


Abstract
Endothelin-1 (ET-1) is a potent vasoconstrictor and growth-promoting mediator that is involved in the maintenance of vascular tone within the healthy circulation. However, a pathogenic role has been implicated by its overproduction in a number of cardiovascular diseases, which include pulmonary hypertension, congestive heart failure, atherosclerosis, and coronary vasospasm. ET-1 mRNA expression and peptide production in human vascular smooth muscle cells (HVSMCs) are markedly increased by exposure to tumor necrosis factor-α and interferon-γ. The intracellular signaling mechanism involved in this pathway is not known. Because the transcription factors nuclear factor-κB (NF-κB), signal transducer and activator of transcription 1 (STAT1), and interferon regulatory factor-1 (IRF-1) often mediate the effects of cytokines in target cells the aim of this study was to determine whether the production of ET-1 after exposure of HVSMCs to cytokines depends upon synergism between NF-κB and STAT1/IRF-1. Immunoblotting showed that cytokine-stimulation of ET-1 release in VSMCs involves nuclear translocation of NF-κB and STAT1. Cytokines also induced an increase in IRF-1 protein expression. Antisense oligonucleotides to NF-κB, STAT1, and IRF-1 significantly inhibited cytokine induced ET-1 release. In conclusion, NF-κB, STAT1, and IRF-1 activation are involved in the stimulation by cytokines of ET-1 release from HVSMCs. However, nuclear run-on assays would provide definitive proof that ET-1 is regulated transcriptionally by cytokines. Because up-regulated production of ET-1 within VSMCs may underlie the causative role of ET-1 in a number of disease states, this finding indicates that NF-κB, STAT1, and IRF-1 within HVSMCs could be central to a number of vascular pathologies and that inhibition of this pathway could be of therapeutic benefit.
Endothelin-1 (ET-1) is a potent vasoconstrictor and growth-promoting mediator that is involved in the maintenance of vascular tone within the healthy circulation (Haynes and Webb, 1994). However, an increased production of ET-1 within the blood vessel has been implicated in the events underlying a number of vascular pathologies (Warner et al., 1996; Parris and Webb, 1997). Indeed, many of the disease states in which ET-1 seems to be involved are associated with elevations in the plasma concentrations of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) (Klemm et al., 1995; Saleh et al., 1997). We have previously demonstrated that ET-1 mRNA expression and peptide production in human vascular smooth muscle cells (HVSMCs) are markedly increased by exposure to cytokines (Woods et al., 1999). Therefore, under the influence of proinflammatory mediators, the vascular smooth muscle can become an important site of ET-1 production. The significance of this finding has been supported by a recent study by Wort et al. (2002). This article provided a possible explanation for the effectiveness of prostacyclin as a therapy for pulmonary hypertension by demonstrating its ability to inhibit ET-1 release from pulmonary vascular smooth muscle cells. This further highlights that vascular smooth muscle cells can be extremely important sources of ET-1 production under inflammatory conditions. The intracellular signaling mechanism by which cytokines bring about this increase in ET-1 release is not presently known. It is immediately apparent, therefore, that it is most important to characterize the processes controlling this ET-1 production and interventions that dampen it down.
Proinflammatory cytokines such as TNF-α and IFN-γ exert their actions via a number of pathways. First, transcription factor nuclear factor-κB (NF-κB) is often the primary effector of TNF-α in target cells (Siebenlist et al., 1994; Thanos and Maniatis, 1995). NF-κB exists in the cell cytosol as an inactive complex of Rel-related factors bound to its inhibitor proteins IκBα and IκBβ. Both IκBβ and IκBα interact with NF-κB through similar domains and both preferentially bind p65 and c-Rel containing dimers. However, there are several differences between these inhibitor proteins. First, IκBα is involved in the transient activation of NF-κB, whereas IκBβ underlies the persistent activation of NF-κB. Second, mitogens and cytokines that activate NF-κB target one inhibitor protein over the other. For example, IκBα is targeted by signaling pathways initiated by TNF, IL-1, or LPS, whereas IκBβ is targeted by pathways initiated by LPS or IL-1 but not by TNF (Baldwin, 1996). In human smooth muscle cells NF-κB is present as a heterodimer of a 65-kDa (p65) and a 50-kDa (p50) subunit bound to either IκBβ or IκBα (Bourcier et al., 1997). Phosphorylation of IκB inhibitory protein and its subsequent degradation by a proteasome-dependent pathway results in activation of NF-κB (Palombella et al., 1994; Chen et al., 1995). NF-κB dimers then translocate to the nucleus and promote transactivation of target genes.
Second, a variety of cytokines activate the signal transducer and activator of transcription (STAT) class of transcription factors (Darnell et al., 1994; Ihle, 1995). IFN-γ uses the Janus kinase/STAT pathway as its primary mode of signaling (Shuai et al., 1992). IFN-γ signaling is initiated when IFN-γ binds to its receptor, thereby promoting receptor dimerization. When this occurs Jak1 and Jak2, which are associated with the cytoplasmic domains of the receptor, are brought into apposition and transphosphorylate each other, resulting in kinase activation. The cytoplasmic domains of the receptors then become tyrosine phosphorylated, allowing for interaction with STAT1. STAT1 is subsequently activated by tyrosine phosphorylation, leading to dimerization, translocation to the nucleus, binding to IFN-γ activation sequences (GAS) in regulatory regions, and transcriptional activation of IFN-γ responsive genes (Darnell et al., 1994). One of the STAT1-induced genes is interferon-regulatory factor-1 (IRF-1) (Pine et al., 1994). IRF-1 is itself a transcription factor that recognizes the sequence termed interferon stimulation response element and plays, like STAT1, a central role in IFN-γ-induced gene expression (Kamijo et al., 1994; Briken et al., 1995).
Here, we have investigated whether activation of NF-κB, STAT1, and IRF-1 transcription factors are involved in the regulation by cytokines of the synthesis of ET-1 from HVSMCs. So by characterizing the components of the intracellular protein kinase signaling cascades involved in this process we may well provide an impetus for new approaches in the use of cardiovascular gene therapy in the treatment of disease conditions associated with cytokine-stimulated elevations in ET-1 release from human vascular smooth muscle.
