


Abstract
Endothelin-1 (ET-1) is the predominant endothelin isopeptide generated by the vascular wall and therefore appears to be the most important peptide involved in regulation of cardiovascular events. Many pathologic conditions are associated with elevations of ET-1 in the blood vessel wall. Because these conditions are often cytokine driven, we examined the effects of a mixture of cytokines on ET-1 production in human vascular smooth muscle cells (VSMCs) derived from internal mammary artery and saphenous vein (SV). Incubation of IMA and SV VSMCs with tumor necrosis factor-α (10 ng/ml) and interferon-γ (1000 U/ml) in combination for up to 48 h markedly elevated the expression of mRNA for prepro-ET-1 and the release of ET-1 into the culture medium. This cytokine-stimulated release of ET-1 was inhibited by a series of dual endothelin-converting enzyme (ECE)/neutral endopeptidase inhibitors, phosphoramidon, CGS 26303, and CGS 26393, with an accompanying increase in big ET-1 release but with no effect on expression of mRNA for prepro-ET-1. These same compounds were 10 times more potent at inhibiting the conversion of exogenously applied big ET-1 to ET-1. ECE-1b/c mRNA is present in SV VSMCs, however no ECE-1a is present in these cells. Thus VSMCs most probably contain, like endothelial cells, an intracellular ECE responsible for the endogenous synthesis of ET-1. Under the influence of pro-inflammatory mediators the vascular smooth muscle can therefore become an important site of ET-1 production, as has already been established for the dilator mediators nitric oxide, prostaglandin I2, and prostaglandin E2.
In recent years it has become clear that following exposure to cytokines and/or endotoxin vascular smooth muscle releases vasoactive mediators normally produced within the vascular endothelium. Most notable, perhaps, among these is nitric oxide (NO), which is produced within the vascular smooth muscle following expression of inducible NO synthase (iNOS) (Wong and Billiar, 1995). Similar stimuli also induce the smooth muscle to express cyclo-oxygenase-2, thereby increasing its capacity to produce the vasodilator prostaglandins E2 and I2 (Bishop-Bailey et al., 1997a,b). Apart from vasodilator mediators, the normal vascular endothelium also produces the potent vasoconstrictor and pro-mitogenic peptide endothelin-1 (ET-1) (Yangisawa et al., 1988; Inoue et al., 1989; Haynes and Webb, 1993; Warner et al., 1996; Parris and Webb, 1997). As for NO and the prostaglandins, an increased production of ET-1 within the blood vessel has been implicated in the events underlying a number of vascular pathologies (Haynes and Webb, 1993; Warner et al., 1996; Parris and Webb, 1997). For example, both specific ET receptor antagonists and ET-1 binding antibodies produce beneficial effects in experimental models of ischemia/reperfusion injury, restenosis, and hypertension as well as in similar human disease states (Haynes and Webb, 1993; Warner et al., 1996; Parris and Webb, 1997). Interestingly, in a number of these vascular pathologies, most obviously in restenosis following balloon angioplasty, there is loss or dysfunction of the endothelium, yet ET-1 still appears to be involved as a causative agent (Haynes and Webb, 1993; Warner et al., 1996; Parris and Webb, 1997). Drawing on our experience of the NO and prostaglandin systems, this suggests that it could be the vascular smooth muscle that produces ET-1 when the endothelium is dysfunctional (Wang et al., 1996). Knowing that it is pro-inflammatory cytokines that induce the expression of NO and prostaglandins in smooth muscle, we might expect that it is these same mediators that induce the expression of ET-1. Indeed, many of the disease states in which ET-1 appears involved are associated with elevations in the plasma concentrations of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) (Warner and Klemm, 1996). In this study, we used vascular smooth muscle cells (VSMCs) derived from human internal mammary artery (IMA) and saphenous vein (SV) to examine the effect of cytokines upon the production of ET-1. Because it appears that in endothelial cells ET-1 is formed from its precursor big ET-1 by the action of an intracellular endothelin-converting enzyme (ECE), we also used inhibitors of ECE to characterize the synthetic pathway present within cytokine-treated VSMCs (Corder et al., 1993; Schmidt et al., 1994;Shimada et al., 1994; Xu et al., 1994). Some of this data was presented to the Fifth International Meeting on Endothelin (Woods et al., 1998).
Experimental Procedures
IMA and SV were obtained from patients undergoing coronary artery bypass surgery.
Cell Culture.
