Kennedy et al., 1998). The consequent reduction of cellular stores of ATP and NAD+ is associated with the development of hyperpermeability of the epithelial monolayer to a fluorescent anionic tracer. Pharmacological inhibition of PARP activity exerts no effect on the development of peroxynitrite-induced DNA single-strand breaks, but it attenuates the decrease in intracellular stores of NAD+ and ATP and the functional loss of intestinal barrier function. Ultimately, this PARP-dependent epithelial dysfunction in circulatory shock or in colitis leads to intestinal hyperpermeability (Liaudet et al., 2000a,b) and increased bacterial translocation through the gut (Taner et al., 2001), thereby further exacerbating the disease condition. However, it seems that the oxidatively damaged intestinal epithelial cells are not in a state of irreversible death. For instance, Jijon and colleagues (2000), using gut preparations from colitic mice, demonstrated that in vitro incubation of the colitic gut segments with the PARP inhibitor 3-aminobenzamide restored its normal permeability values. These epithelial cells were probably in a state of energetic suppression and cell dysfunction, but they had not yet reached the stage of irreversible necrosis.
An additional trigger of PARP activation is activated complement. In mesenteric lymphocytes exposed to sub-lethal concentrations of activated complement (present in zymosan-activated serum, ZAS) the concentration of lymphocyte ATP was dramatically decreased, and the extent of cell death could be significantly reduced by the addition of inhibitors of PARP (Bacurau et al., 2002). Similarly, activated complement-induced vascular injury and local and systemic inflammatory responses can be reduced by pharmacological inhibition of PARP (Cuzzocrea et al., 1997b, 1999; Szabó et al., 1997b).
C. PARP and Proinflammatory Signal Transduction: Implications for Pathophysiology
The following working hypothesis summarizes our current understanding of the role of PARP activation in the development of inflammatory cell injury and the activation of positive feedback cycles of inflammation and reperfusion injury. Ischemia-reperfusion as well as proinflammatory cytokines trigger free-radical formation by stimulating xanthine oxidase activity and also by recruiting activated neutrophils, which express NADPH oxidase as well as a variety of other potential cellular sources. Baseline production of NO from constitutive sources may be supplemented by de novo iNOS expression. As a consequence, the oxidants peroxynitrite, hydrogen peroxide, and hydroxyl radical are formed from the interaction of superoxide and NO. Furthermore, under conditions of oxidative stress, NO may be converted to the more toxic nitroxyl anion (NO−). Oxidant stress generates DNA single-strand breaks. DNA strand breaks then activate PARP, which in turn potentiates NF-κB activation and AP-1 expression, resulting in greater expression of the AP-1- and NF-κB-dependent genes, such as iNOS, ICAM-1, MIP-1α, TNF-α, and C3. Generation of C5a in combination with increased endothelial expression of ICAM-1, recruits a greater number of activated leukocytes to inflammatory foci, producing greater oxidant stress. It is possible that, on a small scale, PARP-mediated necrosis and PARP-mediated proinflammatory gene expression are beneficial or protective processes. For example, NAD+ depletion and cell necrosis may help eliminate “innocent bystander” parenchimal cells having severely damaged DNA (e.g., caused by a nearby occurring neutrophil attack on invading microbes). It is also possible that a low-level, localized inflammatory response may be beneficial in recruiting mononuclear cells to an inflammatory site. For example, invading microorganisms trigger a local neutrophil oxidant burst, and the DNA injury and PARP activation in nearby professional and nonprofessional immune cells triggers proinflammatory cytokine and chemokine production, which recruits additional mononuclear cells to the site of infection to eliminate the invading microorganisms (Fig. 6). It is important to note that the only known mammalian cells that do not contain PARP are the neutrophil and eosinophil granulocytes. It is possible that the presence of PARP in these cells is not compatible with the high levels of local oxidant production that these cells frequently generate.

Proposed physiological functions of PARP activation during the inflammatory process. PARP-mediated NAD+ depletion and cell necrosis may help eliminate “innocent bystander” parenchimal cells with severely damaged DNA (e.g., caused by a nearby-occurring neutrophil attack on invading microbes). A low-level, localized inflammatory response may also be enhanced by PARP. This inflammatory process may be beneficial in recruiting mononuclear cells to an inflammatory site, which will help in eliminating the invading microorganisms.
However, in many pathophysiological states, a multitude of experimental evidence makes us conclude that the above-described feedback cycles amplify themselves beyond what can be considered desirable or controllable by the body's own defense systems. The cycle is renewed by multiple positive-feedback cycles as the increase in oxidant stress triggers more DNA strand breakage. The proposed cycle of inflammatory activation will be augmented in systems in which PARP-dependent MAP kinase activation and NF-κB translocation contribute significantly to free-radical and oxidant formation and granulocyte recruitment (Fig.7). According to this proposed model, PARP occupies a critical position in a positive-feedback loop of inflammatory injury. NAD+ depletion induced by PARP activation is likely to accelerate this positive-feedback cycle by preventing the energy-dependent reduction of oxidized glutathione, the chief intracellular antioxidant and most abundant thiol in eukaryotic cells (Marini et al., 1993). NAD+ is the precursor for NADP, a cofactor that plays a critical role in bioreductive synthetic pathways and the maintenance of reduced glutathione pools. The depletion of reduced glutathione, as a consequence of intracellular energetic failure or overwhelming oxidant exposure, leaves further oxidant stress unopposed, resulting in greater DNA strand breakage. The various oxidants and free radicals produced in inflammation frequently synergize with each other, with respect to PARP activation (Szabo et al., 1997a) as well as other (PARP-independent parallel) oxidant and cytotoxic processes. HOCl, although not inducing DNA single-strand breakage, is still cytotoxic via PARP-independent pathways. At higher concentrations, hypochlorous acid actually inactivates PARP (Van Rensburg et al., 1991), as do high levels of hydrogen peroxide and peroxynitrite (C. Szabó, unpublished observations). This means that under extreme levels of oxidant stress, PARP-independent pathways of cell injury take over. In fact, overwhelming cytotoxicity, both in response to peroxynitrite and hydrogen peroxide, is no longer completely inhibitable with PARP inhibitors or PARP deficiency (Heller et al., 1995; Szabo et al., 1997a). Thus, at extreme levels of oxidant stress, PARP-independent pathways of injury may take over. The concept of combining antioxidants with PARP inhibitors is a viable one and needs to be directly tested in further studies.

Proposed scheme of PARP-dependent and PARP-independent cytotoxic pathways involving nitric oxide (NO⋅), hydroxyl radical (OH⋅), and peroxynitrite (ONOO−) in local and systemic inflammation. Proinflammatory stimuli trigger the release of proinflammatory mediators, which, in turn, induce the expression of the iNOS. NO produced by iNOS and/or by constitutive sources of NO in turn combines with superoxide to yield peroxynitrite. Hydroxyl radical (produced from superoxide via the iron-catalyzed Haber-Weiss reaction) and peroxynitrite or peroxynitrous acid induce the development of DNA single-strand breakage, with consequent activation of PARP. Depletion of the cellular NAD+ leads to the inhibition of cellular ATP-generating pathways, leading to cell necrosis and organ dysfunction. NO alone does not induce DNA single-strand breakage but may combine with superoxide (produced from the mitochondrial chain or from other cellular sources) to yield peroxynitrite. Under conditions of low cellular l-arginine, NOS may produce both superoxide and NO, which then can combine to form peroxynitrite. PARP activation promotes the activation of NF-κB, AP-1, MAP kinases, and the expression of proinflammatory mediators, adhesion molecules, and iNOS. PARP-independent parallel pathways of cellular metabolic inhibition can be activated by NO, hydroxyl radical, superoxide, HOCl, and peroxynitrite.
The above-mentioned scheme, which is also depicted in Fig. 7, unites a multitude of pathways of inflammation and reperfusion injury. The relative importance of the various components of inflammation and cell injury is dependent on the specific disease in question and also on the stage of the disease. For instance, myocardial reperfusion injury and stroke are considered classic oxidant-type reperfusion injury entities rather than inflammatory diseases. Nevertheless, at later stages, an inflammatory component and mononuclear cell recruitment component become evident.
In the following sections, we describe the specific role of PARP in some of the best characterized models of reperfusion injury and inflammation.
D. PARP in Myocardial Reperfusion Injury
Immunohistochemical detection of poly(ADP-ribose) formation demonstrated that PARP is rapidly activated in the reperfused myocardium (Pieper et al., 2000; Liaudet et al., 2001b). The time course of PARP activation is rather prolonged: it is present at 2 h after the start of reperfusion and continues to be present as late as 24 h after reperfusion (Pieper et al., 2000; Liaudet et al., 2001b). This delayed pattern of PARP activation is likely related to the continuing presence of free-radical and oxidant production in the reperfused myocardium. It is also conceivable that a massive, early DNA single-strand breakage, which remains unrepaired for prolonged periods of time, is responsible for the prolonged pattern of PARP activation. The site of the most pronounced PARP activation is the area of necrosis and peri-infarct zone (i.e., area at risk). Most of the poly(ADP-ribose) staining was seen in cardiac myocytes (Pieper et al., 2000; Liaudet et al., 2001b), indicating that the heart tissue itself, rather than the infiltrating mononuclear cells, is the main site of PARP activation. A more diffuse staining pattern can be seen in the area of necrosis: this pattern is likely to reflect the fact that the cellular content (and thus the poly-ADP-ribosylated proteins) is now more-or-less uniformly distributed in the necrotic area because of myocardial necrosis and the associated breakdown of the cell membrane integrity. Because PARP activation triggers cellular necrosis caused by cellular energetic collapse, the primary mode of the PARP inhibitors' cardioprotective effects is related to a direct inhibition of myocyte necrosis. The peri-infarct zone, which contains viable cells, in which PARP is markedly activated is the likely site of the PARP inhibitors' beneficial effects. Activation of PARP has also been demonstrated ex vivo in an isolated perfused heart system after ischemia and reperfusion (Szabados et al., 1999a; Pieper et al., 2000). As discussed earlier, one of the enzymes that undergoes poly(ADP-ribos)ylation is PARP itself (auto-ribosylation). This ischemia-reperfusion-induced self-ADP-ribosylation of PARP can be attenuated by pharmacological inhibitors of PARP (Szabados et al., 2000).
Various cultured cells, including cultured rat cardiac myoblasts, are protected against hydrogen peroxide- and peroxynitrite-mediated cell necrosis by PARP inhibitors (Gilad et al., 1997). From these in vitro observations and from our emerging data implicating the pathogenetic role of PARP activation in circulatory shock (Szabo et al., 1996b), in 1996 we proposed and subsequently evaluated the role of PARP in an acute model of myocardial reperfusion injury in the rat (Zingarelli et al., 1997a). Peroxynitrite formation was evidenced by plasma oxidation of dihydrorhodamine-123 and formation of nitrotyrosine in the ischemic and reperfused portions of the heart (Zingarelli et al., 1997a). Myocardial reperfusion resulted in a marked cellular injury, as measured by an increase of plasma creatine phosphokinase activity and development of a large infarcted area. Pharmacological inhibition of PARP with 3-aminobenzamide significantly improved the outcome of myocardial dysfunction, as evidenced by a reduction in creatine phosphokinase levels, diminished infarct size, and preserved the ATP pools (Zingarelli et al., 1997a). Other investigators confirmed our results in similar experimental models of myocardial reperfusion. In rabbit and pig models of myocardial infarction, pharmacological inhibitors of PARP such as nicotinamide and 3-aminobenzamide dramatically reduced the infarct size (Thiemermann et al., 1997; Bowes et al., 1998b). The cardioprotection afforded by the PARP inhibitors was caused by a selective inhibition of PARP, because the structurally related but inactive agents, such as 3-aminobenzoic acid and nicotinic acid, did not cause a reduction in infarct size (Thiemermann et al., 1997). Over the last several years, a multitude of studies demonstrated the cardioprotective effects of various pharmacological PARP inhibitors in cultured myocytes, in perfused heart systems, and in various in vivo models of myocardial reperfusion injury (Janero et al., 1993;Bhatnagar, 1994, 1997; Gilad et al., 1997; Thiemermann et al., 1997;Zingarelli et al., 1997a, 1998; Bowes et al., 1998a, 1998b, 1999;Docherty et al., 1999; Grupp et al., 1999; Szabados et al., 1999a,2000; Pieper et al., 2000; Yang et al., 2000; Liaudet et al., 2001b;Faro et al., 2002) (Table 2).
Protection against various forms of cardiac injury by pharmacological inhibition or genetic inactivation of PARP in vitro and in vivo
The transgenic mice lacking the functional gene for PARP have provided the unique opportunity to unequivocally define the role of PARP in myocardial injury and also to investigate some of the cellular mechanisms underlying this disease. Using a murine model of myocardial injury after early reperfusion, we found that the absence of a functional PARP gene resulted in a significant prevention of reperfusion injury. Wild-type mice subjected to 1-h ligation and 1-h reperfusion of the left anterior descending branch of the coronary artery induced massive myocardial necrosis and triggered neutrophil infiltration (Zingarelli et al., 1998). When the reperfusion after 1-h ischemia was prolonged to 24 h, wild-type mice also developed high mortality (Yang et al., 2000). In PARP−/− mice, plasma levels of creatine phosphokinase activity were significantly reduced, the histological features of the myocardium were improved, and neutrophil infiltration was reduced and survival improved (Zingarelli et al., 1998; Yang et al., 2000). Protective effects of PARP deficiency can also be demonstrated in isolated perfused hearts. We reported that at the end of the reoxygenation in hearts from wild-type animals, there is a significant suppression in the rate of intraventricular pressure development and in the rate of relaxation (Grupp et al., 1999). In contrast, in the hearts from the PARP knockout animals, no significant suppression of the rate of intraventricular pressure development and relaxation was observed (Grupp et al., 1999). Our findings, both in isolated perfused hearts and in the in vivo models, were recently confirmed by Pieper and colleagues (2002) using PARP-deficient mice. In vivo PARP activation in heart tissue slices was assayed through conversion of [33P]NAD+into poly(ADP)ribose and also was monitored by immunohistochemical staining for poly(ADP-ribose). Cardiac contractility, NO and reactive oxygen species production, and NAD+ and ATP levels were measured (Pieper et al., 2000). Ischemia reperfusion augmented the formation of NO, oxygen-free radicals, and PARP activity. Ischemia reperfusion decreased cardiac contractility and NAD+ levels, effects that were attenuated in PARP-deficient animals (Pieper et al., 2000). The protective effect of PARP deficiency in myocardial infarction has also been repeated using various pharmacological inhibitors of the enzyme. Table 2 gives an overview of the protection afforded by benzamides, isoquinolinones, phenanthridinones, and other classes of PARP inhibitors and compares the effects with that of genetic PARP deficiency. Taken together, there are multiple lines of clear evidence underlining the importance of PARP pathway in the pathogenesis of myocardial ischemia-reperfusion injury. It is likely that both an inhibition of the energetic component of PARP-mediated cell dysfunction (Docherty et al., 1999; Grupp et al., 1999; Szabados et al., 1999a; Pieper et al., 2000) and the suppression of various proinflammatory pathways—including suppression of proinflammatory cytokine and chemokine formation and adhesion receptor expression, as well as prevention of neutrophil recruitment and protection against the loss of endothelial function (Zingarelli et al., 1997a, 1998; Yang et al., 2000; Faro et al., 2002)—contribute to these cardioprotective effects.
Recent work from our laboratory also demonstrates that PARP is necessary for the phenomenon of ischemic myocardial preconditioning (cardioprotective effect of mild ischemic episodes against subsequent ischemia-induced damage of the heart). Using a combined approach (pharmacological inhibition of PARP-proficient and PARP-deficient mice), we observed that the protective effect of preconditioning disappears in PARP−/− mice or in response to the PARP inhibitor 3-aminobenzamide (Liaudet et al., 2001c). The protection against reperfusion injury by preconditioning is associated with partially preserved myocardial NAD+ levels, indicating that PARP activation is attenuated by preconditioning. This conclusion is further strengthened by poly(ADP-ribose) immunohistochemical measurements, demonstrating that ischemic preconditioning markedly inhibits PARP activation during reperfusion (Liaudet et al., 2001c). Because ischemic preconditioning itself induces low levels of nitrosative and oxidative stress (Csonka et al., 2001; Liaudet et al., 2001c) and a low degree of PARP activation, we proposed that the low level of PARP activation during preconditioning may lead to autoribosylation (i.e., autoinhibition) of PARP. This process could, in turn, protect against the deleterious effects of ischemia and reperfusion via the inhibition of the subsequent, massive activation of PARP, which occurs in naive (nonpreconditioned wild-type) animals during reperfusion (Liaudet et al., 2001c).