Materials and Methods
Vascular Smooth Muscle Cell Isolation and Culture. Saphenous vein (SV) was obtained from patients undergoing coronary artery bypass surgery. VSMCs were obtained for culture using the free-floating explant method (Ross, 1971). Vessels were first cleaned, opened longitudinally, and scraped to remove endothelial cells. The smooth muscle was stripped from the adventitia and cut into approximately 2-mm2 pieces before being placed into tissue culture flasks with DMEM containing nonessential amino acids and 10% fetal calf serum (FCS). Cells were then incubated at 37°C in 5% CO2/95% air. After 4 to 6 weeks in culture, sufficient cells had explanted to allow passage of cells for characterization and use in experiments. Smooth muscle cells (passages 2-9) were identified by smooth muscle α-actin staining. Endothelial cell contamination was excluded by the absence of staining for von Willebrand factor.
Stimulation of Cells. Throughout all experiments cells were grown in DMEM containing penicillin (100 U/ml), streptomycin (100 μg/ml), and 10% FCS. HVSMCs were stimulated with a combination of TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml). This combination of cytokines was chosen as it was previously shown to produce maximal stimulation of ET-1 release in VSMCs (Woods et al., 1999).
RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR). SV HVSMCs grown on 10-cm Petri dishes were initially treated with TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml) for 24 h. Cells were subsequently incubated in the presence or absence of actinomycin D (5 μM) for a further 1, 2, or 4 h. In separate experiments cells were either incubated with cycloheximide (10 μM) for 1 or 2 h or actinomycin D (5 μM) for 15 or 30 min after cytokine stimulation for 24 h. Cells were lysed in TRIzol reagent and total RNA was isolated according to the manufacturer's instructions. RT-PCR was carried out as described previously (Woods et al., 1999). Gene-specific primers were selected according to the published sequences in GenBank (Table 1). GAPDH, a constitutively expressed gene, was used as a positive control to correct for intersample variation. RT-PCR was performed with an OmniGene Hybaid thermocycler.
Sequences for oligonucleotide primers used for RT-PCR
Antisense Oligonucleotide (ASO) Experiments. The phosphorothioate oligonucleotides used as antisense for IRF-1, STAT1, and NF-κB were as follows: 5′-TCCGAGTGATGGGCATGTTGG-3′, 5′-GGTGCAGGATGTCTCAGTGG-3′, and 5′-GGGGAACAGTTCATGGC-3′, respectively. ASOs for IRF-1 and STAT1 were designed to encompass the transcriptional start site from published sequences. ASOs for p65 NF-κB were based on published work (Hishikawa et al., 1997). HVSMCs cultured on 24-well plates to reach a confluence of 70 to 80% were incubated with either IRF-1, STAT1, or NF-κB ASO or sense (SO, 0.75 μM) using Transfast reagent (Promega, Madison, WI) at a charge ratio of 3:1 [Transfast/oligodeoxynucleotide (ODN)] in serum-free medium. At the end of a 1-h incubation period, DMEM containing 10% FCS was added. After 24 h, the medium was replaced and cells were treated with a mixture of TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for 24 h. Medium was removed and stored at -70°C until further use.
Measurement of Immunoreactive ET-1. ET-1 levels were measured using specific sandwich immunoassay kits according to the manufacturer's instructions (R & D Systems, Abingdon, UK). Values are reported as mean ± S.E.M. and are representative of cells from four to five patients each assayed in triplicate.
Cellular Respiration/Viability. Cell respiration, an indicator of cell viability, was assessed by the ability of cells to reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan. At the end of each experiment, cells in 24-well plates were incubated with MTT (1 mg/ml) at 37°C for 1 h. After removal of MTT by aspiration, cells were solubilized in dimethyl sulfoxide (200 μl). The extent of reduction of MTT to formazan was quantified by measurement of optical density at 550 nm.
Preparation of Whole Cell and Nuclear Extracts. Confluent monolayers of VSMCs were grown on 10-cm Petri dishes and treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) either alone or in combination for various incubation times. Similarly, for ASO experiments HVSMCs were grown on 10-cm Petri dishes and cells were transfected with ASO following the protocol outlined above. Nuclear extracts were prepared using protocols described previously (Taylor et al., 1999). For the preparation of whole-cell extracts, cells were lysed in phosphate-buffered saline containing 0.1% Triton, 10 mM EDTA, 2 μM leupeptin, 1 μM pepstatin, and 1 mM phenylmethylsulfonyl fluoride.
Western Blotting. Protein concentrations in both nuclear and whole-cell extracts were determined with a Bio-Rad protein assay (Bio-Rad, Hercules, CA). Protein extracts were boiled for 5 min in a 1:1 ratio with gel loading buffer. Equal amounts of protein (20-30μg) were loaded onto gradient gels (10% acrylamide) and subjected to electrophoresis for 1 h at 100 V. The separated proteins were then electrotransferred to nitrocellulose (Hybond C; Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK) at 100 V for 1 h. The blots were incubated in blocking buffer [dried low fat milk 5% (w/v) and Tween 20 0.1% in Tris-buffered saline solution] on an orbital shaker for 3 to 4 h at room temperature. Primary antibodies (rabbit anti-human) diluted appropriately were incubated with the blots overnight at 4°C. After washing for 3 × 5 min, the blots were exposed to the secondary anti-rabbit antibody conjugated to horseradish peroxidase (1:2,000) for 1 h. The immunodetected proteins were visualized using a LumiGLO chemiluminescent reagent (New England BioLabs, Hertfordshire, UK) and exposure to high performance chemiluminescence film (Hyperfilm ECL; Amersham Biosciences).