VSMCs were obtained for culture using the free-floating explant method (Ross, 1971). Vessels were first cleaned and then opened longitudinally and scraped to remove endothelial cells. The smooth muscle was stripped from the adventitia and cut into approximately 2-mm2 pieces before being placed into tissue culture flasks with Dulbecco’s modified Eagles medium containing 2 mM glutamine, nonessential amino acids, sodium pyruvate, and 15% fetal calf serum. Cells were then incubated at 37°C in 5% CO2/95% air. After 4 to 6 weeks in culture, sufficient cells had explanted to allow passage of cells for characterization and use in experiments. Smooth muscle cells (passages 2–9) were identified by smooth muscle α–actin staining. Endothelial cell contamination was excluded by the absence of staining for von Willebrand factor.
Experimental Procedures.
Throughout all experiments cells were grown in Dulbecco’s modified Eagles medium containing 2 mM glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml), and 10% fetal calf serum. In the first series of experiments IMA and SV VSMCs were grown on 96-well plates to confluence (approximately 2500 cells/well) and concentration-response curves to both TNF-α (0.01–100 ng/ml) and IFN-γ (0.1–1000 U/ml) were constructed in IMA and SV. The TNF-α concentration-response curve was carried out in the presence of a fixed concentration of IFN-γ (1000 U/ml) and the IFN-γ concentration-response curve was carried out in the presence of a fixed concentration of TNF-α (10 ng/ml). From these first experiments, cytokine concentrations that produced maximal stimulation of ET-1 release were identified.
In the second series of experiments, VSMCs from IMA and SV grown on 75-cm2 flasks were treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for the final 1, 4, 8, 12, 24, 36, or 48 h of a 48-h incubation period. The medium was retained for assay of ET-1 content and the cells removed for analysis of expression of mRNA for prepro-ET-1, ET-2, and ET-3.
In the third series of experiments, VSMCs grown on 96-well plates were incubated with a combination of TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for 48 h in the presence of a series of ECE and/or neutral endopeptidase 24.11 (NEP) inhibitors; phosphoramidon, CGS 26303, CGS 26393 (a prodrug for CGS 26303), or CGS 24592 (1 nM to 1 mM) (De Lombaert et al.,1994; Trapani et al., 1995).
To examine the effects of ECE inhibitors on the conversion of exogenous big ET-1, VSMCs from both IMA and SV were treated with cytokines as above for 24 h, the medium replaced, and cells incubated with big ET-1 (1 μM) for 2 h in the presence of phosphoramidon, CGS 26303, CGS 26393, or CGS 24592 (1 nM to 1 mM). The effects of ECE inhibitors on expression of prepro-ET-1 mRNA was determined by growing cells on 75-cm2 flasks, which were then treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) in the presence or absence of either phosphoramidon (300 μM), CGS 26393 (300 μM), or CGS 26303 (1 mM) for 48 h. The cells were then removed for analysis of expression of mRNA for prepro-ET-1.
All experiments were carried out in the presence of a cocktail of peptidase inhibitors, captopril (1 μM), bestatin (1 μM), thiorphan (1 μM), and bacitracin (3 μg/ml) to prevent degradation of ET-1 by endogenous peptidases.
Measurement of Immunoreactive ET-1 and Big ET-1.
Medium was removed and stored at −70°C until further use. ET-1 and big ET-1 levels were measured using specific sandwich immunoassay kits following manufacturers’ instructions. Optical density at each appropriate wavelength was measured using a Molecular Devices microplate reader (Molecular Devices, Menlo Park, CA). Values are reported as mean ± S.E.M. and are representative of cells from four to five patients each assayed in triplicate.
Cellular Respiration/Viability.
Cell respiration, an indicator of cell viability, was assessed by the ability of cells to reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan. At the end of each experiment, cells in 96-well plates were incubated with MTT (1 mg/ml) at 37°C for 1 h. Following removal of MTT by aspiration, cells were solubilized in dimethyl sulfoxide (200 μl). The extent of reduction of MTT to formazan was quantified by measurement of optical density at 550 nm.
Expression of mRNA.
VSMCs from both SV and IMA were grown to confluence in 75-cm2 flasks. Cells were lysed in denaturing solution and total RNA was isolated by a guanidinium thiocyanate/isopropanol method with minor modifications (Chomczynski and Sacchi, 1987). Before cDNA synthesis, RNA was treated with commercially available deoxyribonuclease I to eliminate any contaminating DNA. One microgram of total RNA was then converted to cDNA using Ready-To-Go T-primed First Strand Kit. The cDNA obtained from this reaction was used in reverse transcription-polymerase chain reaction (RT-PCR). RT-PCR was performed with an OmniGene Hybaid thermocycler (Hybaid, London, UK. Initial denaturation was performed at 94°C for 2 min, followed by annealing for 45 s at the appropriate temperature and extension at 72°C for 1 min for 35 to 40 cycles, with the final extension period of 7 min at 72°C. The annealing temperatures were set as follows: prepro-ET-1, 53°C; prepro-ET-2, 58°C; prepro-ET-3, 53°C; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 60°C. Gene specific primers were selected according to the published sequences in Genbank (see Table1). GAPDH, a constitutively expressed gene, was used as a positive control to correct for intersample variation.