E. PARP in the Pathogenesis of Cardiomyopathy and Toxic Myocardial Injury
Most published studies in the area of PARP and the heart focused on myocardial injury induced by acute occlusion and reperfusion of the coronary artery. Currently, there is emerging evidence that PARP activation is also present in cardiomyopathy. As a first example, in diabetic cardiomyopathy models that spontaneously develop in the nonobese diabetic mice and in the streptozotocin-induced models of diabetes, the marked depression of myocardial contractile function is associated with a significant increase in poly(ADP-ribosyl)ation in the cardiac myocytes (Pacher et al., 2002b). The myocardial contractile dysfunction can be effectively restored by pharmacological inhibition of PARP (Pacher et al., 2002b). Further work remains to be conducted to determine whether PARP activation also plays a pathogenetic role in other forms of cardiomyopathy, e.g., the one induced by long-term ischemia.
Some information is also available on the activation of PARP in the heart in various experimental models of drug-induced forms of cardiac dysfunction (iatrogenic or toxic cardiomyopathy). In a recent study, 2′,3′-dideoxycytidine and 3′-azido-3′-deoxythymidine were found to induce PARP activation in the heart, and the PARP pathway has been proposed to play a role in the cardiomyopathy induced by these compounds; PARP inhibitors exerted significant therapeutic benefits (Skuta et al., 1999; Szabados et al., 1999b). In addition, the cytotoxic drug doxorubicin induced cardiomyopathy and involved the activation of PARP in the cardiac myocytes. In a recent study (Pacher et al., 2002a) using a dual approach of PARP-1 suppression by genetic deletion or pharmacological inhibition with the phenanthridinone PARP inhibitor PJ34, the role of PARP-1 in the development of cardiac dysfunction induced by doxorubicin was demonstrated. PARP-1+/+ and PARP-1−/−mice received a single injection of doxorubicin. Five days after doxorubicin administration, left-ventricle performance was significantly depressed in PARP-1+/+ mice but only to a smaller extent in PARP-1−/− animals. Similar experiments were conducted in BALB/c mice treated with PJ34 or vehicle. Treatment with PJ34 significantly improved cardiac dysfunction and increased the survival of the animals. In addition, PJ34 significantly reduced the doxorubicin-induced increase in the serum lactate dehydrogenase and creatine kinase activities but not metalloproteinase activation in the heart. Further work is needed to determine whether appropriately selected PARP inhibitors may become useful adjunctive therapies to protect against various forms of iatrogenic cardiomyopathies.
F. PARP in Stroke
The involvement of superoxide and the protective effect of superoxide-neutralizing strategies (Lafon-Cazal et al., 1993; Fagni et al., 1994) and the involvement of NO and the protective effect of NOS inhibition (Tewari et al., 1995) have been well established in various forms of central nervous system injury, including the reperfusion of the ischemic brain (stroke). Infarct volume after vascular stroke is markedly diminished in animals treated with NOS inhibitors and in mice with bNOS gene disruption (Tewari et al., 1995). Using chemical considerations, it was proposed more than a decade ago that peroxynitrite and not NO or superoxide separately is the major cytotoxic mediator in the neuronal injury during stroke (Gu et al., 1995). Reperfusion injury in the central nervous system is associated with the activation of NMDA receptors, which are now believed to play a key role in the pathogenesis of stroke, which triggers the simultaneous production of superoxide and NO. There is now indirect evidence suggesting that activation of the NMDA receptor is associated with a marked increase in a hydroxyl radical-like reactivity in the brain which may be blocked by the inhibition of NOS (Hammer et al., 1993) and which is presumably caused by peroxynitrite generation.
Ischemia-reperfusion injury of the brain can be modeled in the laboratory by exposing primary neuronal cultures to glutamate or its agonists; to various reactive oxygen species, NO donors, or peroxynitrite; or by combined oxygen-glucose deprivation. In cerebellar granule cells, glutamate induces a rapid increase in poly(ADP-ribose) immunoreactivity (Cosi et al., 1994). PARP inhibitors have been shown to protect in these models of brain injury, both in models in which injury was induced by glutamate and in response to chemical compounds that generate NO. The rank order of potency of different classes of PARP inhibitors correlates with the degree of protection (Zhang et al., 1994). Moreover, the protection by PARP inhibition is not associated with changes in calcium influx induced by glutamate. In addition, primary cortical cultures from PARP−/− mice were found to be resistant to toxicity from NMDA, compounds which generate NO, and combined oxygen-glucose deprivation (Wallis et al., 1993, 1996; Cosi et al., 1994; Zhang et al., 1994, 1995; Tewari et al., 1995; Didier et al., 1996; Snyder, 1996; Eliasson et al., 1997). Delaying the treatment of PARP inhibition relative to the stimulus of neuroinjury produced a significant therapeutic window of opportunity in vitro, as demonstrated in a model system consisting of primary rat hippocampal neurons exposed to the NO donor NOC-9 and the peroxynitrite generator compound SIN-1, with nicotinamide being used to block the activity of PARP (Lin et al., 2000). As opposed to NMDA-mediated neuronal injury, which is PARP-dependent, a non-NMDA receptor-mediated form of neuroinjury such as the one induced by the excitatory amino acid AMPA does not involve the PARP pathway (Mandir et al., 2000).
Naturally, the above-discussed in vitro systems only model a component of the complex chain of events initiated in vivo after an ischemic insult or stroke. Nevertheless, the pathophysiological relevance of these observations is supported by the observation that increased poly(ADP-ribosylation) has been demonstrated in the reperfused brain (Endres et al., 1998a). In PARP-1−/− mice, a markedly reduced infarct volume is observed after transient middle cerebral artery occlusion (Eliasson et al., 1997; Endres et al., 1997). The reduction in infarct volume was observed in PARP-1−/− mice that had either a genetic background identical with the wild-type strain (Endres et al., 1997) or a mixed 129/C57B6 genetic background (Eliasson et al., 1997). Thus, the reduction in infarct volume was caused by the absence of the PARP gene product and not by other genetic variables. PARP activation was examined after focal ischemia in the ipsilateral hemisphere by the evaluation of ADP-ribose polymer formation or levels of NAD+. This observation also demonstrates that from the multiple isoforms of PARP, PARP-1 seems to play the main role in the enhanced poly(ADP-ribosylation) in the brain during stroke. ADP-ribose formation was increased and NAD+ was decreased after focal ischemia in wild-type tissue, whereas no ADP-ribose formation was observed in PARP−/−tissue (Eliasson et al., 1997), and NAD+ levels were spared (Endres et al., 1997). PARP activation is mainly related to NO production by the neuronal isoform of NOS because in mice deficient in this enzyme, when subjected to middle cerebral artery occlusion/reperfusion, PARP activation was found to be markedly diminished (Endres et al., 1998a). The protection observed in the PARP−/− mice exceeds the degree of protection reported for any other transgenic model, including the bNOS−/− mice. This observation suggests a common role for PARP activation by other excitotoxic mechanisms, in addition to the production of free radicals and NO. In PARP−/− mice that were also treated with viral transfection of wild-type PARP-1, the protection from middle cerebral artery occlusion was lost with restoration of the gene product (Goto et al., 2002).
The protective effect of PARP deficiency in stroke has also been repeated using various pharmacological inhibitors of the enzyme. Table3 gives an overview of the protection afforded by benzamides, isoquinolinones, phenanthridinones, and other classes of PARP inhibitors and compares their effects with that of genetic PARP deficiency. Taken together, there are multiple lines of clear evidence suggesting the importance of PARP pathway in the pathogenesis of stroke injury. It is important to emphasize that the therapeutic window of intervention is rather large (up to 4–6 h after the onset of ischemia in the middle cerebral artery ischemia-reperfusion models), as demonstrated with the use of both nicotinamide (Ayoub and Maynard, 2002) and a phenanthridinone derivative (Abdelkarim et al., 2001). It is also important to emphasize that the protective effect of PARP inhibition on neurological function is a lasting one that remains significant even after 5 or 21 days of ischemia reperfusion (Ding et al., 2001). In many studies in which more potent PARP inhibitors were used and full dose-response curves were obtained, the protection provided by PARP inhibitors diminishes when the dose of the agent is increased, i.e., a bell-shaped dose-response is observed (Takahashi et al., 1997). This observation may be related to intrinsic neurotoxic effects of the particular compounds used, or it may suggest that the nonselective and complete inhibition of all PARP isoforms may not be a more desirable future therapeutic approach. Although the correlation between cerebrocortical NAD+ and ATP levels and neuronal injury in stroke is far from being understood (Paschen et al., 2000; Plaschke et al., 2000b), it is likely that the energetic/cell necrosis pathway is the main mode of the PARP inhibitors' acute neuroprotective actions in stroke. Findings such as the suppression of NMDA-induced glutamate efflux and overall neurotransmitter dysregulation by PARP inhibitors (Lo et al., 1998) may be a direct consequence of an overall maintenance of cellular energetic status and a reduction of cell necrosis. Nevertheless, it is important to note that there may also be some mild levels of injury (e.g., in a model of 15-min ischemia in the rat) in which a mild degree of PARP activation without NAD depletion may even be beneficial, and its inhibition may not be desirable (Nagayama et al., 2000). Also, although the oxygen-glucose deprivation-induced cell necrosis can be largely prevented by PARP inhibitors (as demonstrated in mixed cortical cell cultures), the same approach is unable to reduce primarily apoptotic-type cell death (such as the CA1 pyramidal cell loss in organotypic hippocampal slices) (Moroni et al., 2001). The notion that PARP inhibition in stroke should be reserved for the most severe forms of the disease—and ones that are associated with predominantly necrotic-type cell death—should certainly be considered and explored further in future studies.
Protection against various forms of neuronal injury by pharmacological inhibition or genetic inactivation of PARP in vitro and in vivo
In addition to the acute neuroprotective effects that are probably mediated by energetic changes and prevention of cell dysfunction and cell death, it is also conceivable that modulation by PARP of inflammatory molecule expression may also contribute to protection. As discussed above, PARP inhibitors suppress the expression of proinflammatory cytokines and chemokines, thereby interrupting positive-feedback cycles of mononuclear cell migration. An additional mechanism specifically relevant for the pathogenesis of central nervous system (CNS) injury may involve the regulation by PARP of the expression of integrin C11a, with subsequent suppression of microglial migration (Ullrich et al., 2001b).
The advantages of PARP inhibitors as agents for the treatment of stroke (as well as myocardial infarction) include the relatively short duration of treatment (an important safety concern when inhibiting an enzyme that regulates nuclear integrity) and the possibility of effective delayed treatment because PARP inhibitors target a delayed process of cell death. Most PARP inhibitors in the therapeutic dose range tend to have little influence on hemodynamic parameters, which can also be considered an advantage. It is clear that PARP is not the only factor involved in the pathogenesis of cell and organ injury in response to oxidant or nitrosative stress. The relative importance of PARP in mediating oxidant injury is dependent on cell type. Furthermore, the protection wanes when cells are challenged with extremely high concentrations of oxidants that trigger cytotoxic effects independent of PARP, such as direct inhibitory effects on the mitochondrial respiratory chain or inhibitory effects on other intracellular energetic or redox processes. Finally, direct interactions of the oxidants with proteins, lipids, arachidonic acid, and other molecules may also play a significant role in the development of cellular injury. It is likely that there are important synergistic interactions between PARP activation and these other cellular processes of cytotoxicity. Nevertheless, from the evidence presented here, it seems that inhibition of PARP alone can “tip the end of the balance” and significantly influence the outcome of stroke, myocardial infarction, and various other ischemia-reperfusion states (see below).
G. PARP in Neurotrauma
There is experimental evidence that PARP activation contributes to the pathogenesis of other forms of brain injury and neurodegenerative disorders. For instance, it has been reported that in spinal cord sections taken after traumatic spinal cord injury, there is evidence of peroxynitrite formation and activation of PARP (Scott et al., 1999). PARP inhibitors provide protection in an in vitro model of traumatic brain injury (Wallis et al., 1993), although at present there are no in vivo studies available on the outcome of spinal cord trauma in animals in which PARP was inhibited or genetically inactivated. On the other hand, there are in vivo studies available showing that genetic inactivation of PARP markedly improves the functional outcome in traumatic brain injury (Whalen et al., 1999, 2000). This protective action has recently been confirmed using a potent novel PARP inhibitor GPI 6150 (La Placa et al., 2001). Although PARP deficiency did not affect the actual size of the primary cortical necrosis, pharmacological inhibition did.
Another area that is directly relevant to neurotrauma is represented by the secondary retinal ganglion cell death model, which develops in response to rat optic nerve transection. Recent studies demonstrated a transient, retinal ganglion cell-specific PARP activation and increased retinal PARP expression early after optic nerve axotomy. In addition, intravitreal injections of 3-aminobenzamide blocked PARP activation in retinal ganglion cells and resulted in an increased number of surviving retinal ganglion cells (Weise et al., 2001). Thus, secondary degeneration of a subset of axotomized retinal ganglion cells results from a necrotic-type cell death mediated by PARP activation. Taken together, a body of evidence indicates that in addition to stroke, PARP inhibition may constitute a relevant strategy for the clinical treatment of various forms of traumatic brain injury.
H. PARP in Reperfusion Injury of the Gut, Eye, Kidney, and Skeletal Muscle
Probably because of the relative simplicity of the experimental design involved, the role of PARP activation was investigated fairly early on in splanchnic occlusion-reperfusion, a convenient experimental model which, nevertheless, does not represent a medical problem comparable to heart attacks or stroke in humans. In a series of studies in anesthetized rodents, splanchnic occlusion shock was induced by clamping both the superior mesenteric artery and the celiac trunk followed by release of the clamp (reperfusion). There was a marked increase in the oxidation of dihydrorhodamine-123, which is a marker of oxidative processes induced by peroxynitrite, in the plasma of these animals after reperfusion, but not during ischemia alone (Cuzzocrea et al., 1997a; Liaudet et al., 2000b). Immunohistochemical examination showed a marked increase in the immunoreactivity to nitrotyrosine, indicating the presence of peroxynitrite, in the reperfused necrotic ileum and demonstrated the activation of PARP both by enzyme activity measurements and by immunohistochemical techniques (Cuzzocrea et al., 1997a; Liaudet et al., 2000b). In addition, in ex vivo measurements in aortic rings from shocked rats, there was a PARP-dependent reduction in the contractions caused by norepinephrine and also impaired responsiveness to the relaxant effect of acetylcholine, which means vascular hyporeactivity and endothelial dysfunction, respectively. PARP knockout mice subjected to splanchnic occlusion and reperfusion also showed reduced alterations in remote organs, as shown by lesser lipid peroxidation (malondialdehyde formation) and neutrophil infiltration in the lung and liver when compared with their wild-type counterparts (Liaudet et al., 2000b).
Splanchnic artery ischemia and reperfusion also resulted in a marked increase in epithelial permeability. Inhibition of PARP with 3-aminobenzamide or PJ34 or its genetic inactivation significantly reduced ischemia/reperfusion injury in the bowel as evaluated by histological examination and functional measurements (Cuzzocrea et al., 1997a; Liaudet et al., 2000b; Jagtap et al., 2002). PARP inhibition also prevented the infiltration of neutrophils into the reperfused intestine, as evidenced by reduced myeloperoxidase activity, and improved the histological status of the reperfused tissues. The free radical/oxidant basis of PARP activation in this model is underlined by studies showing that free-radical scavengers such as tempol andN-acetylcysteine inhibit the activation of PARP in the reperfused gut (Cuzzocrea et al., 2000a,c). Taken together, the results demonstrate that PARP inhibition exerts multiple protective effects in splanchnic artery occlusion/reperfusion injury and suggest that peroxynitrite and/or hydroxyl radical, produced during the reperfusion phase, cause DNA strand breaks, PARP activation, and subsequent cellular dysfunction. The vascular endothelium may be an important site of protection by PARP inhibition in shock caused by splanchnic occlusion. The reduced neutrophil infiltration and the reduced nitrotyrosine after inhibition or inactivation of PARP may be related to the interruption of positive feedback cycles, similar to that already described in relation to the reperfused heart.
Relatively less information is available on the role of PARP in other ischemic and reperfused organs. Immunohistochemical studies showed elevated poly(ADP-ribose) levels at the retinal ganglion cell layer and the inner nuclear layer in the ischemic-reperfused retina (Chiang and Lam, 2000). Intracameral infusion of 3-aminobenzamide dose-dependently reduced the retinal injury in albino Lewis rats (Kupper et al., 1995). In a follow-up study, intravital administration of 3-aminobenzamide was able to improve the morphology of the retina even when the start of treatment was delayed to 12 and 18 h after reperfusion, which can be considered a remarkable therapeutic window (Chiang and Lam, 2000). In addition to the reperfusion injury of the retina, there is also some circumstantial in vitro evidence implicating the pathogenic contribution of PARP activation in hyperbaric oxygen-induced eye damage (Padgaonkar et al., 1993).