Materials. DMEM plus sodium pyruvate, penicillin, streptomycin, nonessential amino acids, and TRIzol reagent were supplied by Invitrogen (Paisley, UK). FCS, actinomycin D, and cycloheximide were obtained from Sigma Chemical (Poole, Dorset, UK). Human recombinant TNF-α, IFN-γ, and human ET-1 immunoassay kit were purchased from R & D Systems. Human anti-NF-κB p65 and IκBβ antibody were purchased from BIOMOL Research Laboratories (Plymouth Meeting, MA). Human anti-IRF-1 antibody was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit affinity-purified polyclonal antibodies to IκBα, STAT1, phospho-STAT1(Tyr701), and Phototope-horseradish peroxidase Western detection kit were obtained from New England BioLabs. Phosphorothioate oligonucleotides were synthesized by Amersham Biosciences.
Data Analysis. Results are expressed as mean ± S.E.M. Data were analyzed using the Prism software (GraphPad Software Inc., San Diego, CA). Band intensities from RT-PCR and Western blotting were analyzed densitometrically by the Molecular Analyst Software (Bio-Rad). All data were compared by repeated measures analysis of variance followed by a Dunnett's multiple comparison test.
Results
Actinomycin D Inhibits Cytokine-Stimulated Up-Regulation of Prepro-ET-1 mRNA in HVSMCs. To determine whether cytokine-stimulated up-regulation of prepro-ET-1 mRNA is caused by increased transcriptional rate of the ET-1 gene, the effects of actinomycin D, an inhibitor of transcription, were investigated. Stimulation of HVSMCs with TNF-α and IFN-γ for 24 h resulted in a significant increase in prepro-ET-1 mRNA in comparison with unstimulated cells. Subsequent treatment of cytokine-stimulated HVSMCs with actinomycin D (5 μM) for 1, 2, and 4 h resulted in prepro-ET-1 mRNA being barely detectable after 4 h (Fig. 1). Incubation with actinomycin D for 30 min also significantly reduced cytokine-stimulated increases in prepro-ET-1 mRNA. In contrast, steady-state levels of prepro-ET-1 mRNA were maintained in cells in the absence of actinomycin (data not shown).

Actinomycin D inhibits cytokine-stimulated increases in prepro-ET-1 mRNA, whereas cycloheximide has no effect. SV HVSMCs grown on 10-cm Petri dishes were prestimulated with TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml) for 24 h followed by incubation with actinomycin D (5 μM) for 1, 2, and 4 h (A). In separate experiments after prestimulation with cytokines for 24 h, cells were incubated with either actinomycin D (5 μM) for 15 or 30 min or cycloheximide (10 μM) for 1 or 2 h (B). Extraction of total RNA and RT-PCR were carried out. Expression of mRNA for GAPDH was used as a positive control. In A, lane 1 represents control unstimulated cells; lane 2, HVSMCs exposed to TNF-α and IFN-γ for 24 h; and lanes 3, 4, and 5, HVSMCs exposed to TNF-α and IFN-γ for 24 h followed by treatment with actinomycin D (5 μM) for 1, 2, and 4 h, respectively. Results shown are representative of n = 3 experiments.
Cycloheximide Does Not Effect Cytokine-Stimulated Induction of Prepro-ET-1 mRNA. HVSMCs were incubated with cycloheximide (10 μM), a protein synthesis inhibitor, to determine whether TNF-α and IFN-γ could act through synthesis of a new protein to alter stability of preproET-1 mRNA. Cycloheximide did not significantly alter cytokine-stimulated prepro-ET-1 mRNA (ratio ET-1/GAPDH in arbitrary units: TNF-α/IFN-γ, 1.42 ± 0.33; cycloheximide 1 h, 1.61 ± 0.44; cycloheximide 2 h, 1.66 ± 0.61; n = 3 for each).
TNF-α and IFN-γ Induce the Nuclear Translocation of NF-κB in HVSMCs. Exposure of SV HVSMCs to a combination of TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml) resulted in an increased nuclear accumulation of p65 NF-κB, as determined by densitometric analysis of Western blots using a specific p65 antibody. Increased levels were seen within 0.25 h and were maintained for up to 2 h. Levels of immunodetectable NF-κB protein present in the nuclear extracts of unstimulated cells were less than in cells treated with TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml), consistent with basal levels of NF-κB activity (Fig. 2, A and B). Densitometric analysis did not confirm that cytosolic levels of p65 changed significantly over time after stimulation with cytokines (Fig. 2B).