Sequences for oligonucleotide primers used for RT-PCR
For the ECE mRNA studies, total cellular RNA was extracted using RNAzol B. ECE-1 and GAPDH mRNA expression was measured using the Access RT-PCR kit. Total RNA (100 ng) was added to each reaction. RT-PCR primers were designed on the basis of published human cDNA sequences for human ECE-1a and human ECE-1c. The ECE-1c primer would not discriminate between ECE-1b and ECE-1c and is, therefore, subsequently referred to as ECE-1b/c (see Table 1). A common antisense primer was used for ECE-1a and ECE-1b/c. PCR conditions were as follows: denaturation, 94°C for 30 s; annealing, 1 min at 60°C; and extension, 68°C for 2 min for 25 cycles. For GAPDH annealing occurred at 65°C and extension 72°C, for 20 cycles. A final extension for 10 min at 72°C was then performed.
RT-PCR products were then size fractionated through a 0.8% agarose gel and the bands visualized with ethidium bromide and photographed. The relative levels of mRNA encoding for prepro-ET-1 were quantified by densitometry using Kodak Biomax 1D analysis software (Anachem, Bedfordshire, UK) for Macintosh and normalized to GAPDH mRNA.
Materials.
DMEM plus sodium pyruvate, fetal calf serum, penicillin, streptomycin, glutamine, nonessential amino acids, sodium pyruvate, and deoxyribonuclease I were supplied by Gibco BRL (Paisley, UK) and bacterial endotoxin were obtained from Sigma Chemical Co. (Poole, UK). Human recombinant TNF-α, IFN-γ, interleukin-1β, interleukin-6, and human ET-1 immunoassay kit were purchased from R&D Systems (Abingdon, UK). Human big ET-1 peptide and phosphoramidon were obtained from The Peptide Institute (Osaka, Japan). Big ET-1 immunoassay kits were purchased from Assay Designs, Inc. (Ann Arbor, MI). CGS 26303 ((S)-2-biphenyl-4yl-1-(1H-tetrazol-5-yl)-ethylamino-methyl phosphonic acid), CGS 26393 (prodrug for CGS 26303), and CGS 24592, ((S)-N-[2-(phosphonomethylamino-3-(4-biphenyl)-propionyl]-3-aminopropionic acid) were a generous gift from Dr. Arco Jeng (Novartis, Summit, NJ). Total RNA extraction kit was purchased from Clontech Laboratories, Inc. (Palo Alto, CA). Access RT-PCR kit was purchased from Promega (Southhampton, UK) and the RNAzol B was purchased from Biogenesis (Poole, UK). The first-strand cDNA synthesis kit (Ready-To-Go T-Primed First Strand Kit) was obtained from Pharmacia Biotech (Hertfordshire, UK). Primer sequences were synthesized by Severn Biotech Ltd. (Worcestershire, UK).
Statistical Analysis.
Data is expressed as mean ± S.E.M. Statistical comparisons were made by ANOVA or unpairedt test and values were considered to be significant atp < .05.
Results
Concentration-Dependent Effects of TNF-α and IFN-γ on Production of ET-1 by Human IMA and SV VSMCs.
TNF-α, in the presence of a fixed concentration of IFN-γ (1000 U/ml) stimulated ET-1 release in a concentration-dependent manner with maximal stimulation occurring at 10 ng/ml in both IMA (Fig.1A) and SV (Fig. 1B) VSMCs. This concentration of TNF-α was selected for use in further experiments. IFN-γ in the presence of a fixed concentration of TNF-α (10 ng/ml) similarly stimulated ET-1 release in a concentration-dependent manner, with maximal stimulation occurring at 1000 U/ml in both IMA and SV VSMCs (data not shown). As with TNF-α, this concentration of IFN-γ was selected for all further experiments. In other experiments, addition of interleukin-1β (up to 10 ng/ml), interleukin-6 (up to 10 ng/ml), and bacterial endotoxin (up to 10 μg/ml) either separately or in combination produced no additional increase in the production of ET-1 by either IMA or SV VSMCs.

Effects of TNF-α (0.01–100 ng/ml) in the presence of IFN-γ (1000 U/ml) on release over 48 h of ET-1 from cultured human IMA (A) and SV (B) VSMCs. Accumulation of ET-1 in culture medium was measured by specific sandwich enzyme-linked immunoassay. Data represents mean ± S.E.M. for cells from three patients each assayed in triplicate.