The kidney is known to be extremely sensitive to various forms of ischemic and oxidant-mediated injury. In vitro studies in various experimental models demonstrated the importance of PARP activation in the dysfunction and death of kidney epithelial cells in vitro. In rat renal proximal tubular cell cultures, the hydrogen peroxide (but not the tert-butyl hydroperoxide) -induced cell death was blocked by various inhibitors of PARP (Chatterjee et al., 1999a; Jung et al., 2000; Min et al., 2000). In LLC-PK1-cultured renal epithelial cells, the mode of hydrogen peroxide-induced cell death was investigated in detail, and it was confirmed that it is the necrotic- and not the apoptotic-type cell death which is blockable by PARP inhibition (Filipovic et al., 1999). With respect to the molecular mode of protection: in Madin-Darby canine kidney cells exposed to hydrogen peroxide PARP inhibitors prevented the occludin junctional damage (Cuzzocrea et al., 2000b). In the LLC-PK1, the improvement in cell survival seen with 3-aminobenzamide has been suggested to be related to inhibition of histone H3 phosphorylation (Tikoo et al., 2001). The role of the PARP pathway has also been demonstrated in kidney dysfunction induced by ischemia and reperfusion in vivo. In rat models of renal ischemia-reperfusion injury, PARP inhibitors accelerated the recovery of normal renal function, as assessed by monitoring the levels of plasma creatinine and blood urea nitrogen and fractional excretion of sodium; prevented the ischemia-reperfusion-induced reduction in glomerular filtration rate; increased cell proliferation at 1 day postinjury, as assessed by proliferating cell nuclear antigen; improved the histopathological appearance of kidneys examined at 7 days postinjury; and increased ATP levels measured at 24 h postischemia (Martin et al., 2000). The free-radical/oxidant basis of PARP activation in this model is underlined by studies showing that free radical scavengers such as tempol and desferrioxamine inhibit the activation of PARP in the reperfused kidney (Chatterjee et al., 2000). The therapeutic window of intervention has not yet been investigated in this model.
There is also some evidence to implicate the role of PARP activation and the protective effect of PARP inhibition in the reperfused skeletal muscle (Thiemermann et al., 1997), liver (Chen et al., 2001), and cochlea (Tabuchi et al., 2001).
I. PARP in Arthritis
Recent studies have clearly demonstrated the role of PARP activation in various forms of local inflammation induced by the prototypical inflammatory stimuli zymosan and carrageenan. For example, in carrageenan-induced paw edema, inhibition of PARP with 3-aminobenzamide reduced paw swelling and inhibited the infiltration of neutrophils into the inflamed paw (Szabo et al., 1997b). Furthermore, in a model of acute local inflammation (carrageenan-induced pleurisy), 3-aminobenzamide inhibited the inflammatory response (pleural exudate formation, mononuclear cell infiltration, histological injury) (Cuzzocrea et al., 1998a,b). Similar to the effect of the pharmacological inhibitors, PARP−/− animals were found to be resistant against zymosan-induced inflammation and multiple organ failure when compared with the response of wild-type mice (Szabo et al., 1997b). GPI 6150, a novel potent PARP inhibitor, was also found to be very effective in attenuating joint swelling and various parameters of inflammation in rodent models of carrageenan-induced paw edema and zymosan-induced multiple organ failure (Mazzon et al., 2001).
Inhibition of PARP also reduced the formation of nitrotyrosine, an indicator of the formation of peroxynitrite, in the inflamed tissues (Szabo et al., 1997b; Cuzzocrea et al., 1998a). This finding was at first unexpected because PARP activation is distal to the generation of oxidants. The explanation for this finding is likely related to the fact that PARP−/− phenotype or pharmacological inhibition of PARP reduces the infiltration of neutrophils into inflammatory sites (Szabo et al., 1997b, 1998c; Cuzzocrea et al., 1998a). Thus the reduction in tissue injury by PARP inhibitors may result from a decreased inflammatory infiltrate, which would be associated with a reduction in both oxygen- and nitrogen-centered free-radical production (hence, reduced nitrotyrosine staining). The basis for PARP-inhibitable neutrophil infiltration is not yet defined, but it may relate to the effect of PARP activation on the expression of intercellular adhesion molecules and/or to the modulation by PARP of a postadhesion event (Roebuck et al., 1995; Szabo et al., 1997b;Cuzzocrea et al., 1998a,b; Mazzon et al., 2001). Other mechanisms by which PARP modulates neutrophil tissue infiltration cannot be excluded, including an effect on endothelial integrity (Szabo et al., 1997a;Soriano et al., 2001c).
Oxygen-derived free radicals and oxidants are massively overproduced in arthritis (Oyanagui, 1994; Santos and Tipping, 1994; Kaur et al., 1996). Furthermore, several lines of evidence suggest a role for NO overproduction in the pathogenesis of arthritis. The expression of iNOS and the production of large amounts of NO have been demonstrated in chondrocytes from experimental animals and humans (Hauselmann et al., 1994; Murrell et al., 1995; Sakurai et al., 1995; Grabowski et al., 1996; Hayashi et al., 1997). An increase in the circulating levels of nitrite/nitrate (the breakdown products of NO) has been demonstrated in patients with arthritis (Farrell et al., 1992; Stichtenoth et al., 1995). Increased plasma and synovial fluid levels of nitrotyrosine, a marker of peroxynitrite formation have been demonstrated in patients with arthritis (Kaur and Halliwell, 1994). Similarly, increased nitrotyrosine formation was observed in the joints of mice suffering from collagen-induced arthritis (Szabo et al., 1998c). The development of the disease has been shown to be ameliorated by various nonisoform-selective inhibitors of NO synthase in various animal models of adjuvant-induced arthritis (Ialenti et al., 1993; McCartney-Francis et al., 1993; Stefanovic-Racic et al., 1994, 1995; Weinberg et al., 1994). Mercaptoethylguanidine, an anti-inflammatory agent with a combined mechanism of action (inhibition of the inducible isoform of NO synthase, scavenging peroxynitrite, and inhibition of cyclooxygenase) also provided marked beneficial effects in collagen-induced arthritis (Brahn et al., 1998).
Several series of experiments directly implicate the key role of PARP activation in the pathophysiology of arthritis. In murine models of arthritis, inhibition of PARP with nicotinamide reduced the onset of the disease (Ehrlich et al., 1995; Miesel et al., 1995; Kroger et al., 1996b). The onset, progression, and remission of arthritis positively correlated with the phorbol ester-activated respiratory burst of neutrophils and monocytes (Miesel et al., 1995). Inhibition of PARP not only prevented the development of arthritis, it also inhibited the progress of established collagen-induced arthritis (Kroger et al., 1996b). The combined application of thalidomide (as a drug that inhibits tumor necrosis factor α expression in arthritis) and nicotinamide provided a powerful synergistic inhibition of arthritis (Kroger et al., 1996b). Furthermore, studies with 5-iodo-6-amino-1,2-benzopyrone, a novel PARP inhibitor that lacks oxyradical scavenging properties, also protected in a mouse model of collagen-induced arthritis: the PARP inhibitor reduces both the incidence of arthritis and the severity of the disease throughout the experimental period (Szabo et al., 1998c). Histological evaluation of the paws in the vehicle-treated arthritic animals revealed signs of severe suppurative arthritis with massive mixed (neutrophil, macrophage, and lymphocyte) infiltration. In the animals treated with the PARP inhibitors, the degree of arthritis was significantly reduced: a moderate, primarily neutrophil infiltration into several of the larger joints, coupled with mild-to-moderate necrosis and hyperplasia of the synovium (Szabo et al., 1998c). GPI 6150, another potent poly(ADP-ribose) polymerase inhibitor, was also found to be highly effective in a rodent model of adjuvant-induced arthritis (Mazzon et al., 2001). PJ34, another potent novel PARP inhibitor, was found to be highly effective in a murine model of collagen-induced arthritis (Mabley et al., 2001a). Finally, PARP-1-deficient mice were found to be resistant against collagen-induced joint swelling and inflammation when compared with wild-type animals (J.G. Mabley and C. Szabo, unpublished data). As in the other forms of inflammation, hydroxyl radical and peroxynitrite are the most likely triggers of PARP activation in arthritis.
PARP activation has not yet been directly demonstrated in samples from arthritic patients. Nevertheless, in a human study analyzing DNA single-strand breakage in peripheral mononuclear cells from arthritic patients, a significant elevation was found when compared with healthy volunteers (Bhusate et al., 1992). Studies conducted approximately a decade ago in humans reported increased frequency of circulating antibodies against PARP, chiefly in patients with systemic lupus erythematosus as well as rheumatoid arthritis (Negri et al., 1990; Lee and Axelrod, 1995; Decker et al., 1998). It is likely that these antibodies do not directly reflect on the potential PARP activation in these patients but, rather, may be related to disease-associated increased cell necrosis, followed by systemic spillage of PARP from the nucleus and sensitization against PARP (as well as numerous other components of nuclear and cytoplasmatic debris) in these patients. In another study, patients with systemic lupus erythematosus showed an approximately 70% decrease in poly(ADP-ribose) synthesis; this decreased synthesis persisted even with the addition of histones or DNase (Decker et al., 1998). The findings of increased DNA strand breakage coupled with decreased ex vivo PARP activity may be related to either an earlier increase in PARP activation followed by auto-ADP-ribosylation of the enzyme and eventual inactivation or to massive cleavage and inactivation of PARP by caspases during the apoptotic process followed by the release of the cellular content via postapoptotic necrosis (Wu et al., 2001).
J. PARP in Inflammatory Bowel Disease
Recent studies in a variety of rodent models of experimental colitis [induced by trinitrobenzene sulfonic acid, dextrane sulfate solution or genetic interleukin (IL)-10 deficiency] support the role of PARP activation in the pathogenesis of the disease (Zingarelli et al., 1999; Jijon et al., 2000; Mabley et al., 2001a). Intraluminal administration of the trinitrobenzene sulfonic acid in 50% ethanol induced mucosal erosion and ulceration associated with increased neutrophil infiltration, lipid peroxidation, an intense staining for nitrotyrosine, and progressive weight loss. Genetic ablation of the PARP gene or pharmacological inhibition of PARP with 3-aminobenzamide resulted in significant resistance to the damage induced by trinitrobenzene sulfonic acid administration, reduced nitrotyrosine formation and tissue levels of malondialdehyde, and reduced neutrophil recruitment into the injured tissue (Zingarelli et al., 1999). Similarly, PJ34 exerts marked protective effects against the histological damage, lipid peroxidation, neutrophil infiltration, and mortality in a dextrane sulfate colitis model in the mouse (Mabley et al., 2001a). These in vivo data are in good agreement with in vitro studies demonstrating protection by pharmacological inhibition of PARP against intestinal epithelial cell injury (necrosis) induced by hydrogen peroxide (Watson et al., 1995) or peroxynitrite (Kennedy et al., 1998). One recent study assessed the role of PARP in the colitis seen in IL-10 gene-deficient mice. IL-10 gene-deficient mice demonstrated significant alterations in colonic cellular energy status in conjunction with increased permeability, proinflammatory cytokine release, and nitrosative stress (Jijon et al., 2000). After 14 days of treatment with 3-aminobenzamide, IL-10 gene-deficient mice demonstrated normalized colonic permeability; reduced TNF-α and IFN-γ secretion, iNOS expression, and nitrotyrosine levels; and significantly attenuated inflammation. Time-course studies showed that 3-aminobenzamide rapidly altered cellular metabolic activity and decreased cellular lactate levels. This was associated with the normalization of colonic permeability and was followed by a down-regulation of proinflammatory cytokine release (Jijon et al., 2000). Importantly and unexpectedly, not only the deterioration of the intestinal epithelial function was prevented by PARP inhibition in vitro, but ex vivo incubation of intestinal segments was able to restore some of the intestinal epithelial function (Jijon et al., 2000). As discussed earlier, this latter finding may indicate that the intestinal epithelial cell population is not yet necrotic or dead, but it rather persists in a state of metabolic and functional suppression that can be reversed by inhibition of PARP.
K. PARP in Inflammatory Diseases of the Central Nervous System: Allergic Encephalomyelitis to Multiple Sclerosis
Increased oxygen-derived free-radical production and oxidative injury has been reported in central nervous system tissues from animals subjected to experimental allergic encephalomyelitis (EAE), and oxidative and nitrosative injury has been implicated in the pathogenesis of chronic CNS inflammatory disorders such as multiple sclerosis. The overproduction of NO and oxyradicals in EAE leads to the generation of peroxynitrite. Accordingly, increased nitrotyrosine staining has been reported in humans with multiple sclerosis (MS), as well as in the active EAE lesions (Koprowski et al., 1993; Cross et al., 1997; van der Veen et al., 1997). Furthermore, putative peroxynitrite scavengers have been shown to improve the outcome of EAE in mice (Hooper et al., 1998, 2000; Cross et al., 2000). Emerging data directly implicate the role of the peroxynitrite-PARP axis in the pathogenesis of EAE. In a rat model of EAE in male Lewis rats, 3-aminobenzamide and 5-iodo-6-amino-1,2-benzopyrone delayed the course of the disease (Scott et al., 2001). PARP inhibition resulted in both a delay in the onset and a reduction in the incidence and severity of disease signs. Increased poly(ADP-ribose) immunoreactivity was associated with the development of the brain lesions in vehicle-treated rats, whereas inhibition of PARP with 5-iodo-6-amino-1,2-benzopyrone eliminated the development of the lesions and abolished poly(ADP-ribose) immunoreactivity (Scott et al., 2001). Similarly, pharmacological inhibition of PARP with the novel potent PARP inhibitor PJ34 potently reduces neurological signs and improves survival in a murine model of EAE (G. S. Scott, D. C. Hooper, and C. Szabo, unpublished data). The mechanism by which the inhibition of PARP suppresses the course of EAE has not been clarified. Undoubtedly, one of the major features of MS and EAE is demyelination. EAE (and presumably MS) is triggered and amplified by a variety of interrelated immunological events. Immunological, clinical, and pathological studies suggest that T lymphocytes directed against myelin antigens are involved in the pathogenesis of MS. It is now clear that myelin-basic protein-specific T cells mediate the destruction of CNS myelin in EAE. Although the autoimmune disease is initiated by antigen-specific autoreactive T cells, there is accumulating evidence that CNS injury is essentially mediated by CNS-infiltrating inflammatory cells, and inhibition of cell infiltration can suppress the course of EAE (Kent et al., 1995; Eng et al., 1996; Miyagishi et al., 1997). In addition, it is established that activated inflammatory mononuclear cells contribute to tissue damage in several inflammatory diseases by releasing highly reactive oxygen metabolites (Malfroy et al., 1997), nitrogen metabolites (see above), and subsequent activation of matrix metalloproteinases (Gijbels et al., 1994). It is therefore possible that demyelination associated with EAE and MS results from oxidative injury caused by a cascade of reactive oxygen and nitrogen metabolites produced by CNS-infiltrating activated macrophages and other inflammatory cells. The infiltration of mononuclear cells into the CNS is a process that is closely linked to the breakdown of the blood-brain barrier, a process related to the production of oxidants and free radicals in EAE (Tewari et al., 1995). Once mononuclear cells infiltrate the CNS, and myelin degradation begins, a variety of positive-feedforward cycles initiate. For instance, phagocytosis of opsonized myelin can trigger the expression of iNOS in macrophages, which can, in turn, further enhance the process of demyelination during multiple sclerosis or EAE (van der Laan et al., 1996). The induction of iNOS and induction of proinflammatory cytokines may enhance each other during EAE. This is supported by the finding that aminoguanidine, an inhibitor of the inducible NO synthase, reduced the expression of TNF-α in EAE (Brenner et al., 1997). Although the exact cell types involved have not yet been identified, recent data indicate that the activation of NMDA receptors also plays an important role in the pathogenesis of EAE. In fact, antagonists of these receptors has been shown to suppress the course of disease (Kupper et al., 1995; Wallstrom et al., 1996). Possibly this process is related to the decreased metabolism of glutamate in astrocytes during EAE (Hardin-Pouzet et al., 1997). Considering the abundant evidence for a role of PARP activation in the pathogenesis of NMDA-mediated neuroinjury (see Section III.F.), the above-mentioned studies lend further support to our working hypothesis that PARP activation plays a role in EAE, and the inhibition of PARP has beneficial effects.
Currently, the cellular and molecular targets in which the inhibition of PARP would interrupt the inflammatory cascade leading to demyelination in EAE are unclear. Nevertheless, several possibilities can be considered by which PARP inhibition prevents myelin degradation in EAE: 1) protection against oligodendrocyte death and improved myelin synthesis; 2) protection against astrocyte death; 3) protection against the breakdown of the blood-brain barrier and the related 4) inhibition of mononuclear cell infiltration into the CNS; 5) inhibition of the expression of the inducible NO synthase during EAE and 6) inhibition of NMDA activation-related cell injury; and finally, as discussed earlier (see Section III.F.) 7) PARP inhibition may suppress microglial migration (Ullrich et al., 2001b) and can thereby down-regulate the consequent encephalitogenic T-cell proliferation. With respect to oligodendrocyte death, there are direct in vitro data showing that oligodendrocytes and astrocytes are susceptible to NO-induced or hydrogen peroxide-induced mitochondrial damage and death, and this cell death can be partially inhibited by PARP inhibitors (Mitrovic et al., 1994; Ying and Swanson, 2000). It is also noteworthy that the turnover of oxidatively damaged nuclear proteins in microglial cells has been shown to be linked to the activation state of poly(ADP-ribose) polymerase (Ullrich et al., 2001a). Taken together, it is conceivable that oligodendrocytes and astrocytes would be injured and develop dysfunction in a PARP-dependent fashion during the course of EAE.