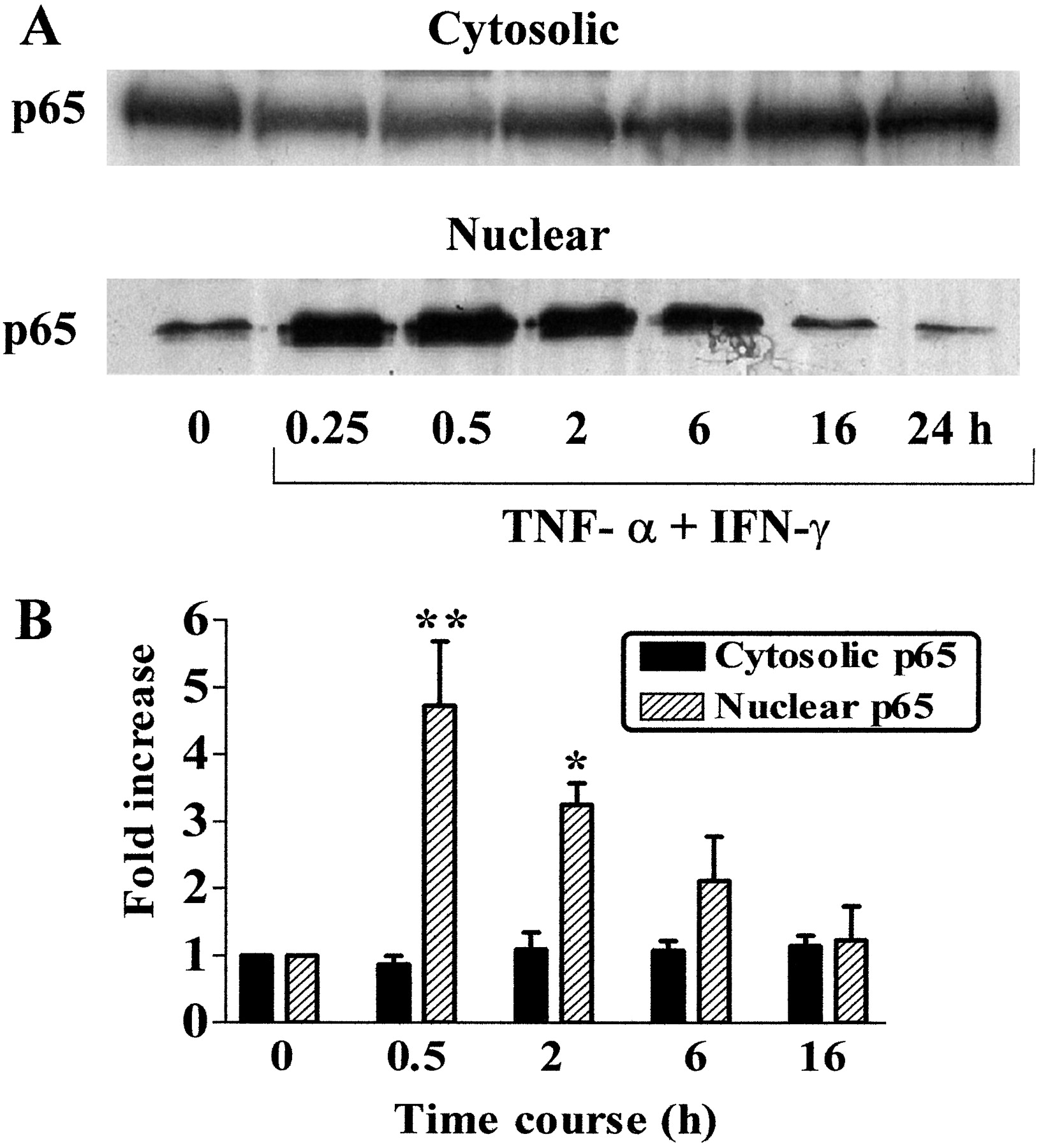
TNF-α and IFN-γ induce the nuclear translocation of NF-κB in HVSMCs. HVSMCs were incubated with TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml) for 0.25, 0.5, 2, 6, 16, or 24 h. Nuclear and cytosolic extracts were prepared and Western blotting was carried out. Blot A shows levels of NF-κB p65 in both cytosolic and nuclear extracts from SV HVSMCs over 24 h, respectively. B represents densitometric analysis of Western blots obtained for both nuclear and cytosolic fractions in whole-cell extracts. Each column represents results of three separate experiments. **, p < 0.01; *, p < 0.05 compared with untreated control cells.
Cytokines Induce Loss of IκBα in HVSMCs but Do Not Alter Levels of IκBβ over Time. The inhibitor protein IκBα is an excellent marker of NF-κB activity, because its cytokine-induced phosphorylation and subsequent proteasome-linked degradation result in the release of NF-κB. IκBα exists in the cytosol of unstimulated HVSMCs as a 37-kDa protein (Fig. 3A). After stimulation with cytokines, IκBα disappeared rapidly in less than 0.5 h, and returned to detectable levels at 2 h (Fig. 3C). TNF-α alone stimulated degradation of IκBα protein within 0.5 h. The time course observed was very similar to that seen with TNF-α and IFN-γ together. Unexpectedly, IFN-γ alone also significantly induced loss of IκBα protein within 0.5 h (Fig. 3B). Cytokine treatment of SV VSMCs did not significantly alter levels of IκBβ at times of up to 24 h as determined by densitometry (n = 3; Fig. 3A).

Cytokines induce loss of IκBα in HVSMCs but do not alter levels of IκBβ over time. SV HVSMCs were incubated with TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml) either alone or in combination for 0.25, 0.5, 2, 6, 16, or 24 h. Whole-cell extracts were prepared, and Western blotting was performed. Immunoblots in A and B show levels of either IκBα or IκBβ in whole-cell extracts of SV HVSMCs treated with a combination of cytokines over 24 h. Results shown are representative of n = 3 to 4 experiments. C, densitometric analysis of Western blots obtained for IκBα in whole-cell extracts. Each column represents results of three separate experiments. *, p < 0.05 compared with untreated control cells.
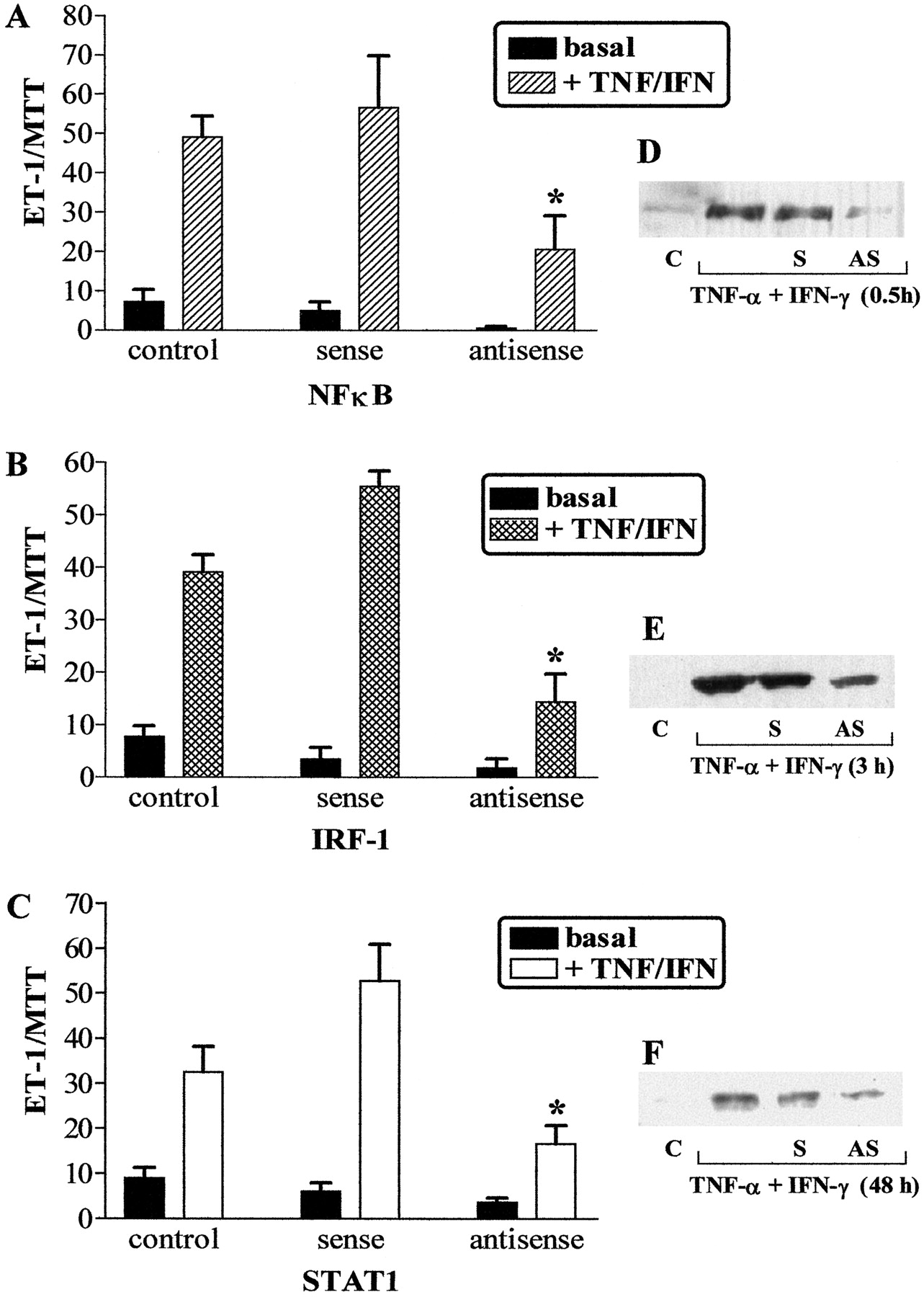
ASOs to NF-κB p65 Inhibit Cytokine-Stimulated ET-1 Release in SV HVSMCs. Treatment of SV HVSMCs with NF-κB p65 ASOs significantly reduced TNF-α- and IFN-γ-stimulated ET-1 release by 68.3 ± 6.5% (n= 4) compared with cytokine-stimulated SO control. SOs or scrambled oligonucleotide controls did not effect cytokine-induced ET-1 release. Basal release of ET-1 was not significantly altered by treatment with SOs or ASOs (Fig. 4A). Decreased expression of NF-κB protein in cytokine treated ASO-transfected cells was confirmed by Western blot analyses (Fig. 4D).

NFκB, IRF-1, and STAT1 ASOs inhibit cytokine-stimulated ET-1 release from SV HVSMCs. A-C, effects of ASOs on cytokine-stimulated ET-1 peptide release over 24 h. Results are expressed as ratio of ET-1/MTT to account for effects of transfection of ASOs on cell viability. Each column represents results from four separate experiments obtained from four independent cell cultures. *, p < 0.05 compared with cytokine-treated cells. Right, effects of ASO treatment on NF-κB (D), STAT1 (E), and IRF-1 (F) protein expression as determined by Western blotting of whole-cell extracts. C, untreated cells; S, sense ODN; AS, antisense ODN.
Cytokines Stimulate STAT1 Phosphorylation and Translocation into the Nucleus in HVSMCs. STAT proteins are latent transcription factors that become activated by phosphorylation of specific tyrosine residues, resulting in their translocation to the nucleus and subsequent transactivation of target genes. To evaluate whether TNF-α and IFN-γ stimulated phosphorylation of STAT1, both whole-cell and nuclear extracts of unstimulated and cytokine-treated cells were analyzed. Treatment of SV HVSMCs with TNF-α and IFN-γ did not affect levels of total STAT1 protein up to 24 h in whole-cell extracts (Fig. 5, A and E). In contrast in the absence of cytokines there was no phosphorylated-STAT1 present in the cells. However, after stimulation with cytokines there was a marked increase in the level of phosphorylated STAT1 within 0.25 to 0.5 h, and this was sustained for up to 6 h (Fig. 5, B and E). Phosphorylated STAT1 was also found to be present in the nucleus within 0.5 h and was less detectable at 16 h (Fig. 5C). IFN-γ alone was capable of stimulating phosphorylation of STAT1 (Fig. 5D), whereas TNF-α alone was without effect (data not shown).

Cytokines stimulate STAT1 phosphorylation and nuclear translocation in HVSMCs. Cells were incubated with TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml) for various time points. Nuclear and whole and cell extracts were prepared and Western blotting was performed. Immunoblots in A and B show levels of STAT1 and phosphorylated-STAT1 in cytokine-stimulated SV VSMCs over 24 h. C, levels of phosphorylated-STAT1 in nuclear extracts from SV VSMCs stimulated with cytokines over 16 h. D, demonstrates the effect of IFN-γ alone on STAT1 phosphorylation in HVSMCs. Data shown are representative of n = 3 experiments. E, represents densitometric analysis of Western blots obtained for both STAT1 and P-STAT1 in whole-cell extracts. Each column represents results of three separate experiments. *, p < 0.05 compared with untreated control cells.
STAT1 ASOs Inhibit Cytokine-Stimulated ET-1 Release from SV. To evaluate the involvement of STAT1 in the synergistic actions of TNF-α and IFN-γ, cells were transfected with STAT1 ASOs. STAT1 ASOs significantly reduced cytokine stimulation of ET-1 release by 67 ± 6.5% (n = 3), whereas STAT1 SOs did not affect cytokine-stimulated ET-1 release. Basal release of ET-1 was not significantly altered by treatment with STAT1 SOs or ASOs (Fig. 4C). STAT1 ASOs reduced the expression of STAT1 protein, confirming selectivity of the ASO for its target protein (Fig. 4F).