L. PARP in Systemic Inflammation and Circulatory Shock
Circulatory shock is associated with the enhanced formation of oxyradicals and with the expression of iNOS, resulting in the overproduction of NO. NO and superoxide react to form peroxynitrite, and all three species have been implicated in the pathogenesis of cardiovascular dysfunction and multiple organ failure in various forms of systemic inflammation and shock. In isolated cells and tissues, authentic peroxynitrite is capable of mimicking many of the pathophysiological alterations associated with shock (endothelial and epithelial dysfunction, vascular hyporeactivity, and cellular dysfunction), and these alterations are, in part, related to PARP activation (Tewari et al., 1995).
In studies in anesthetized rats, the inhibition of PARP with 3-aminobenzamide and nicotinamide reduced the suppression of the vascular contractility of the thoracic aorta ex vivo (Szabo et al., 1996b; Zingarelli et al., 1996b). In another recent study in pigs injected with Escherichia coli endotoxin, pretreatment with 3-aminobenzamide eliminated the LPS-induced increase in pulmonary and total respiratory resistance, indicating that PARP activation plays an important role in the changes of lung mechanics associated with endotoxin-induced acute lung injury (Albertini et al., 2000).
Peroxynitrite production has been suggested to contribute to endothelial injury in circulatory shock (Zingarelli et al., 1997b). Peroxynitrite can impair the endothelium-dependent relaxations (Villa et al., 1994). Data demonstrating the protective effects of 3-aminobenzamide against the development of endothelial dysfunction in vascular rings obtained from rats with endotoxic shock (Szabo et al., 1996b) suggest that DNA strand breakage and PARP activation occur in endothelial cells during shock and that the subsequent energetic failure reduces the ability of the cells to generate NO in response to acetylcholine-induced activation of the muscarinic receptors on the endothelial membrane. It is possible that this impairment is related to endothelial depletion of NADPH (an essential cofactor of NO synthase) because of PARP overactivation (Soriano et al., 2001b,c,d).
Activation of the PARP pathway has also been implicated in the pathophysiology of the cellular energetic failure associated with endotoxin shock by demonstration of increased DNA strand breakage, decreased intracellular NAD+ and ATP levels, and mitochondrial respiration in peritoneal macrophages obtained from rats subjected to endotoxin shock (Zingarelli et al., 1996a,b). This cellular energetic failure was reduced by pretreatment of the animals with the PARP inhibitors 3-aminobenzamide or nicotinamide (Zingarelli et al., 1996a,b). In contrast to these results in peritoneal macrophages, it seems that the PARP pathway only plays a limited role in the hepatic dysfunction associated with endotoxin shock. In an endotoxic shock model in the rat, the inhibition of PARP with 3-aminobenzamide and nicotinamide did not affect the alterations in most parameters of liver injury. Inhibition of PARP with 1,5-dihydroxyisoquinoline resulted in a small protective effect (Wray et al., 1998), whereas PJ34 treatment resulted in significant protection against liver and kidney dysfunction in endotoxic shock in the rat (Jagtap et al., 2002). Another water-soluble, potent PARP inhibitor, 5-aminoisoquinolinone, significantly reduced the circulating aspartate aminotransferase, alanine aminotransferase, and γ-glutamyl-transferase levels (indicators of liver injury and dysfunction) in hemorrhagic shock (McDonald et al., 2000).
There is a clear and pronounced protection by PARP inhibition against the shock-induced intestinal epithelial permeability changes. In endotoxic shock in rats and mice, inhibition of PARP activation by 3-aminobenzamide or by PJ34 protects against the intestinal hyperpermeability, and so does genetic depletion of PARP in hemorrhagic shock (Liaudet et al., 2000a; Jagtap et al., 2002).
The role of PARP activation in the pathogenesis of hemorrhagic shock was recently further investigated in a murine model by comparing the response to hemorrhage and resuscitation in wild-type and PARP-deficient mice (Liaudet et al., 2000a). Animals were bled to a low but tolerable mean blood pressure of 45 mm Hg and subsequently resuscitated with isotonic saline. There was a massive activation of PARP, detected by poly(ADP-ribose) immunohistochemistry, which localized in the areas of the most severe intestinal injury, i.e., the necrotic epithelial cells at the tip of the intestinal villi, and colocalized with tyrosine nitration, which is an index of peroxynitrite generation (Liaudet et al., 2000a). The finding that in various forms of shock, most of the PARP activation localizes to the tip of the villi is especially interesting because it markedly contrasts with the normal (baseline, noninflammatory) conditions, where the majority of PARP activation in intestinal villi is localized to the lower villi: nondividing but differentiating and maturing cells in the upper crypts and on the villi contain no more than approximately 10% of the synthetase activity of lower-crypt cell nuclei (Porteous et al., 1979). Intestinal PARP activation during hemorrhagic shock results in gut hyperpermeability, which developed in the wild-type but not in the PARP-deficient mice. PARP-deficient mice were also protected from the rapid decrease in blood pressure after resuscitation and showed an increased survival time, as well as reduced pulmonary neutrophil sequestration (Liaudet et al., 2000a). The beneficial effects of PARP suppression were not related to a modulation of the NO pathway or to a modulation of signaling through IL-6, which increased to the same degree in both wild-type and knockout mice exposed to hemorrhagic shock. There was no evidence of severe oxidant stress (no increase in tissue malondialdehyde levels and no depletion in reduced glutathione levels in any of the organs studied) (Liaudet et al., 2000a), which was indicative that PARP activation does not require prior oxidant stress or low antioxidant status in this particular experimental model, and it seems to develop at a relatively early stage of shock. In a large animal model of hemorrhagic shock, treatment with 3-aminobenzamide significantly ameliorated the decrease in blood pressure, cardiac output, and stroke work; slightly increased left atrial pressure during resuscitation; and significantly prolonged survival (Szabo et al., 1998a). PARP activation and associated cell injury probably plays crucial roles in the pathogenesis of intestinal injury, cardiovascular failure, and multiple organ damage associated with endotoxic and septic shock, as well as resuscitated hemorrhagic shock.
Pharmacological inhibition of PARP, either with 3-aminobenzamide (Szabo et al., 1998a), 5-iodo-6-amino-1,2,-benzopyrone (Szabo et al., 1997c), or PJ34 (Jagtap et al., 2002) improves survival rate in mice challenged with high-dose endotoxin. Also, several recent studies compared the survival times of wild-type and PARP-deficient mice in response to high-dose endotoxin and compared the degree and nature of liver damage in the two experimental groups. In one study, all PARP-deficient animals survived high-dose (20 mg/kg) LPS-mediated shock, which killed 60% of wild-type animals (Kuhnle et al., 1999). Similar results were obtained by another independent group led by de Murcia (Oliver et al., 1999). Moreover, LPS-induced necrotic liver damage was significantly reduced in the PARP-deficient mice (Kuhnle et al., 1999). In contrast, when apoptotic liver damage was induced via injection of low concentrations of LPS (30 μg/kg) intod-galactosamine-sensitized mice or via activation of hepatic cell-death receptors, PARP-deficient animals were not protected (Kuhnle et al., 1999). Thus, PARP activation is involved in systemic LPS toxicity, whereas it plays a minor role in apoptotic liver damage mediated by TNF or CD95.
As far as the mechanism of the protective effects of PARP inhibitors are concerned, two main pathways have been identified: PARP, in its basal, constitutive state, can act as a coinducer of proinflammatory gene expression in shock; and PARP activation can mediate inflammatory cell dysfunction and ultimately cell necrosis (see above). These two pathways may actually be interrelated in shock, because by reducing the production of inflammatory chemokines and cytokines, PARP inhibition actually reduces the amount of tissue-infiltrating mononuclear cells and thus results in the production of less genotoxic oxidants and free radicals, thereby attenuating the degree of DNA single-strand breakage and PARP activation and ultimately preventing cell dysfunction and/or necrosis (Fig. 7).
Most of the above-mentioned studies of endotoxic and hemorrhagic shock used pharmacological inhibitors of PARP. These studies are inherently problematic because of dosing issues, nonspecific actions, and vehicle problems. For instance, in a study by Wray and colleagues (1998), the PARP inhibitor 1,5-dihydroxyisoquinoline seemed to have exerted beneficial effects, but similar protective effects were also seen in the vehicle-controlled animals treated with the drug vehicle dimethyl sulfoxide alone. Recently, it became apparent that dimethyl sulfoxide is actually an inhibitor of PARP, an effect which may have influenced the outcome of the above-referenced study (Banasik and Ueda, 1999). Also, as discussed earlier, 3-aminobenzamide and nicotinamide possess antioxidant effects that are unrelated to PARP inhibition. These effects may have contributed to some of the beneficial effects observed in prior studies. The pharmacological studies using various PARP inhibitors were followed by more recent studies comparing wild-type and PARP-deficient mice subjected to circulatory shock. These latter studies have confirmed these findings and demonstrated that PARP-deficient mice are markedly protected from the lethality and organ dysfunction induced by these insults (Kuhnle et al., 1999; Liaudet et al. 2000a; Oliver et al., 1999).
Because endotoxin-induced shock does not share many characteristics of human sepsis—in fact, in many instances, novel therapeutic strategies that were protective in endotoxic shock were subsequently found to be not protective or even detrimental in sepsis induced by live bacteria—the next logical step of the investigation was to explore the potential role of the PARP pathway in sepsis induced by live bacteria. Using PJ34, we have observed that pharmacological inhibition of PARP improves survival in a porcine model of severe hypodynamic sepsis induced by E. coli clot implantation (Jagtap et al., 2002;Goldfarb et al., 2002). PJ34 is also effective in rodent models of endotoxic shock, as well as in a rodent model of cecal ligation and puncture (CLP)-induced mortality (Soriano et al., 2001a; Jagtap et al., 2002). In addition, we have recently compared the response to cecal ligation and puncture-induced severe polymicrobial sepsis in wild-type and PARP-deficient mice (Soriano et al., 2002). We have found that mice genetically deficient in PARP had significantly lower plasma levels of various cytokines (TNF-α, IL-6, IL-10) and exhibited a reduced degree of organ inflammation, indicated by decreased myeloperoxidase activity in gut and lung. Furthermore, there was a significant improvement of the survival rates of PARP−/− mice subjected to CLP when compared with the wild-type controls (Soriano et al., 2002). In contrast to severe models of septic shock, in a mild model of resuscitated CLP-induced septic shock in rats (characterized by no hemodynamic alterations, undetectable degree of oxidative and nitrosative stress, no changes in organ NAD+ and ATP levels, and low level of organ failure), inhibition of PARP with 3-aminobenzamide did not affect any of the outcome variables studied (Baechtold et al., 2001). As discussed earlier [seePoly(ADP-Ribose) Polymerase in Stroke], it is possible that PARP only becomes activated in the most severe forms of disease, and this is when its inhibition can be expected to offer significant therapeutic benefit.
Although it has been hypothesized for several years that PARP activation and the related energetic impairments contribute to the diminished tissue-oxygen extraction in shock (e.g., Szabo, 1996;Liaudet et al., 2001a; Fink, 2002), the first direct evidence to prove this hypothesis appeared recently and only in a reductionist in vitro system. In immunostimulated CaCo-2 intestinal epithelial cells, the impairment of cellular oxygen use has been shown to be preventable by inhibition of PARP with PJ34 (Khan et al., 2002). Similar studies in various experimental models of circulatory shock are required to directly test this attractive hypothesis.
From the above-described observations, one can conclude that in response to pharmacological inhibition or genetic deletion of PARP, the improved hemodynamic status in shock and sepsis is caused by improved cardiac and vascular function and possibly by the improved cellular energetic status in some organs. These improvements, in turn, result in an overall survival benefit in this condition. Despite the impressive number of published studies, it is obvious that much further work is required to delineate the exact role of PARP in sepsis. For example, to date, there are no reports with PARP inhibitors in large animal models of hyperdynamic sepsis. We believe that future work in this area of research should use appropriate, clinically relevant, and fairly severe models of bacterial sepsis rather than ones with mild injury. Subjects to be addressed in these further studies should include the best dosing regimen to be used and the identification of the therapies that work additively or synergistically when coadministered with PARP inhibitors. Also, future studies must examine the degree, tissue and disease heterogeneity, and time course of PARP activation in humans.
M. PARP in the Pathogenesis of Diabetes
Type 1 or insulin-dependent diabetes mellitus is an autoimmune disease occurring predominantly in children and young adults resulting in the destruction of the pancreatic β cells but not the other endocrine islet cells. The actual trigger for the process of β-cell destruction is poorly characterized but is either an external factor (viral, chemical) or an internal stimulus (cytokines, free radical) that damages a proportion of the β cells leading to the release of specific β-cell proteins, which can be taken up by antigen-presenting cells and processed to antigenic peptides. The process involves the transcription of cytokine genes including interferon-γ, which can feed back onto the antigen-presenting cells to increase expression of IL-1β and TNF-α. The T-helper cells also activate B-lymphocytes, which produce islet cell autoantibodies, and this is followed by cytotoxicity by killer cell activation. In the serum of a large proportion of individuals having an increased risk of developing type 1 diabetes, autoantibodies against specific β-cell antigens such as insulin (Atkinson et al., 1986), proinsulin (Kuglin et al., 1988), and glutamic acid decarboxylase (Baekkeskov et al., 1990) have been detected. Cytotoxic T-lymphocytes are also activated via the T-helper cells. The products of immune cell activation including cytokines and free radicals are the direct participants in inducing β-cell death. The final event in the autoimmune process is the removal of cell debris by macrophages.
The pathogenesis of type 1 diabetes has been mostly investigated in rodent models of type 1 diabetes. Initial models used β-cell toxins such as streptozotocin, which was found to be able to induce diabetes either directly or via an immune cell-mediated mechanism depending on whether a single large dose (Rerup, 1970) or multiple low doses (Rossini et al., 1977; Kolb, 1987) were administered. The latter model is widely known as multiple-low-dose streptozotocin (MLDS) diabetes. Destruction of the β cells in diabetes has been attributed to the production of various immune-cell mediators such as cytokines and free radicals produced in the islet itself. Major mediators of β-cell death seem to be NO and various related free-radical and oxidant species. High concentrations of NO are produced in the islet directly from the infiltrating macrophages (Kleemann et al., 1993) and indirectly from the induction of iNOS in various cell types of the islet after exposure to immune cell-derived proinflammatory cytokines. The induction of iNOS and production of free-radical species have been implicated in the development of diabetes in many of the animal models, with iNOS-deficient mice being protected from streptozotocin-induced diabetes (Flodstrom et al., 1999) and iNOS being detected in the pancreas of the nonobese diabetic (NOD) mouse (Rabinovitch et al., 1996) along with evidence of peroxynitrite formation (Suarez-Pinzon et al., 1997). Isolated rat, mouse, and human islets of Langerhans have frequently been used to identify the events involved in β-cell dysfunction and death in diabetes. Exposure of islets to appropriate combinations of proinflammatory cytokines results in the inhibition of insulin secretion (Southern et al., 1990; Corbett et al., 1993; Eizirik et al., 1994), which is mediated by NOS induction and subsequent free-radical formation (Southern et al., 1990; Corbett et al., 1993). Cytokines also inhibit islet DNA synthesis (Khatim et al., 1988) and glucose oxidation through the inhibition of the mitochondrial enzyme aconitase (Green et al., 1994). Cytokines were also found to decrease islet ATP (Green et al., 1994), cAMP (Green et al., 1994), and NAD+ levels (Fehsel et al., 1993; Bolaffi et al., 1994; Radons et al., 1994) and to inhibit insulin biosynthesis (Green et al., 1994). Cytokine combinations also cause DNA damage, an effect that is mediated by NO and related radicals and oxidants (Delaney et al., 1993; Fehsel et al., 1993). The final outcome of exposure of islets to cytokines is cell death: necrosis (Kroncke et al., 1991) and/or apoptosis (Kaneto et al., 1995). Many of the effects induced in islet cells by cytokine treatment have been duplicated using chemically generated free radicals such as NO, oxygen-free radicals, and peroxynitrite. Inhibition of insulin secretion (Cunningham et al., 1994; Eizirik et al., 1996), induction of DNA damage (Delaney et al., 1993; Fehsel et al., 1993; Hadjivassiliou et al., 1998), and islet cell lysis (Heller et al., 1994) and apoptosis (Kaneto et al., 1995) have all been observed in rat and human islets exposed to NO, reactive oxygen species, or peroxynitrite in vitro. The consequences to the β-cell of this cellular dysfunction are multiple and involve a variety of oxidant-mediated protein and lipid modifications.