IRF-1 Protein Synthesis Is Stimulated by TNF-α and IFN-γ The IRF-1 gene is induced by a number of stimuli, including IFN-γ. We investigated the isolated and combined effects of TNF-α and IFN-γ on IRF-1 synthesis. In the absence of cytokines there was no IRF-1 protein detectable in the cells. After stimulation with cytokines, there was a significant increase in IRF-1 protein levels present in the cells within 1 h as determined by densitometry. The amount of protein increased up to 2 h and was less detectable at 4 h (Fig. 6A). IFN-γ and TNF-α alone induced IRF-1 protein synthesis, although the effect of TNF-α was not as great as that of IFN-γ (Fig. 6B).

Cytokines stimulate IRF-1 protein synthesis in HVSMCs. Cells were incubated with TNF-α (10 ng/ml) and IFN-γ (1,000 U/ml) for various time points. Whole-cell extracts were prepared and Western blotting was performed. A, levels of IRF-1 in whole-cell extracts up to 4 h. B, effect of each cytokine alone and in combination on IRF-1 protein synthesis. C, effect of NF-κB ASO/SO and STAT ASO/SO on cytokine-stimulated IRF-1 protein. Data shown is representative of n = 3 experiments.
IRF-1 ASOs Reduce Cytokine-Stimulated ET-1 Release in SV HVSMCs. To investigate whether IRF-1 may be functioning as a signal mediator downstream of STAT1, we investigated the effect of IRF-1 ASOs on cytokine-stimulated ET-1 release. The inhibition of IFN-γ-mediated IRF-1 induction by ASOs significantly reduced TNF-α and IFN-γ stimulated ET-1 release by 65.3 ± 7.7% (n = 4), whereas IRF-1 SOs did not affect cytokine-stimulated ET-1 release. Basal release of ET-1 was not altered by treatment with IRF-1 SOs or ASOs (Fig. 4B). In the presence of IRF-1 ASOs IRF-1 protein expression was decreased compared with that in the presence of IRF-1 SOs but not completely abolished (Fig. 4E).
NFκB and STAT1 ASOs Reduce Cytokine-Stimulated Induction of IRF-1 in SV HVSMCs. The ability of NF-κB and STAT1 ASOs to inhibit cytokine-stimulated induction of IRF-1 was investigated. NF-κB AS significantly reduced cytokine stimulated IRF-1 induction to 37 ± 4.3% (p < 0.01 compared with cytokine-treated cells; n = 3), whereas NF-κB sense had no significant effect. Similarly, STAT1 antisense inhibited TNF-α and IFN-γ induction of IRF-1, reducing protein levels to 45.5 ± 2.5% (p < 0.01 compared with cytokine-treated cells; n = 3), whereas STAT1 sense had no effect (Fig. 6, C and D).
Discussion
We have previously reported that TNF-α and IFN-γ stimulated the up-regulation of prepro-ET-1 mRNA and ET-1 release in a concentration-dependent manner in both human internal mammary artery and SV HVSMCs (Woods et al., 1999). This finding has relevance in vivo, because unregulated production of ET-1 by the vascular smooth muscle after loss or dysfunction of the endothelium may underlie the causative role of ET-1 in a number of disease state pathologies (Warner et al., 1996; Parris and Webb, 1997). Most anti-endothelin therapeutic strategies have so far focused on the development of potent ET receptor antagonists and inhibitors of ET-1 synthesis (endothelin converting enzyme inhibitors) to lessen the adverse effects of excess ET-1. However, another therapeutic approach could be to interfere with the intracellular signaling mechanisms leading to increased prepro-ET-1 gene transcription. Unfortunately, no data have yet been presented on the regulatory pathway involved in cytokine-stimulated ET-1 release. Here, therefore, we aimed to elucidate the signal transduction pathways underlying this response in the belief that inhibition of this pathway could be therapeutically beneficial.
First, we wanted to establish whether cytokine-stimulated up-regulation of prepro-ET-1 mRNA is caused by an increased transcriptional rate of the ET-1 gene. To determine this, we investigated the effects of actinomycin D, an inhibitor of transcription. Actinomycin D inhibited cytokine-stimulated increases in prepro-ET-1 mRNA, suggesting that cytokines are having a direct effect on transcription of ET-1 gene. Similar findings have been obtained in bovine aortic endothelial cells in which TNF-α increases the transcriptional rate of the ET-1 gene without affecting the stability of prepro-ET-1 mRNA (Marsden and Brenner, 1992). In an attempt to further demonstrate that transcriptional and not post-transcriptional mechanisms are responsible for the up-regulation of ET-1 mRNA by TNF-α and IFN-γ, we performed experiments using the protein synthesis inhibitor cycloheximide. If the inhibition of TNF-α- and IFN-γ-stimulated prepro-ET-1 mRNA obtained with actinomycin D were caused by suppression of new mRNA synthesis second to synthesis of a new protein that was capable of altering stability of prepro-ET-1 mRNA, cycloheximide should also reduce cytokine-stimulated induction of ET-1 mRNA by inhibiting synthesis of this new protein. However, treatment of cells with cycloheximide had no effect on cytokine-induced induction of prepro-ET-1. This indicates that the effects seen upon prepro-ET-1 mRNA are most unlikely to be associated with synthesis of a new protein that alters the stability of prepro ET-1 mRNA. However, nuclear run-on assays would provide definitive proof that ET-1 is regulated transcriptionally by cytokines.
To study the mechanism underlying the synergistic effect of TNF-α and IFN-γ on ET-1 release, we investigated the role of a number of transcription factors as detailed below. Because transcription factor NF-κB often mediates the effects of TNF-α in target cells it is the most likely candidate to be involved in mediating the effects of TNF-α on ET-1 release in HVSMCs. HVSMCs in vitro express multiple Rel family members with dimers of p65 and p50 comprising basal and inducible NF-κB binding activities (Bourcier et al., 1997). We used a number of different approaches to determine whether activation of NF-κB is involved in TNF-α and IFN-γ stimulation of ET-1 release. To begin with, we examined whether the combination of TNF-α and IFN-γ could stimulate nuclear translocation of NF-κB in HVSMCs. We found that after stimulation of HVSMCs with a combination of TNF-α and IFN-γ, there was an increased nuclear accumulation of p65 NF-κB. These findings are in accordance with the previous observation that TNF-α stimulation of SV HVSMCs induces transient NF-κB binding activity that peaks by 1 h and declines to near basal levels by 6 h (Bourcier et al., 1997). The inhibitor protein IκBα is an excellent marker of NF-κB activity because its cytokine-induced phosphorylation and subsequent degradation results in the release of active NF-κB (Henkel et al., 1993). After stimulation with cytokines IκBα disappeared rapidly. This time course for the cytokine-induced loss of IκBα paralleled the time course observed for increased NF-κB activity. IκBβ protein levels in SV were unaffected by cytokine treatment. Indeed, because IκBβ is normally targeted only by pathways initiated by LPS or IL-1, one would not expect this protein to be affected.
Having established that the combination of cytokines used to induce ET-1 release also stimulates nuclear translocation of NF-κB, we wanted to determine whether inhibition of this transcription factor could affect ET-1 release. We inhibited NF-κB at a molecular level using antisense specifically directed against p65 NF-κB mRNA. Treatment with p65 NF-κB ASOs significantly inhibited cytokine-stimulated ET-1 release, whereas treatment with SOs had no significant effect. Western blotting confirmed the specificity of ODNs used as transfection of cells, with p65 NF-κB ASOs reducing expression of p65 NF-κB protein and SOs having no effect. These results obtained with ASOs confirm the involvement of p65 NF-κB in mediating cytokine-stimulated ET-1 release. We have previously demonstrated that BAY 11-7082 (an inhibitor of cytokine-induced IκB phosphorylation) inhibited cytokine-induced up-regulation of prepro-ET-1 mRNA expression and the production of ET-1 peptide, further supporting a role for NF-κB in mediating these effects of both TNF-α and IFN-γ (Woods et al., 2000). We can therefore propose a role for NF-κB in mediating the stimulatory effects of TNF-α and IFN-γ on ET-1 release in HVSMCs. In corroboration with our findings, a previous study has shown that ET-1 transcription is controlled by NF-κB in advanced glycation end product-stimulated cultured endothelial cells (Quehenberger et al., 2000).
Because IFN-γ uses the Janus kinase/STAT pathway as its primary mode of signaling (Shuai et al., 1992), we looked at the effect of IFN-γ on STAT1 phosphorylation in HVSMCs. Stimulation of HVSMCs with TNF-α and IFN-γ resulted in a rapid phosphorylation of STAT1 and its translocation into the nucleus. This time course of STAT1 activation is similar to that reported in other studies (Shuai et al., 1992). We again used the antisense technique to investigate the involvement of STAT1 in mediating ET-1 release from cytokine-stimulated VSMCs. ET-1 release was significantly down-regulated after exposure of cells to STAT1 ASOs. These findings suggest that the JAK/STAT1 pathway is involved in cytokine-stimulated ET-1 release from HVSMCs.
As previously mentioned, IFN-γ-induced activation of STAT1 can lead to a production of IRF-1, an IFN-γ primary response gene. IRFs constitute a growing family of transcription factors that contains, among others, IRF-1 and IRF-2 (Mamane et al., 1999). The IRF family of transcription factors has been demonstrated to play a major role in the regulation of several inflammatory genes, including inducible nitric-oxide synthase (Saura et al., 1999). IRF-1 can be induced early by IFN-γ through a STAT1-dependent pathway. We therefore investigated the effects of cytokine stimulation on induction of IRF-1. In the absence of cytokines, there was no IRF-1 protein detectable in the cells. After stimulation with cytokines there was an increase in IRF-1 protein levels present in the cells. IRF-1 ASO pretreatment significantly reduced cytokine-stimulated ET-1 release and Western blotting confirmed a reduction in IRF-1 protein by IRF-1 ASOs. SOs were without effect on either IRF-1 protein or ET-1 release. Interestingly, we have also demonstrated that NF-κB ASO and STAT1 ASO reduce the induction of IRF-1 protein by TNF-α and IFN-γ. This suggests that up-regulation of ET-1 mRNA by cytokines is mediated by NF-κB- and STAT1-dependent induction of expression of IRF-1. Together, these findings suggest that IRF-1 is a key downstream mediator involved in TNF-α/IFN-γ synergy.
We have previously established that incubation of HVSMCs with TNF-α and IFN-γ is necessary to stimulate ET-1 release (Woods et al., 1998). Synergistic effects of TNF-α and IFN-γ are known to occur in a number of cell types (Fish et al., 1999; Paludan, 2000). The majority of promoters induced synergistically by IFN-γ and TNF-α contain binding sites for STAT1 or IRF-1 and NF-κB (Paludan, 2000). A number of studies have demonstrated synergy between STAT1 and NF-κB (Jahnke and Johnson, 1995; Krzesz et al., 1999). Indeed, a recent study demonstrated that in rat aortic smooth muscle cells TNF-α, via activation of NF-κB, and IFN-γ, via activation of STAT, up-regulate CD40 gene expression (Krzesz et al., 1999). Synergy between NF-κB and IRF-1 has also been previously reported. For example, TNF-α and IFN-γ synergistically activate the RANTES promoter through NF-κB and IRF-1 transcription factors (Lee et al., 2000). An interaction between IRF-1 and NF-κB during activation of inducible nitric-oxide synthase transcription has also been demonstrated (Saura et al., 1999). Collectively, our data suggest that the synergistic activity of IFN-γ and TNF-α in SV HVSMCs is dependent mainly upon cooperation between IFN-γ activated STAT1/IRF-1 and TNF-α-activated NF-κB. The molecular mechanism underlying the synergistic action of IRF-1 and NF-κB has been extensively studied by others, and there is evidence from different experimental approaches that IRF-1 and NF-κB interact physically (Drew et al., 1995; Neish et al., 1995). However, the study reported here produced no evidence to support this.