Okamoto and his coworkers (Uchigata et al., 1982) proposed that the primary DNA damage and subsequent decrease in cellular NAD+ levels are linked by activation of PARP. They proposed that the decrease in NAD+ levels is responsible for the loss of cellular ATP and leads to the inhibition of proinsulin biosynthesis, ultimately resulting in the loss of β-cell viability and cell death (Uchigata et al., 1982). For the last two decades, the role of PARP activation in the process of β-cell death has been subject to extensive investigations in vitro. Islets of Langerhans isolated from rat, mouse, or human are all functionally inhibited and ultimately destroyed by inflammatory cell mediators such as cytokines and free radicals, as well as chemical β-cell toxins such as streptozotocin and alloxan. The role of PARP in these processes has been investigated in a multitude of studies using various pharmacological enzyme inhibitors. The application of streptozotocin to isolated mouse and rat islets results in the formation of DNA strand breaks (Wilson et al., 1988), activation of PARP (Wilson et al., 1988), and a decrease in β-cell NAD+ (Bolaffi et al., 1987) and proinsulin content (Sandler et al., 1983), along with an inhibition of insulin secretion (Masiello et al., 1985, 1990). Application of nicotinamide, 3-aminobenzamide, or thymidine, although not preventing the DNA damage induced by streptozotocin, protected the islets' functionality by preventing the decrease in NAD+ and proinsulin as well as by partially reversing the inhibition of insulin secretion (Masiello et al., 1985,1990; Bolaffi et al., 1987). The reversibility of the streptozotocin-induced damage in β-cells by PARP inhibitors is a function of the degree of preservation of intracellular NAD+ pools. The role of PARP in the deleterious effects of cytokines on isolated islets is not as clear as with the chemically induced damage. This finding is possibly caused by the use of nicotinamide for these studies, because at the doses used, nicotinamide also acts as a protein synthesis inhibitor and free radical scavenger (in addition to being a PARP inhibitor). Nicotinamide has been shown to have either no effect or a partial protective effect against cytokine-inhibited insulin release or stimulation of NO formation from isolated islets (Fehsel et al., 1993; Reddy et al., 1995). The use of a more potent PARP inhibitor, INH2BP, does reverse cytokine-mediated inhibition of glucose-stimulated insulin secretion, an effect found to be independent of the inhibition of NO formation (Mabley et al., 2001b). Nicotinamide also reverses the inhibitory effects of IL-1β on accumulated insulin release and NO production (Andersen et al., 1994). Exposure of human islets to cytokine combinations also causes islet cell destruction, as determined by islet DNA and insulin contents which decrease dramatically (Rabinovitch et al., 1994); again, these effects are reversed by nicotinamide (Rabinovitch et al., 1994). Nicotinamide prevented cytokine-mediated lysis of mouse islet cells (Yamada et al., 1990b) but was unable to protect rat, mouse, or human islets from cytokine-mediated apoptosis (Hoorens and Pipeleers, 1999). This observation is in line with the evidence that the cell-death pathway triggered by PARP activation and NAD+depletion is necrosis rather than apoptosis. NO has been shown to be the major mediator of cytokine-induced damage, and the induction of NOS in islet cells by cytokines is totally dependent on NF-κB activation (Flodstrom et al., 1996). Reports have implicated PARP in promoting the activation of NF-κB (Oliver et al., 1999) but in the context of islet cell function, this promotion seems to be due to the physical presence of the enzyme (through molecular scaffolding functions and physical association) rather than its catalytic activity; thus the activation of NF-κB is less affected by pharmacological inhibitors than by genetic disruption of the PARP gene (Andrade et al., 1996; Oliver et al., 1999;Soriano et al., 2001c).
Exposure of isolated islets to NO, reactive oxygen species, or hydrogen peroxide, either generated chemically or from activated macrophages, results in the inhibition of insulin secretion, decrease in NAD+ and proinsulin levels, and eventual islet cell lysis—effects associated with the activation of PARP. Although nicotinamide failed to prevent the formation of primary DNA damage in radical exposed cells, the presence of the compound effectively inhibited the activation of PARP, as assessed by the complete lack of poly(ADP-ribose) formation and by the preservation of intracellular NAD+ concentrations. Nicotinamide and 3-aminobenzamide both improved the survival of rat islet cells exposed to chemically generated NO (Kallmann et al., 1992; Radons et al., 1994) or to reactive oxygen species generated by xanthine oxidase (Burkart et al., 1992; Heller et al., 1994). Nicotinamide also protected human islets from hydrogen peroxide-induced necrosis (Hoorens and Pipeleers, 1999). Nicotinamide was even able to protect rat islet cells cocultured with syngeneic-activated macrophages from cell lysis via a nitric oxide-dependent mechanism (Burkart and Kolb, 1993). It was also found that nicotinamide was able to inhibit iNOS mRNA induction in activated macrophages (Pellat-Deceunynck et al., 1994), an effect of the compound which may have also contributed to the β-cell protection. A recent study showed that peripheral blood from newly diagnosed diabetic patients incubated with nicotinamide produced significantly less IL-12 and TNF-α (Kretowski et al., 2000). This observation suggests that nicotinamide is able to influence monocyte/macrophage function in peripheral human blood, thus providing another potential mechanism of protection against the development of diabetes.
The combined results from the in vivo and in vitro studies using PARP inhibitors indicate that protection from diabetes development, as observed after administration of PARP inhibitors in rodents, is mediated by direct protection against the necrotic damage of the β cells and may be related to the suppression of proinflammatory mediator production. The particularly large concentrations of the PARP inhibitors used in vitro to produce the protective effects, in the millimolar range, give rise to many nonspecific pharmacological effects other than PARP inhibition. Nicotinamide at 5- to 50-mM concentrations used in vitro to protect the islets is able to inhibit enzymes other than PARP (Kolb and Burkart, 1999) as well as to interfere with transcriptional (Pellat-Deceunynck et al., 1994) and translational processes (Hauschildt et al., 1991, 1992). Although the more potent PARP inhibitors (which are active in the micromolar or nanomolar range) exert protective effects similar to those of nicotinamide and 3-aminobenzamide against cytokine-mediated inhibition of insulin secretion and islet cell destruction. To truly define the role of PARP in diabetes, the engineering of a mouse deficient in the PARP gene was required. The development of a PARP-deficient mouse has allowed for the direct examination of the role of PARP in type 1 diabetes. In 1995, Kolb and colleagues (Heller et al., 1995) isolated islets from the newly engineered PARP-deficient mouse (Wang et al., 1995) and exposed them to nitric oxide or reactive oxygen species. The islets isolated from PARP−/− mice had no NAD+ depletion and were resistant to both NO and reactive oxygen species toxicity, providing evidence that PARP activation is responsible for most of the loss of NAD+ after such treatment. However, PARP−/− islets were not completely resistant to lysis induced by nitric oxide and reactive oxygen species, especially at higher concentrations. Furthermore, application of a PARP inhibitor, 3-aminobenzamide, failed to protect against the islet lysis induced by extremely high levels of NO or reactive oxygen species, indicating the presence of an alternative pathway of cell death independent of PARP activation and NAD+ depletion.
Thus, the hypothesis was formulated that primary DNA damage in the β-cell leads to activation of PARP with subsequent depletion of intracellular NAD leading to the inhibition of proinsulin synthesis, β-cell necrosis, and diabetes. Subsequently, various pharmacological inhibitors of PARP were tested in all animal models of diabetes. By far the most common inhibitor used is nicotinamide. Other PARP inhibitors have also been used, including benzamide analogs such as 3-aminobenzamide, and more recently isoquinolines, benzopyrones, and phenanthridinones. Many in vivo studies used chemically induced animal models of diabetes in which diabetes is induced in the rat by a single intravenous or intraperitoneal injection of streptozotocin. One of the earliest studies with nicotinamide was conducted by Schein et al. (1967). These investigators found that intraperitoneal injection of 500 mg/kg nicotinamide preserved β-cell function and prevented the development of diabetes in rats treated with a single high dose of streptozotocin. This protection from hyperglycemia was coupled with a preservation of islet NAD+ levels and prevention of β-cell destruction. Inhibitors of PARP, including nicotinamide and 3-aminobenzamide injected intravenously to rats, were found to prevent both alloxan- and streptozotocin-induced depletion of NAD+ and inhibition of proinsulin synthesis (Yamamoto and Okamoto, 1980; Uchigata et al., 1982, 1983; Okamoto and Yamamoto, 1983). Further studies with 3-aminobenzamide indicated that this compound prevented the development of diabetes in streptozotocin-treated rats (Masiello et al., 1985). PARP inhibitors have been far less potent in protecting against the MLDS model of diabetes, which is usually induced in mice rather than in rats. The induction of diabetes in the MLDS model involves not only the direct destruction of β cells by streptozotocin, but the indirect destruction by induction of an autoimmune response against the β cell. Nicotinamide protected MLDS-treated mice from hyperglycemia (Rossini et al., 1977; Bouix et al., 1995) but only had a weak effect on the development of insulitis (Mendola et al., 1989b). The more potent inhibitors of PARP such as the benzopyrone INH2BP or the phenanthridinone derivative PJ34 (Soriano et al., 2001c; Mabley et al., 2001b) exert much more dramatic protective effects than nicotinamide. These observations suggest that PARP inhibition greatly protects pancreatic β cells from both streptozotocin and immune cell-mediated destruction.
Nicotinamide has also been used to influence the onset of diabetes in the two genetic models of type 1 diabetes with varying degrees of success. In the NOD model, recent studies provided evidence for the presence of peroxynitrite (Suarez-Pinzon et al., 1997), a potent NO-derived oxidant, which is able to induce pancreatic β cell dysfunction and death in vitro and which is a potent activator of PARP because of its ability to induce DNA single-strand breakage (see above). By using specific antibodies and immunohistochemical methods, it was found that cells positive for nitrotyrosine (an indicator of peroxynitrite generation) were found significantly more frequently in islets from acutely diabetic NOD mice than in islets from normoglycemic NOD mice and control BALB/c mice. The nitrotyrosine-positive cells in islets were identified to be macrophages and also β cells. Most of the β cells in islets from acutely diabetic NOD mice were nitrotyrosine-positive, whereas significantly fewer β cells were nitrotyrosine-positive in islets from normoglycemic NOD mice and BALB/c mice. Also, the percentage of β cells in islets from NOD mice (normoglycemic and diabetic) correlated inversely with the frequency of nitrotyrosine positive β cells (Suarez-Pinzon et al., 1997). In the NOD mouse nicotinamide prevents the development of both the spontaneous (Yamada et al., 1982; Reddy et al., 1990) and cyclophosphamide-accelerated (O'Brien et al., 2000) models of type 1 diabetes. Unlike the MLDS model of diabetes, in the NOD model, nicotinamide was able to suppress insulitis by a significant degree (Reddy et al., 1990) as well as reduce major histocompatibility complex class II protein expression in the pancreas of NOD mice (Papaccio et al., 1999). These observations indicate a role for PARP in immune-cell infiltration in inflammatory conditions, an effect also observed in other diseases (Szabo et al., 1997b). In fact, the treatment of NOD mice with nicotinamide for only 5 weeks at a very early age is able to suppress diabetes over the following 25 weeks (Kim et al., 1997), indicating that PARP plays a fundamental role in the early stages of diabetes development. In recent studies, long-term treatment of NOD mice with the potent PARP inhibitor PJ34 protected against the cyclophosphamide-accelerated induction of diabetes, and when PJ34 treatment was started at 5 weeks of age, it markedly suppressed the spontaneous development of diabetes (Mabley et al., 2001a). The protective effects of PARP inhibitors in the NOD model of diabetes are possibly related to inhibiting β-cell necrosis and preventing immune-cell activation by proteins released from necrotizing islet cells.
In 1999, three independent groups reported (Burkart et al., 1999;Masutani et al., 1999; Pieper et al., 1999) in short succession the massive resistance of PARP−/− mice to streptozotocin-induced diabetes. In all three studies, PARP−/− mice were injected with a single dose of streptozotocin, and in all cases, the control mice developed a severe hyperglycemia within 1 week of the injection. This action was coupled with a marked decrease in pancreatic insulin levels and morphological analysis showing significant atrophy of the islets caused by a marked loss of β-cells. In contrast, the PARP−/− mice had normal blood glucose concentration levels, with no evidence of islet atrophy, β-cell loss, or altered cellular arrangement. In fact, the islets exhibited a histological appearance that was similar to that of the vehicle-treated nondiabetic mice. The complete protection of mice lacking the PARP gene from streptozotocin-induced diabetes is remarkable and identifies PARP as an essential downstream executor in the development of diabetes in this animal model. The susceptibility of PARP−/− mice to the MLDS model of diabetes has also been investigated recently by our group (Mabley et al., 2001b). As noted above, the MLDS model has a significant immune-cell component in the β-cell destruction and is characterized by a progressive hyperglycemia and an insulitis similar to that observed in patients with recent-onset type 1 diabetes. PARP−/− mice are significantly less susceptible to MLDS-induced diabetes, with a lower disease incidence and decreased hyperglycemia than the wild-type animals. This resistance against diabetes is coupled with a higher insulin content and therefore increased β-cell mass in MLDS-treated PARP−/− mice, as compared with treated PARP+/+ mice. However, it is notable that no complete protection was observed when these mice were treated with a single high dose of streptozotocin, indicating that other pathways activated via the immune system can also lead to β-cell destruction (Mabley et al., 2001b). This finding is consistent with the in vitro data (Heller et al., 1995), demonstrating the presence of a PARP-independent pathway of islet cell death in response to nitric oxide and reactive oxygen species.
It is interesting to note that a recent study reported that after the introgression of a disrupted PARP-1 allele onto the autoimmune diabetes-prone NOD mouse strain, these mice were protected neither from spontaneous nor from cyclophosphamide-accelerated diabetes (Gonzalez et al., 2002). Surprisingly, they were also highly sensitive to the diabetes induced by a single high dose of streptozotocin (Gonzalez et al., 2002), standing in sharp contrast with C57BL/6 mice that bear the same inactivated PARP-1 allele (see above). Although the mechanism of this unexpected behavior remains clarified, these results suggest that NOD mice are characterized not only by their immune dysfunction but also by a peculiarity of their islets leading to a PARP-1-independent mechanism of streptozotocin-induced β-cell death.
The studies with the PARP-deficient mice have provided strong evidence for the depletion of NAD+ resulting from PARP activation as the dominant metabolic event in the destruction of the β-cell after DNA damage. PARP inhibition improves the resistance of the β-cell toward the deleterious effects of proinflammatory mediators such as cytokines and free radicals and presents a clear therapeutic target for the prevention of type 1 diabetes. Accordingly, the concept of protecting β-cells from inflammatory damage by nicotinamide was introduced into human clinical trials early on. The early small-scale promising findings (Mendola et al., 1989a; Pozzilli et al., 1989, 1996; Elliott and Chase, 1991; Elliott et al., 1993; Kolb and Burkart, 1999) were followed by subsequent smaller (Chase et al., 1990; Pozzilli et al., 1994a,b; Visalli et al., 1999; Vidal et al., 2000) and larger (Lampeter, 1993, 1998; Gale, 1996) studies that failed to show significant therapeutic efficacy. It is important to note that even if PARP is a valid target to prevent type 1 diabetes, nicotinamide may not be a potent enough pharmacological agent to prevent disease development. The development of more potent and specific inhibitors of PARP may mean that true prevention of type 1 diabetes in humans occurs only when these inhibitors are available for human use.
N. PARP in the Pathogenesis of Diabetic Cardiovascular Dysfunction
In established diabetic patients, the quality of life and life expectations are determined by the complications of diabetes rather than by the primary disease. Among these complications, vasculopathies affecting both the micro- and macrocirculation (evidenced clinically, among others, by accelerated atherosclerosis, dysfunction of the eye and kidney, diminished blood flow to extremities, and increased risk of developing a variety of cardiovascular diseases) are probably the most dominant factors. The major cause of mortality in diabetes is macrovascular disease affecting the cardiac and cerebrovascular circulation, which seems to have a more complex pathogenesis (Keen et al., 1999; Tooke and Goh, 1999; De Vriese et al., 2000; Schaper et al., 2000; Standl and Schnell, 2000). The processes involved in atherothrombotic disease are complex and include variation in lipid metabolism, vascular responses, cell-cell interactions, and the fluid and cellular phases of coagulation and fibrinolysis. The complex interactions between all of these processes are crucially altered by the metabolic milieu that characterizes diabetes mellitus, tipping the delicate balance toward atheroma formation, platelet aggregation, and thrombus formation (Keen et al., 1999; Tooke and Goh, 1999; De Vriese et al., 2000; Schaper et al., 2000; Standl and Schnell, 2000). In contrast to macrovascular alterations, diabetes-associated microvascular disease has been strongly related to glycemic control. Hyperglycemic episodes occur even in the most balanced forms of diabetes mellitus and are closely associated with the development of vascular failure. A significant portion of diabetic patients will eventually develop some degree of vascular failure.
The vascular tone is regulated by various neurohumoral mediators and mechanical forces acting on the innermost layer of blood vessels, the endothelium. The main pathway of vasoregulation involves the activation of the endothelial isoform of NO synthase (eNOS) resulting in NO production (Furchgott, 1999; Ignarro, 1999). Endothelium-dependent vasodilatation is frequently used as a reproducible and accessible parameter to probe endothelial function in various pathophysiological conditions (Furchgott, 1999; Ignarro, 1999; De Caterina, 2000). It is well established that endothelial dysfunction, in many diseases, precedes and predicts as well as predisposes for the subsequent, more severe vascular alterations. Endothelial dysfunction has been documented in various forms of diabetes and has many pathogenetic components, including increased polyol pathway flux, altered cellular redox state, increased formation of diacylglycerol, the subsequent activation of specific protein kinase C isoforms, and accelerated nonenzymatic formation of advanced glycation end products (King, 1996;Carter and Grant, 1997). In addition, recent studies have established that oxygen- and nitrogen-derived oxidants and free radicals play a significant role in diabetes-associated endothelial dysfunction (Giugliano et al., 1996; Honing et al., 1998; Lyall et al., 1998; Cai and Harrison, 2000). The cellular sources of reactive oxygen are multiple and include advanced glycation end products, NADH/NADPH oxidases, and the mitochondrial electron transport chain (Nishikawa et al., 2000). We recently found that high glucose-induced oxidative stress leads to DNA single-strand breakage and PARP activation in murine and human endothelial cells (Soriano et al., 2001c). The involvement of oxyradicals and NO-derived reactive species in PARP activation and the evidence for nitrated tyrosine residues both suggested that peroxynitrite may be one of the final mediators responsible for single strand-breakage and subsequent PARP activation (Soriano et al., 2001c). To test the relevance of a PARP-dependent mechanism for the high glucose-induced cell dysfunction, we decided to measure cellular pyridine nucleotide concentrations in the endothelial cells exposed to high glucose. There was a severe suppression of cellular high-energy phosphate levels as well as a suppression of NAD+ and NADPH levels in endothelial cells exposed to high glucose levels for 1 to 2 days. These effects were prevented by PARP inhibition or by PARP−/−phenotype (Soriano et al., 2001c). Because eNOS is a NADPH-dependent enzyme, we proposed that the cellular depletion of NADPH in endothelial cells exposed to high glucose is directly responsible for the suppression of eNOS activity and the reduction in the diabetic vessels' endothelium-dependent relaxant ability (Fig.8). In support of this hypothesis, we subsequently demonstrated that there is a PARP-dependent suppression of vascular NADPH levels in diabetic blood vessels in vivo (Soriano et al., 2001c). In these latter studies, we induced diabetes in mice and rats by streptozotocin treatment, and PARP inhibition treatment by PJ34 was delayed to limit its interference with the primary process of islet cell destruction. The activation of PARP in the blood vessels was already apparent 2 weeks after the onset of diabetes, and thus it slightly preceded the occurrence of the endothelial dysfunction, which developed between the second and fourth week of diabetes (Soriano et al., 2001b). Ex vivo experiments demonstrated the loss of endothelial function, as measured by the relaxant responsiveness of precontracted vascular rings to acetylcholine. Delayed treatment with the PARP inhibitor, starting 1 week after streptozotocin, ameliorated vascular poly(ADP-ribose) accumulation and restored normal vascular function without altering systemic glucose levels, plasma-glycated hemoglobin levels, or pancreatic insulin content (Soriano et al., 2001c). Furthermore, delayed treatment of the animals with the PARP inhibitor restored the established diabetic endothelial dysfunction, and even in vitro incubation of diabetic dysfunctional blood vessels with PARP inhibitors of various structural classes (an aminobenzamide, an isoquinoine, and a phenanthridinone derivative) was able to improve the endothelium-dependent relaxant responsiveness (Soriano et al., 2001b). A recent study extended our knowledge on the role of PARP activation in the development of diabetic endothelial dysfunction into the area of human investigations: in forearm skin biopsies from healthy subjects, healthy individuals with parental history of type 2 diabetes, subjects with impaired glucose tolerance, and a group of patients with type 2 diabetes, it was found that the percentage of PARP-positive endothelial nuclei was higher in the group with parental history of type 2 diabetes and diabetic patients when compared with the control patients. Immunoreactivity for nitrotyrosine (a marker of reactive nitrogen species) was also higher in the diabetic group when compared with all other groups (p < 0.01). No differences in the expression of eNOS and the receptor for advanced glycation end products were found among all four groups. The polymorphism of the eNOS gene was also studied and was not found to influence eNOS expression or microvascular functional measurements. Thus, one can conclude that in humans, PARP activation is present in healthy subjects at risk of developing diabetes, as well as in patients with established type 2 diabetes, and it is associated with impairments in the vascular reactivity in the skin microcirculation (Komjati et al., 2002).
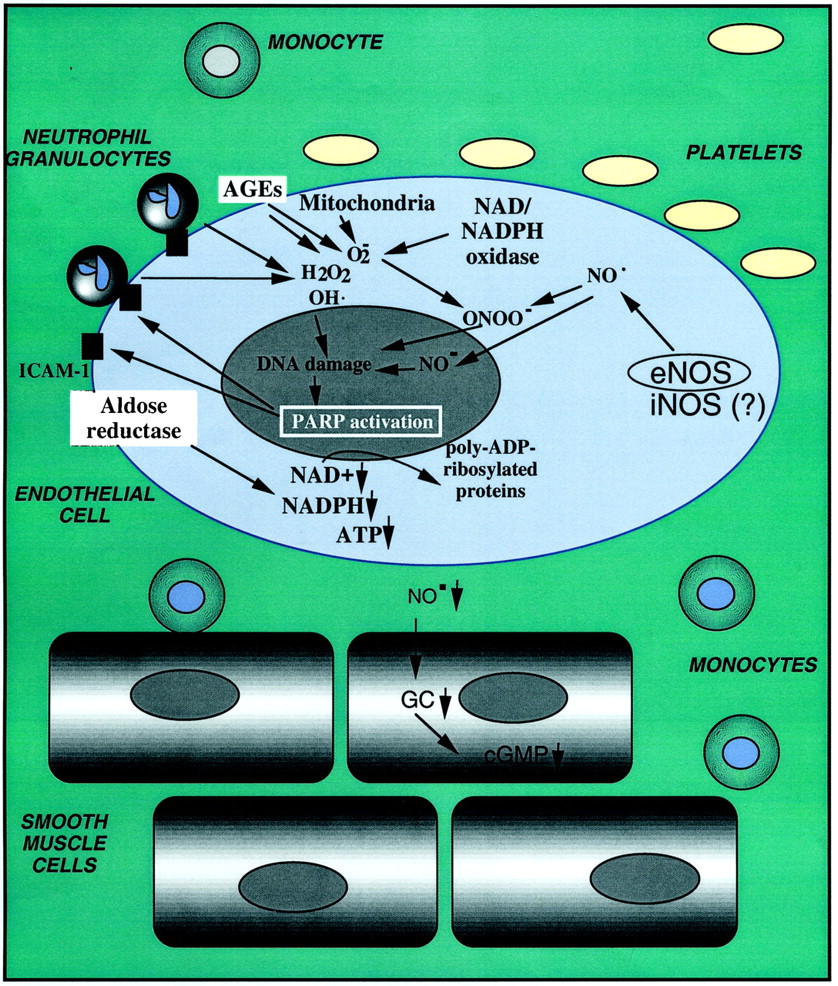
Role of PARP in the development of endothelial cell dysfunction in diabetes mellitus. PARP-dependent and PARP-independent cytotoxic pathways involving nitric oxide (NO⋅), hydroxyl radical (OH⋅), and peroxynitrite (ONOO−) in the context of diabetic endothelial dysfunction. The vascular tone is regulated by various neurohumoral mediators such as acetylcholine, ATP, ADP, and bradykinin and mechanical forces (e.g., pulsatile flow and shear stress) physiologically activating the innermost layer of blood vessels, the endothelium. The main pathway of vasoregulation involves the mobilization of intracellular calcium in endothelial cells, followed by the activation of eNOS, resulting in NO production. The enzyme uses the substrates l-arginine and molecular oxygen to produce NO and is dependent on a variety of substrates, including NADPH and tetrahydrobiopterin. NO produced by eNOS then diffuses to the smooth muscles, activates soluble guanylyl cyclase (GC) and thereby triggers a cGMP-mediated relaxation of the vascular smooth muscle. The NO released from the endothelium also maintains an antiatherogenic intravascular surface and inhibits the adhesion and activation of neutrophils, monocytes, and platelets. In diabetes, transiently of persistently elevated high-circulating glucose in diabetes interrupts the normal homeostatic functions of the vascular endothelium. First, hyperglycemia triggers the release of oxidant mediators from the mitochondrial electron transport chain, from NADH/NADPH oxidase from advanced glycation end products (AGEs), and from other sources. High glucose may also up-regulate eNOS expression or may trigger the expression of iNOS in the endothelium. NO, in turn, combines with superoxide to yield peroxynitrite. Hydroxyl radical (produced from superoxide via the iron-catalyzed Haber-Weiss reaction) and peroxynitrite or peroxynitrous acid induce the development of DNA strand breakage, with consequent activation of PARP. Depletion of the cellular NAD+ leads to the inhibition of cellular ATP-generating pathways, leading to a global cellular dysfunction. This energetic failure may culminate in endothelial cell necrosis. The PARP-triggered depletion of cellular NADPH directly impairs the endothelium-dependent relaxations. The effects of elevated glucose are also exacerbated by increased aldose reductase activity, leading to the depletion of NADPH and generation of reactive oxidants. NO alone does not induce DNA single-strand breakage, but it may combine with superoxide (produced from the mitochondrial chain or from other cellular sources) to yield peroxynitrite. PARP activation, via a manner that is not yet characterized, promotes the activation of NF-κB, AP-1, MAP kinases, and the expression of proinflammatory mediators, adhesion molecules such as ICAM-1, and iNOS. PARP-independent parallel pathways of cellular metabolic inhibition can be activated by NO, hydroxyl radical, superoxide, and peroxynitrite. By promoting neutrophil recruitment and oxidant generation, positive-feedback cycles are triggered. PARP activation and mitochondrial oxidant production form another positive-feed-back cycle. Ultimately, the reduced NO output from the endothelial cells reduces the antithrombotic properties of the endothelial surface and triggers the adhesion and activation of platelets. The reduced endothelial NO output reduces the basal vasodilatory tone of the vascular smooth muscle, leading to transient occurrence of chronic vasospasm, end-organ ischemia, and increased incidence of cardiovascular events such as coronary vasospasm, myocardial infarction, or stroke. Activation and intravascular migration of mononuclear cells may also promote atherogenesis.
Although most of the studies on the role of PARP in the pathogenesis of diabetic endothelial dysfunction, as discussed above, originated in macrovessels, there is circumstantial evidence that similar processes are operative for the pathogenesis of diabetic microvascular injury (retinopathy and nephropathy). For example, our group has recently provided evidence for PARP activation in the microvessels of the diabetic retina (Szabo et al., 2001a). In addition, a study more than a decade ago demonstrated that the presence of glomerular depositions (mesangial distribution) of IgG, as evaluated with immunofluorescence technique, was significantly reduced in streptozotocin-diabetic rats treated with nicotinamide for 6 months (Wahlberg et al., 1985). Further studies using potent and specific inhibitors of PARP are needed to further delineate the role of PARP in the pathogenesis of diabetic retinopathy, neuropathy, and nephropathy.
The presence of myocardial dysfunction independent of coronary artery disease in diabetes mellitus has been well documented in both humans and animals (Fein, 1990; Illan et al., 1992; Regan et al., 1994; Bell, 1995; Joffe et al., 1999). This diabetic cardiomyopathy is characterized by an early diastolic dysfunction and a late systolic one, with intracellular retention of calcium and sodium and loss of potassium. The mechanism of diastolic dysfunction remains unknown, but it does not seem to be caused by changes in blood pressure, microvascular complications, or elevated circulating glycated hemoglobin levels (Bell, 1995; Gough et al., 1995). Recent data demonstrate that the PARP pathway also plays a role in the pathogenesis of diabetic cardiomyopathy. Cardiac dysfunction was noted both in the streptozotocin-induced and genetic (nonobese diabetic) models of diabetes mellitus in rats and mice. Development of diabetes was accompanied by hyperglycemia, PARP activation in the diabetic myocardium, a selective loss of endothelium-dependent vasodilation in the thoracic aorta, and an early diastolic dysfunction of the heart. Treatment with the phenanthridinone-based PARP inhibitor PJ34 starting 1 week after the onset of diabetes restored normal vascular responsiveness and significantly improved cardiac dysfunction, despite the persistence of severe hyperglycemia. The beneficial effect of PARP inhibition persisted even after several weeks of discontinuation of the treatment (Pacher et al., 2002b). It is possible that the diabetic endothelial PARP pathway and the diabetic cardiomyopathy are interrelated: an impairment of the endothelial function may lead to global or regional myocardial ischemia, which may secondarily impair cardiac performance. It is noteworthy that the protective effect of PARP inhibition against diabetic cardiac dysfunction extended several weeks beyond the discontinuation of treatment; this observation may have important implications for the design of future clinical trials with PARP inhibitors. The pharmacokinetic profile of PJ34 supports the view that the prolonged persistence of the effect of PJ34 is not related to the continued presence of the inhibitor, but it may be related to the permanent interruption by the PARP inhibitor of positive-feedback cycles of cardiac injury. Previous studies in various pathophysiological conditions have demonstrated that PARP inhibitors suppress positive-feedback cycles of adhesion-receptor expression and mononuclear cell infiltration, as well as intracellular oxidant generation (see Sections III.D. and III.F.). It is also conceivable that the degree of PARP activation may be more pronounced at the onset of the development of diabetic cardiovascular complications (as compared with a later stage of the disease), and when PARP is inhibited at an earlier time, this may result in more sustained beneficial effects. The beneficial effect of PJ34 on myocardial function is not related to an anabolic effect because PJ34 treatment did not influence the body and heart weight loss in diabetic animals, whereas it dramatically improved cardiac function.
The role of PARP activation in diabetes is not limited to the primary disease (islet cell death) and the development of various forms of cardiovascular dysfunction. A vast body of evidence supports the role of PARP activation in the process of islet cell regeneration (Yonemura et al., 1984, 1988; Terazono et al., 1988; Sugiyama et al., 1991;Akiyama et al., 2001; Bernard-Kargar and Ktorza, 2001). Furthermore, PARP activation seems to play a pathogenetic role in the rejection of transplanted islet cells, and PARP inhibitors may prolong the half-life of pancreatic-protected islets from dysfunction after transplantation (Nomikos et al., 1986; Sandler and Andersson, 1988; Kenmochi et al., 1994; Marquet et al., 1994; Otonkoski et al., 1997, 1999).
There may be a significant role of PARP activation in the pathogenesis of diabetic neuronap conductance (diabetic neuropathy). Structurally diverse PARS inhibitors, 3-aminobenzamide and 1,5-isoquinolinediol, correct established nerve blood flow and conduction deficit and energy deficiency in diabetic rats. In addition, when put onto a galactosamine-containing diet PARS+/+ mice slowly developed sciatic motor and hindlimb digital sensory nerve conduction deficits, whereas PARS−/− mice on the same diet preserved normal motor and sensory nerve conduction. Nerve energy state, assessed from phosphocreatine concentrations and phosphocreatine/creatine ratios as well as reduced glutathione concentration were compromised in the wild-type group, but remained stable in the PARP-deficient mice. The findings support a role for PARP activation in functional and metabolic deficits characteristic for, at least, early diabetes-like neuropathy (Obrosova et al., 2002). Further studies are required to determine whether PARP inhibitors can also restore neuronal function (as opposed to preventing its development).
Taken together, multiple lines of evidence support the view that PARP activation plays a crucial role in multiple interrelated aspects of diabetes and its complication, and it is justified to expect that potent, bioavailable, and nontoxic PARP inhibitors, when available, will exert beneficial effects against the development of both the primary diabetes and its cardiovascular complications.