Other mechanisms may underlie the synergistic effects we see, because few of these pathways are mutually exclusive and independent of each other. Synergy is dependent upon different mechanisms cross talking and potentially amplifying each other. To investigate this synergism further, we investigated the effect of each cytokine alone on activation of each transcription factor. TNF-α alone induced degradation of IκBα (an indicator of NF-κB activation) but did not stimulate phosphorylation of STAT1. However, TNF-α alone did induce IRF-1 protein expression, although the effect of TNF-α is not as potent as that of IFN-γ. Previous studies have also reported that TNF-α can induce IRF-1 in some cell types (Pine et al., 1994; Ohmori et al., 1997). Therefore, given that TNF-α also activates NF-κB and that IRF-1 and NF-κB synergize, enhanced IRF-1 expression will further augment the degree of synergism between IFN-γ and TNF-α.
IFN-γ alone stimulated STAT1-phosphorylation, IRF-1 protein expression and also induced the degradation of IκBα inhibitor. The time course for this IFN-γ-induced loss of IκBα was very similar to that of TNF-α. IFN-γ does not typically activate NF-κB, although it has been reported to in one study that explored the effects of IFN-γ on TNF-α induced NF-κB activity. In particular, Cheshire and Baldwin (1997) showed that IFN-γ synergistically enhanced TNF-α induced NF-κB nuclear translocation via a mechanism involving the enhanced degradation of IκBα and de novo IκBβ degradation. Therefore, IFN-γ-enhanced degradation of IκBα inhibitor protein may partly account for the synergism observed between TNF-α and IFN-γ in HVSMCs.
To determine whether TNF-α and IFN-γ could activate NF-κB, STAT1, and IRF-1 that bind to sites on the prepro-ET-1 gene and subsequently activate transcription, we set out to establish whether there are any binding sites for these transcription factors in the promoter region of the prepro-ET-1 gene. We analyzed the 5′-flanking region of the human prepro-ET-1 gene (GenBank accession no. J05008) by using the TRANSFAC database and identified four putative NF-κB p65 binding sites between positions -882 and -2179 downstream of the transcription initiation start site (Inoue et al., 1989; Heinemeyer et al., 1998). A previous study has confirmed a functional binding site for NF-κB at position -2090 (Quehenberger et al., 2000). We also identified STAT and IRF-1 binding sites at positions -2175 and -2344, respectively. Although further investigation is necessary to determine whether these putative bindings sites are functionally relevant, our results suggest that in HVSMCs, TNF-α and IFN-γ stimulate the activation of NF-κB and STAT1/IRF-1, which cooperatively induce transcription of the prepro-ET-1 gene. This study therefore provides data suggesting a novel signaling pathway for cytokine-stimulated ET-1 release from HVSMCs.
Much research is currently aimed at developing the potential of cardiovascular gene therapy, whereby applications of DNA technology are used to regulate transcription of disease-related genes (Levi and Coronel, 1997). Recently, cis-element double-stranded ODNs, referred to as “decoy” ODNs, have been reported to have the potential to be powerful antigene strategies (Sawa et al., 1997; Morishita et al., 1998). Transfection of double-stranded ODNs leads to a modulation of gene expression by reducing authentic cis-trans interaction. Does this gene therapy have any bearings upon our findings? It would seem most probably, yes. As previously mentioned, proinflammatory cytokines have been found to be associated with elevated expression of ET-1 and endothelin-converting enzyme in a number of pathological conditions (Saleh et al., 1997). A recent study has detected immunoreactive ET-1 in the tunica media of different types of arteries obtained from patients with coronary artery disease and/or portal hypertension, whereas in vessels from healthy organ donors immunoreactive ET-1 was detectable almost exclusively in endothelial cells. This study suggests that in pathological conditions in which endothelial damage occurs ET-1 can be synthesized in VSMCs of these patients (Rossi et al., 1999). This study adds further credence to the importance of our findings. Also, a recent article that provided a possible explanation for the effectiveness of prostacyclin as a therapy for pulmonary hypertension by demonstrating its ability to inhibit ET-1 release from pulmonary vascular smooth muscle cells also clearly emphasizes the significance of our study (Wort et al., 2002). Here, we have shown the transcription factors NF-κB, STAT1, and IRF-1 to underlie cytokine-induction of increased vascular smooth muscle ET-1 levels. Application of transcription factor decoy strategy aimed at these transcription factors may prove to be extremely useful in the treatment of disease states associated with cytokine-stimulated elevations in ET-1.
Footnotes
-
This work was supported by a grant from The British Heart Foundation (PG/99001). T.D.W. holds a British Heart Foundation Lectureship (BS/95003).
-
This work was presented previously in Woods M, Wood EG, Mitchell JA, and Warner TD (2000) Signal transduction pathways involved in cytokine stimulation of endothelin-1 release from human vascular smooth muscle cells. J Cardiovasc Pharmacol 36(Suppl 1):S407-S409.
-
ABBREVIATIONS: ET-1, endothelin-1; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; HVSMC, human vascular smooth muscle cell; VSMC, vascular smooth muscle cell; NF-κB, nuclear factor-κB; IκBα, inhibitor κBα; IL-1, interleukin-1; LPS, lipopolysaccharide; STAT, signal transducer and activator of transcription; IRF-1, interferon regulatory factor-1; SV, saphenous vein; DMEM, Dulbecco's modified Eagle's medium; FCS, fetal calf serum; RT-PCR, reverse transcription-polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ASO, antisense oligonucleotides; SO, sense oligonucleotide; ODN, oligodeoxynucleotide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; BAY 11-7082, (E)-3-(4-methylphenylsulfonyl)-2-propenenenitrile.
- Received January 9, 2003.
- Accepted July 7, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}