O. PARP Inhibitors as Adjuvant Therapeutics for the Treatment of Various Forms of Cancer
Tumor cells derived from benign or malignant tumors have been shown to have perturbations in poly(ADP-ribose) metabolism. For example, Tomoda and colleagues (1991) found enhanced expression for the PARP-1 gene in all five malignant lymphomas tested, but no increase in the level of the mRNA was observed in any reactive proliferative cases or normal lymph nodes. Furthermore, in low-grade malignant non-Hodgkin lymphoma cells, a high cellular PARP content (DNase-induced PARP activity), unchanged basal PARP activity, and low level of protein-bound ADP-ribose were found compared with normal lymphocytes (Wielckens et al., 1980). In benign adenomatous colon polyps and colon cancers, alterations of polymer length have been described (Hirai et al., 1983). Recently, increased PARP activity has been reported in hepatocellular carcinomas as compared with healthy liver tissue (Shiobara et al., 2001). Because tumor cells represent an undifferentiated phenotype and because PARP activity seems to show an inverse correlation with cell differentiation, it is not surprising that most tumors have accelerated poly(ADP-ribose) metabolism. The question arises as to what effect PARP inhibition may have on the growth and viability of tumor cells. Although there are data available in the literature showing direct toxic effect of PARP inhibitors on tumor cells (Mendeleyev et al., 1995), most studies focus on the potentiating effect of PARP inhibitors on alkylating agent- or ionizing radiation-induced tumor cell death. The exposure of cells to ionizing radiation leads to hydroxyl radical-mediated DNA injury, whereas alkylating agents directly damage DNA. Other types of cytotoxic drugs such as topoisomerase inhibitors may also lead to increased DNA breakage (Li and Liu, 2001). Some topoisomerase poisons such as camptothecin do not interfere with the DNA nicking function of the enzyme but do inhibit DNA rejoining, thereby converting topoisomerase I into DNase (Li and Liu, 2001). Thus ionizing radiation, alkylating agents, and topoisomerase inhibitors cause DNA damage and PARP activation. According to our current understanding of the role of PARP in DNA-damage signaling, inhibition of PARP in irradiated or alkylated cells would delay DNA repair and would thereby divert cells from route 1 to route 2 (see Fig. 5) by indirectly facilitating DNA damage-induced apoptotic cell death, a process orchestrated mainly by the tetrameric anti-oncogene p53. Indeed, several lines of evidence indicate that tumor cells can be sensitized by PARP inhibitors toN-methyl-N-nitrosourea, bleomycin, campthotecin, and ionizing radiation-induced cytotoxicity (Nduka et al., 1980; Weltin et al., 1994, 1997; Boulton et al., 1995; Bowman et al., 1998, 2001;Griffin et al., 1998; Delaney et al., 2000; Tentori et al., 2001c). This effect of PARP inhibitors is specific, for identical results were obtained by using other approaches to inhibit PARP such as dominant-negative PARP inhibition by overexpression of the DNA-binding PARP domain or by using PARP-deficient cells (Kupper et al., 1995;Rudat et al., 1998).
With respect to PARP and radiosensitization, recent studies from Berger's laboratory showed that exponentially growing ADPRT54 and ADPRT351 cells (i.e., PARP-deficient lines) were hypersensitive to X-radiation compared with the parental V79 cells. Under this condition of growth, although the parental V79 cells exhibit G1 arrest in response to X-irradiation, the PARP-deficient cells do not undergo this specific p53-dependent cell-cycle arrest. In contrast, all the cell lines showed similar sensitivity to X-radiation under growth-arrested conditions. Furthermore, all the cell lines were equally proficient in performing potentially lethal damage repair. These findings suggest the following: 1) PARP is involved in X-ray-induced damage repair in replicating cells; 2) PARP is not required for X-ray-induced damage repair in quiescent cells; 3) PARP does not participate in potentially lethal damage repair; and 4) deficiency of PARP may potentiate the cytotoxicity of X-irradiation by interfering with the p53-dependent G1 block that occurs after X-irradiation (Chatterjee and Berger, 2000).
As mentioned above, with the use of PARP-deficient cell lines, the question of whether the experimental observations are related to the physical absence of PARP or to the lack of its catalytic activity cannot be directly addressed. In the context of tumor radiosensitization, this question has recently been addressed by Poirier's group. Extracts prepared from wild-type cells or cells lacking PARP-1 were compared in their ability to repair plasmid DNA damaged by either X-rays (single-strand DNA breaks) or by N:-methyl-N:′-nitro-N:-nitrosoguanidine (methylated bases). The extracts behaved the same way. Therefore, it was concluded that the hypersensitivity of PARP-1 null mutant cells to γ-irradiation and alkylating agents is not directly caused by a defect in DNA repair itself, but rather by results from greatly reduced poly(ADP-ribose) formation during base-excision repair in these cells (Vodenicharov et al., 2000). This finding supports previous work in which pharmacological inhibition of PARP reduced DNA repair (Cristovao and Rueff, 1996; Griffin et al., 1998; Boulton et al., 1999; Schlicker et al., 1999), indicating that the catalytic activity of the enzyme, rather than the physical presence or absence of the PARP protein, is the relevant factor in this respect.
Limited information is also available on the use of PARP inhibitors for radiation sensitization in vivo. Even these limited studies must be interpreted with great caution, because the inhibitor used (nicotinamide) is extremely weak, and the degree of intratumor PARP inhibition, in response to the systemic administration of the vitamin, has not been evaluated and must be a partial inhibition at best. Nevertheless, when nicotinamide (50–500 mg/kg) was injected intraperitoneally into CDF1 or C3H mice and radiosensitization was measured in tumors and healthy tissues after local irradiation, irradiating tumors at peak times resulted in enhancement ratios of 1.27 (C3H), 1.75 (KHT), and 1.45 (SCCVII) with high nicotinamide doses and 1.27 (C3H), 1.28 (KHT) and 1.36 (SCCVII) with low doses. [Tumor response was assessed using either growth delay (C3H) or clonogenic survival (KHT/SCCVII)]. Irradiating healthy tissues at peak times after injecting 100 to 200 mg/kg nicotinamide gave enhancement ratios of 1.20 (skin), 0.90 (bladder), and 1.02 (lung). [Normal tissue toxicities evaluated included early-responding skin (development of moist desquamation of the foot) and late-responding bladder (reservoir function estimated by cystometry) and lung (ventilation rate measured by plethysmography)]. This study confirmed the differential sensitivity of healthy tissues and tumor cells for the radiosensitizing effect of PARP inhibition in vivo and reached the important conclusion that appropriate doses of nicotinamide will enhance tumor radiation damage while having minimal effects in healthy tissues. It also concluded that, at least for nicotinamide, the best tumor-effect radiation should be given at the time of peak plasma drug concentrations (Horsman et al., 1997). This study is not in contrast with the findings of de Murcia and colleagues, in which the PARP−/− mice were found to be more sensitive to radiation toxicity than were the wild-type counterparts (see above), but rather indicates that an optimally timed and dosed pharmacological suppression (and possibly not the complete inhibition) is the direction to pursue for the purpose of introducing PARP inhibitors into the experimental cancer radiotherapy.
Important for a future stage of drug development may be the finding that further enhancement of the PARP inhibition induced radiation sensitization in vivo can be achieved by the combination of the PARP inhibition and the radiation therapy with carbogen administration (Bernier et al., 1999; Bussink et al., 1999). Hyperthermia may also enhance the radiosensitizing action of PARP inhibition (Kjellen et al., 1986).
P. Antiretroviral Effect of PARP Inhibitors
An increasing body of evidence suggests the involvement of PARP in HIV infection. During the life cycle of the HIV-1 virus within the infected cell, the RNA genome of the virus is reverse-transcribed into double-stranded DNA by reverse transcriptase. The proviral DNA, in turn, enters the nucleus, where the virion-associated viral enzyme integrase catalyzes the integration of the viral double-stranded DNA into the host genome. This process requires nicking of both DNA strands and may therefore lead to PARP activation. Indeed, increased PARP activity of HIV-infected cells has been reported by Furlini and colleagues (1991), indicating that the HIV life cycle may require poly(ADP-ribosylation). Furthermore, two studies (Cole et al., 1991;Krasil'nikov et al., 1991) independently showed that benzopyrone derivatives, trisubstituted benzamides, and nicotinamide possessed potent antiviral effects in HIV-infected cells. Another study that used three different PARP inhibitory approaches (chemical inhibition, antisense, and dominant-negative inhibition by DBD) also reported similar results (Gaken et al., 1996). A possible role of poly(ADP-ribosylation) in HIV replication is also supported by findings from Tanaka and coworkers (1995a), showing that sensitivity of subclones of human promyelocytic cell line U937 inversely correlated with the PARP content of the cells. Subclones with high HIV sensitivity contained 4- to 7-fold less PARP as compared with low-sensitivity clones. The same group recently demonstrated that phorbol ester-induced HIV-promoter activity was almost abolished in mutant l-1210 cells, which express only 8% of PARP of the wild-type cells (Kameoka et al., 1999). Similar results were obtained in human Jurkat and J111 cells, which were cotransfected with the reporter plasmid and a plasmid expressing a PARP-antisense RNA (Kameoka et al., 1999). However, in the same system, pharmacological inhibition did not inhibit HIV-promoter activity (Kameoka et al., 1999). These data point toward a regulatory mechanism whereby protein-protein interaction between PARP and a yet-elusive phorbol 12-myristate 13-acetate-induced transcription factor (possibly NF-κB) is required for HIV-promoter activity. Another study by Yamagoe and associates (1991) reported suppression by PARP inhibitors of ultraviolet light-induced HIV-1 transcriptional activity. Using a construct in which the chloramphenicol acetyltransferase gene was placed under the control of the HIV-1 long-terminal repeat, they found that in HeLa cells, 3-aminobenzamide and nicotinamide suppressed UV-induced HIV-1 gene expression but not tat-mediated expression. They also found that suppression occurred at the posttranscriptional level (Yamagoe et al., 1991). Recently, in an elegant study, Snyder's group demonstrated that PARP-deficient fibroblasts cannot be infected with pseudotyped HIV-1, whereas the virus has efficiently infected PARP-proficient fibroblasts (Ha et al., 2001). Moreover, the same study showed that the lack of HIV infection in PARP-deficient fibroblasts is caused by defective viral integration.
At present, it seems that PARP regulates HIV infection at two levels: integration and transcription. Although data with pharmacological inhibitors are inconclusive, most reports support the hypothesis that PARP inhibition may be a viable strategy to control HIV infection. To explain why in some cases pharmacological PARP inhibitors failed to block HIV infection, it was proposed that a near-complete PARP inhibition may be necessary for the antiviral effect, whereas partial inhibition of the enzyme is ineffective (Ha et al., 2001). Using the recently emerging novel potent PARP inhibitors, this hypothesis can be readily tested. In hepatitis B virus (HBV) infection, the role of PARP seems to differ from that reported in HIV infection (Dandri et al., 2002). Integration of HBV into the genome of HepG2 hepatoma cells was found to increase upon DNA damage induction by hydrogen peroxide. The integration-promoting effect of hydrogen peroxide was likely to be counteracted by PARP activation because PARP inhibitors further increased HBV integration, indicating that PARP-1 may function to limit the occurrence of de novo HBV integration (Dandri et al., 2002). The role of PARP in controlling other viral infections may also be worth investigating. There are data available in the literature showing increased PARP activity associated with cytomegalovirus replication (Furlini et al., 1984). Whether PARP inhibitors can block cytomegalovirus infection remains to be explored.
Q. PARP in the Pathogenesis of Other Diseases
It has been suggested that PARP activation contributes to the pathogenesis of other forms of brain injury and neurodegenerative disorders. For instance, PARP activation has been implicated in the pathogenesis of Parkinson's disease, a chronic progressive neurologic disorder related to the degeneration of the neurons of the substantia nigra which contain melanin; this is another disease in the pathogenesis of which the activation of NMDA receptors plays a crucial role. The synthetic heroin analog MPTP can selectively damage neurons in the nigrostriatal dopaminergic pathway and produce Parkinsonism in experimental animals (Przedborski and Jackson-Lewis, 1998; Blum et al., 2001). There is evidence for both the production of reactive oxygen intermediates (Przedborski et al.,; Cassarino et al., 1997; Hung and Lee, 1998) and NO-derived radicals/oxidants (Schulz et al., 1995a,b,c; Ara et al., 1998; Beal, 1998; Ferrante et al., 1999;Liberatore et al., 1999) in the pathogenesis of MPTP neurotoxicity. In brain injury induced by MPTP, the neuronal NO synthase is the source of cytotoxic NO and peroxynitrite. Accordingly, protection is provided by the neuronal NO synthase inhibitors 7-nitroindazole orS-methylthiocitrulline (Schulz et al., 1995c; Przedborski et al., 1996; Matthews et al., 1997b; Ferrante et al., 1999). Furthermore, genetically engineered mice that lack the bNOS gene are resistant to toxicity induced by MPTP as compared with wild-type littermates (Matthews et al., 1997a). Direct evidence for the involvement of PARP in the pathogenesis of toxicity induced by MPTP comes from a mouse model of Parkinson's disease. MPTP treatment reduces striatal dopamine and cortical norepinephrine levels by more than 50% in these animals, whereas simultaneous treatment with each of five different inhibitors of PARP ameliorates the catecholamine depletion induced by MPTP (Cosi et al., 1996). The protective potency of benzamide and its derivatives parallels their efficacy as enzyme inhibitors (Cosi et al., 1996). Furthermore, recent studies have demonstrated that mice lacking functional PARP are also resistant against MPTP neurotoxicity (Mandir et al., 1999).
As outlined in Table 4, additional diseases with a PARP-related pathogenetic component include acute respiratory distress syndrome of various etiologies. With respect to acute respiratory distress syndrome, it is noteworthy that in in vitro experiments, it has been shown that inhibition of PARP preserves normal permeability (Szabó et al., 1997c) and surfactant synthesis in oxidatively damaged pulmonary epithelial cells (Hudak et al., 1995). Likewise, there seems to be a PARP-related component in the homocysteine-induced endothelial cell injury, at least in vitro (Blundell et al., 1996), with potential implications for a variety of cardiovascular diseases (Mangoni and Jackson, 2002). PARP seems to play a role in the endothelial dysfunction associated with aging and hypertension (Pacher et al., 2002c,d). Moreover, recent studies implicated PARP activation in the process of dexamethasone-induced (i.e., stress-related) immune suppression (Drazen et al., 2001). Additional PARP-dependent diseases include multiple organ failure of various etiologies, acetaminophen-induced and other forms of toxic liver injury, and sulfur mustard-induced dermal necrosis (Table 4). Preliminary data also indicate the beneficial effects of PARP inhibitors in rodent models of asthma (Virág et al., 2002b), contact hypersensitivity (Bakondi et al., 2002b), and periodontal inflammation (Lohinai et al., 2001). Increased poly(ADP-ribosylation) has also been demonstrated in a variety of other diseases including Alzheimer's disease (Love et al., 1999), microwave-induced tissue injury (Singh et al., 1994), contact dermatitis (Szabó et al., 2001b; Virág et al., 2002a), and sunburn-related dermal inflammation (Balard and Giacomoni, 1989; Jacobson et al., 2001; Farkas et al., 2002), but the causative role of PARP in these conditions has not yet been addressed in detail. Nevertheless, topical administration of a nicotinamide-derivative PARP inhibitor BPG-15 has demonstrated significant protective effects in a rodent model of sunburn injury (Farkas et al., 2002). In persons affected with ataxia telangiectasia (A-T), associated mutations in the ataxia telangiectasia-mutated gene render cells unable to cope with the genotoxic stresses from ionizing radiation and oxidative damage, thus resulting in a higher concentration of unrepaired DNA damage and the activation of PARP in an uncontrolled manner. From in vitro studies in fibroblasts, it seems that there is an improvement of cellular growth and NAD+ levels in A-T cells with PARP inhibition, suggesting that the cellular metabolic status of A-T cells is compromised and the inhibition of PARP may relieve some of the drain on cellular pyridine nucleotides and ATP. Thus, it is possible that therapy using PARP inhibitors may provide a benefit for individuals affected with A-T (Marecki and McCord, 2002).
Effect of PARP inhibition in selected animal models of disease
IV. Conclusions and Future Directions
Over the last decade, a multitude of studies have verified the role of PARP activation in a wide range of pathophysiological conditions. Furthermore, a series of animal experiments have proved that PARP-inhibition therapy represents an effective approach to treating a variety of diseases. The key to this remarkable effectiveness lies in the fact that PARP inhibition targets a relatively late event of oxidative cell injury. Therefore, the therapeutic window of intervention is quite wide, as indicated by the success of posttreatment regimens in some models. The wide variety of disease models in which PARP inhibition proved beneficial also indicates that PARP inhibitors block a common pathway(s) of tissue injury, such as NF-κB activation or oxidative stress-induced cytotoxicity. Further work needs to establish the exact in vivo mechanism of action of PARP inhibitors. It must be emphasized that data obtained from knockout studies cannot always be extrapolated to situations in which PARP is present but is inhibited by pharmacological agents. For example, it has been shown that PARP-deficient cells have disturbed cell-cycle progression and contain a tetraploid population, a finding that could not be reproduced in wild-type cells with PARP inhibition (Simbulan-Rosenthal et al., 2000, 2001a). Moreover, topoisomerase I activity is enhanced by protein-protein interaction with PARP when NAD+ is not present. However, in the presence of NAD+ ,topoisomerase I activity is down-regulated by poly(ADP-ribosylation), indicating that the mere presence of PARP may exert biological effects which may be opposite to the effects of the catalytic activity of PARP (Bauer et al., 2000).
An exciting development in PARP research has been the discovery of new poly(ADP-ribosylating) enzymes. Given their intracellular localization, dependence, or nondependence for activation on DNA damage, the novel PARP enzymes may have distinct biological functions. It is not known at present how PARP-inhibition therapy affects the function of these minor isoforms and whether inhibition of PARP-2 to PARP-7 contributes to the well-established in vivo effects of PARP inhibitors. Bearing in mind that results from most pharmacological studies could be reproduced by using PARP-1-deficient animals and cells, we conclude that PARP-1 is the major target of PARP inhibitors in inflammation, reperfusion injury, and HIV infection. The development of isoform-selective PARP inhibitors and generation of knockout mice deficient in the novel PARP enzymes will clarify the biological roles of new PARP homologs.
The marked beneficial effect of PARP inhibitors in many animal models of various diseases suggests that PARP inhibitors can be exploited to treat human diseases. However, before potent PARP inhibitors can be used in humans, crucial safety issues must be addressed. Because PARP has been implicated in DNA repair and maintenance of genomic integrity, one possible risk associated with long-term PARP inhibition might be increased mutation rate and cancer formation. It is encouraging that PARP-deficient mice have not been reported to have an increased occurrence of spontaneous tumors. However, an increased number of chemically induced tumors has been observed in PARP-deficient mice as compared with wild-type ones (Tsutsumi et al., 2001). The crossing of the PARP-deficient mice with the p53-deficient mice was recently conducted by two independent groups and yielded conflicting results (Conde et al., 2001; Tong et al., 2001). Carcinogenesis studies have not been published in mice that were treated long-term with PARP inhibitors. It is possible that enhanced environmental carcinogenesis is caused by the lack of PARP protein but not by decreased poly(ADP-ribosylation), because several examples showed a dissociation of these two functions (see above). If poly(ADP-ribosylation) is required to prevent carcinogenesis, appropriate dosage may provide a level of PARP inhibition sufficient to improve disease signs with residual PARP activity that is enough to boost DNA repair. When considering the risk-benefit ratios associated with the development of PARP inhibitors for therapeutic purposes, clear distinctions must be made between short-term and long-term treatments as well as between considerations for the treatment of life-threatening diseases versus other disease indications.
There may exist a novel alternative approach to inhibiting PARP that may not interfere with DNA repair. Inhibition of PARG leads to the accumulation of poly(ADP-ribosylated) proteins. Because PARP-1 is the major acceptor of poly(ADP-ribose), PARG inhibition and poly(ADP-ribosylation) is inhibitory to PARP, and nontransient auto-poly(ADP-ribosylation) of PARP results in the suppression of PARP activity. The viability of this approach is indicated by recent reports demonstrating the neuroprotective effect of the PARG inhibitor gallotannin from hydrogen peroxide-induced cytotoxicity (Ying and Swanson, 2000; Ying et al., 2001). Further work is required to develop potent and specific PARG inhibitors and to generate PARG-deficient mice to learn more about the biological role of PARG and the effects of PARG inhibition.
Acknowledgments
We thank Prof. Gilbert de Murcia for critically reading the manuscript. The work in the authors' laboratories is supported by grants from the National Institutes of Health (R01GM60915), the Hungarian National Science Research Fund (OTKA T035182), and the Hungarian Ministry of Health (ETT-046/2001). L.V. is supported by a Bolyai Fellowship from the Hungarian Academy of Sciences.
Footnotes
Address for correspondence: Csaba Szabó M.D., Ph.D., Inotek Pharmaceutical Corp., 100 Cummings Center, Suite 419E, Beverly, MA 01915. E-mail: szabocsaba{at}aol.com
Abbreviations
- PARP-1
- poly(ADP-ribose) polymerase-1
- A-T
- ataxia telangiectasia
- bNOS
- brain nitric-oxide synthase
- BRCT
- breast cancer susceptibility protein C terminus
- caspase
- cysteinyl aspartate-specific protease
- CLP
- cecal ligation and puncture
- CNS
- central nervous system
- DBD
- DNA binding domain
- EAE
- experimental allergic encephalomyelitis
- eNOS
- endothelial nitric-oxide synthase
- GPI 6150
- 1,11b-dihydro-[2H]benzopyrano[4,3,2-de]isoquinolin-3-one
- HBV
- hepatitis B virus
- HIV
- human immunodeficiency virus
- HOCl
- hypochlorous acid
- ICAM-1
- intercellular adhesive molecule 1
- IFN
- interferon
- IL
- interleukin
- INH2BP
- 5-iodo-6-amino-1,2-benzopyrone
- iNOS
- inducible nitric-oxide synthase
- LPS
- bacterial lipopolysaccharide (endotoxin)
- MAP
- mitogen-activated protein
- MLDS
- multiple low-dose streptozotocin
- AIF
- apoptosis-inducing factor
- MNNG
- N-methyl-N′-nitro-N-nitrosoguanidine
- NMDA
- N-methyl-d-aspartate
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MRC
- multiprotein replication complex
- MS
- multiple sclerosis
- NF-κB
- nuclear factor κB
- NLS
- nuclear localization signal
- NO
- nitric oxide
- NOD
- nonobese diabetic
- NOS
- nitric-oxide synthase
- PARG
- poly(ADP-ribose) glycohydrolase
- PARP
- poly(ADP-ribose) polymerase
- PARP−/− cells/mice
- cells/mice homozygous for disrupted poly(ADP-ribose) polymerase genes
- PARP+/+ cells/mice
- cells/mice with undisrupted poly(ADP-ribose) polymerase genes
- PJ34
- the hydrochloride salt ofN-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide
- TNF-α
- tumor necrosis factor α
- TRF
- telomere repeat-binding factor
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
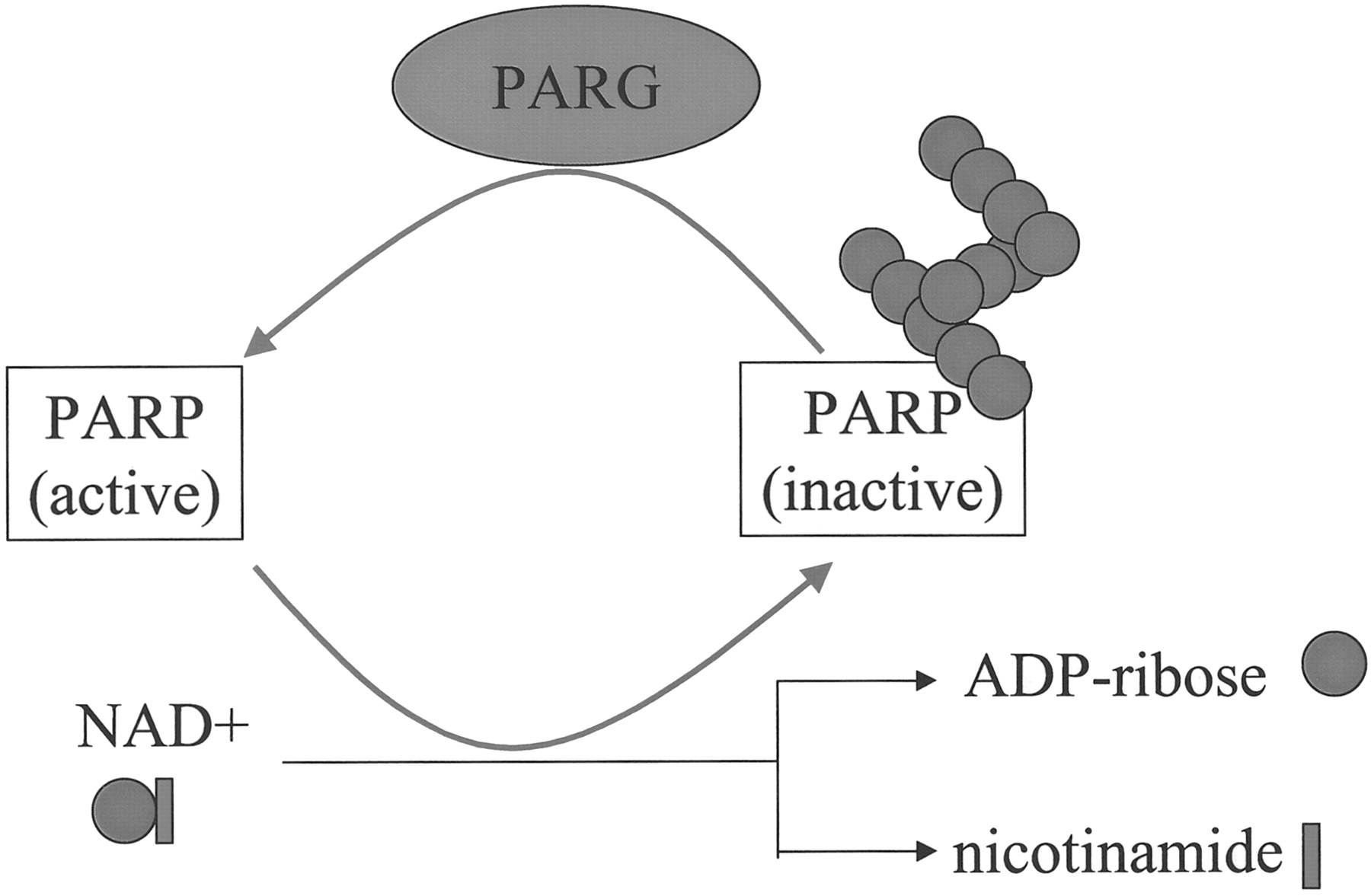{kind=link}
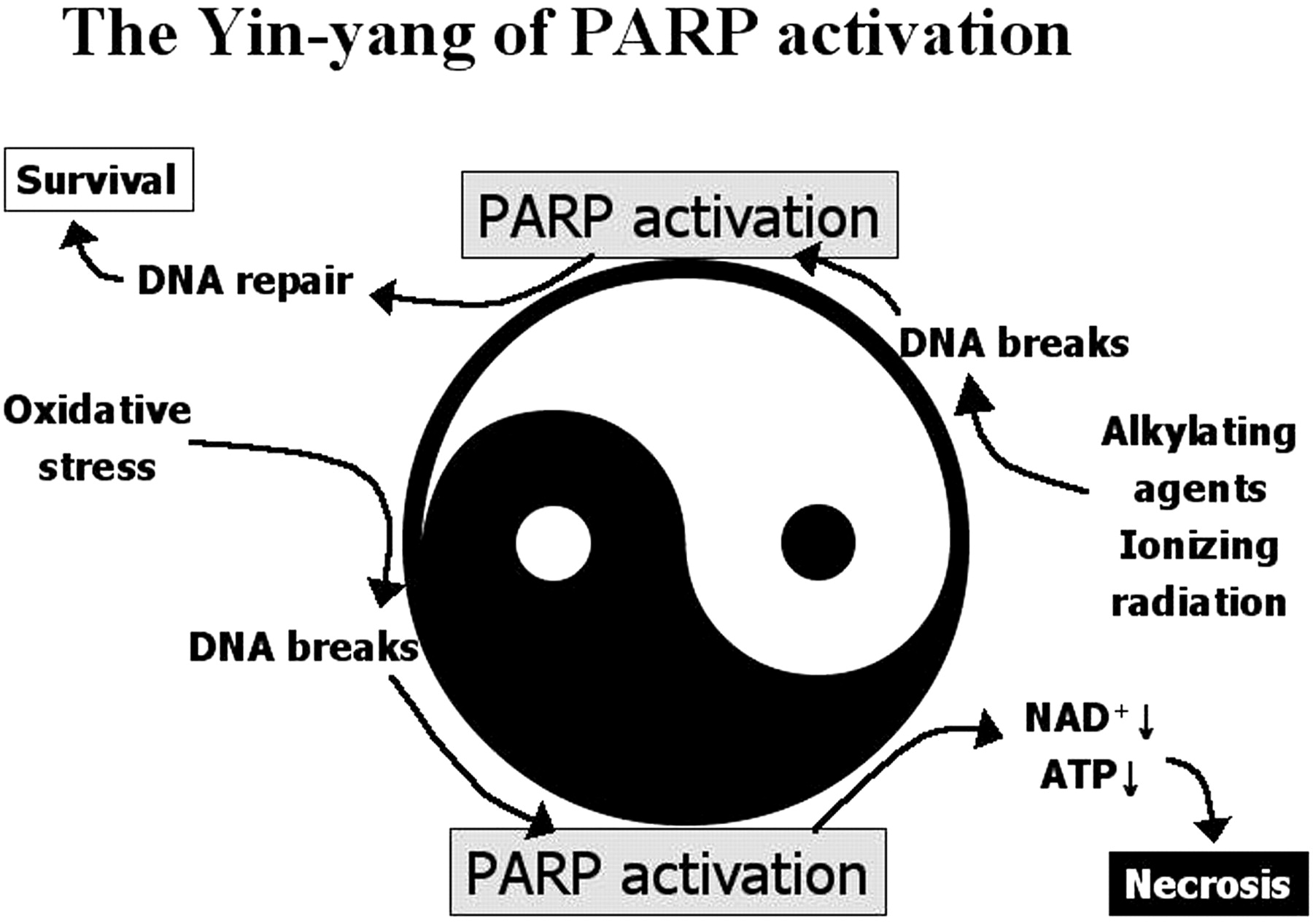{kind=link}
{kind=link}
{kind=link}
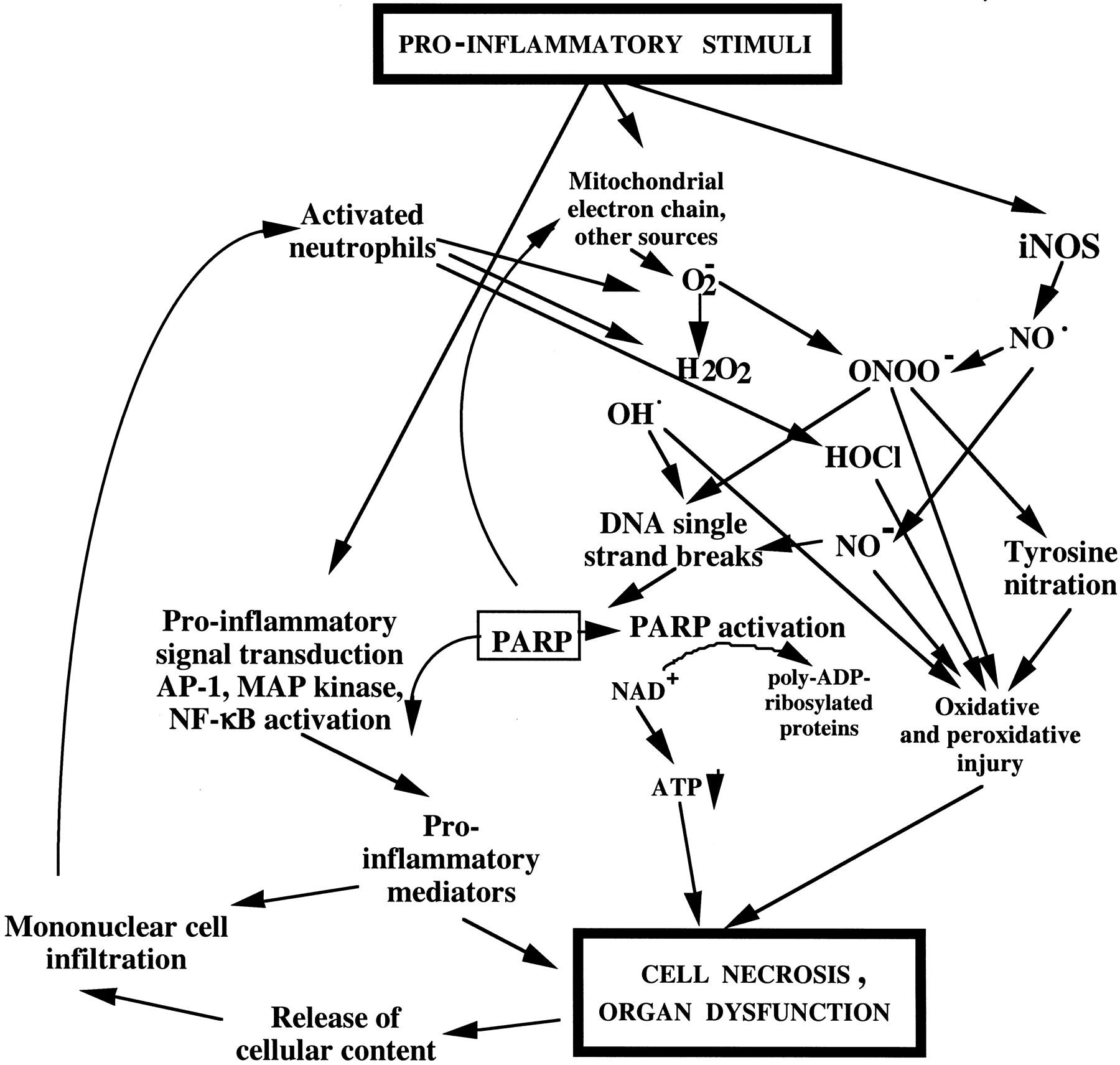{kind=link}
{kind=link}