Time-Dependent Effect of TNF-α and IFN-γ on Expression and Production of ET-1 by Human IMA and SV VSMCs.
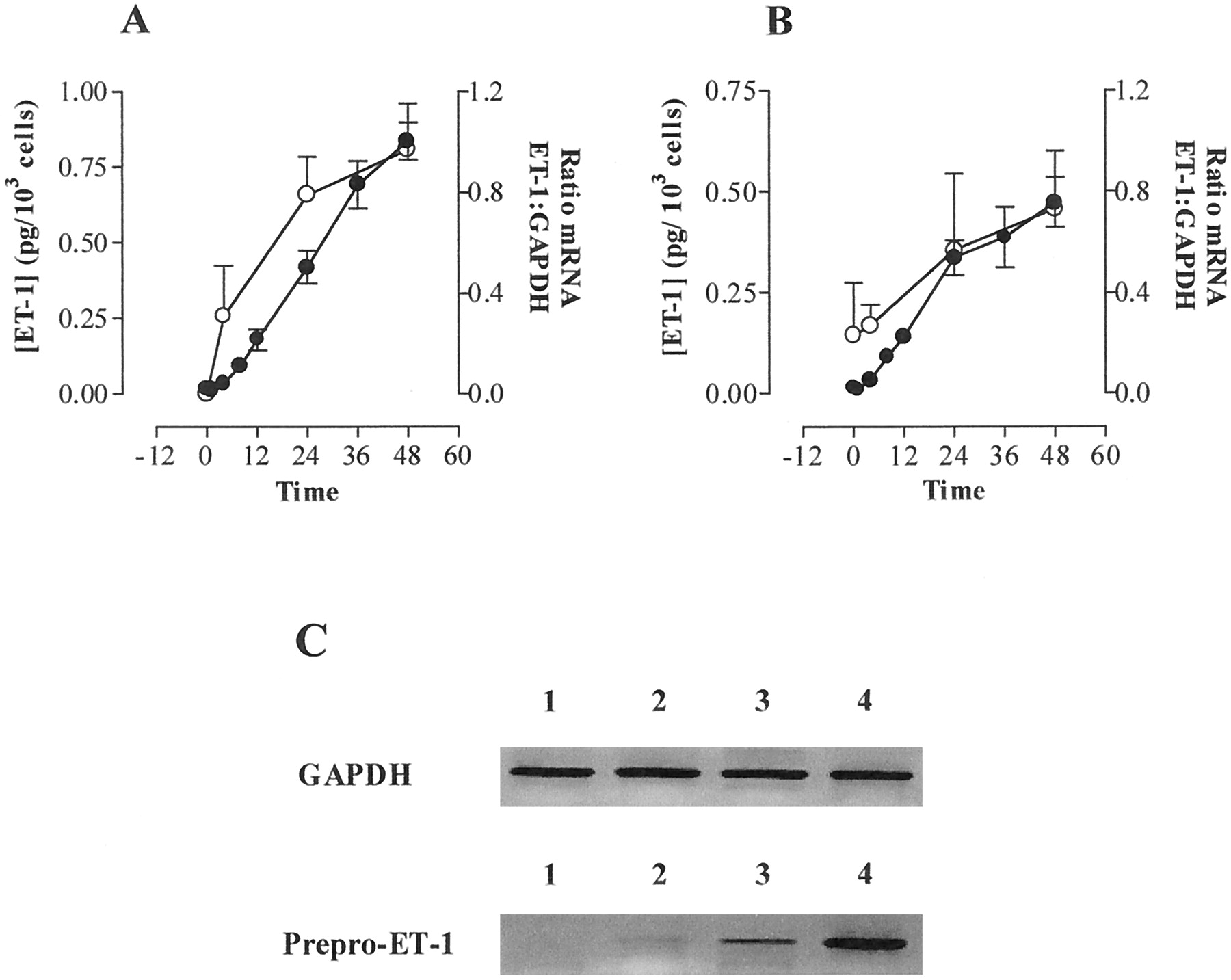
The combination of TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) caused a sustained release of ET-1 over 48 h from both IMA and SV VSMCs that was associated with a very marked increase in expression of mRNA for prepro-ET-1, with no changes in GAPDH (Fig. 2C). Importantly, the increase in prepro-ET-1 mRNA preceded the elevation in concentration of ET-1 within the culture medium (Fig. 2, A and B), supporting the idea that (de novo) synthesis of ET-1 was occurring. mRNA for prepro-ET-2 and prepro-ET-3 was undetectable.
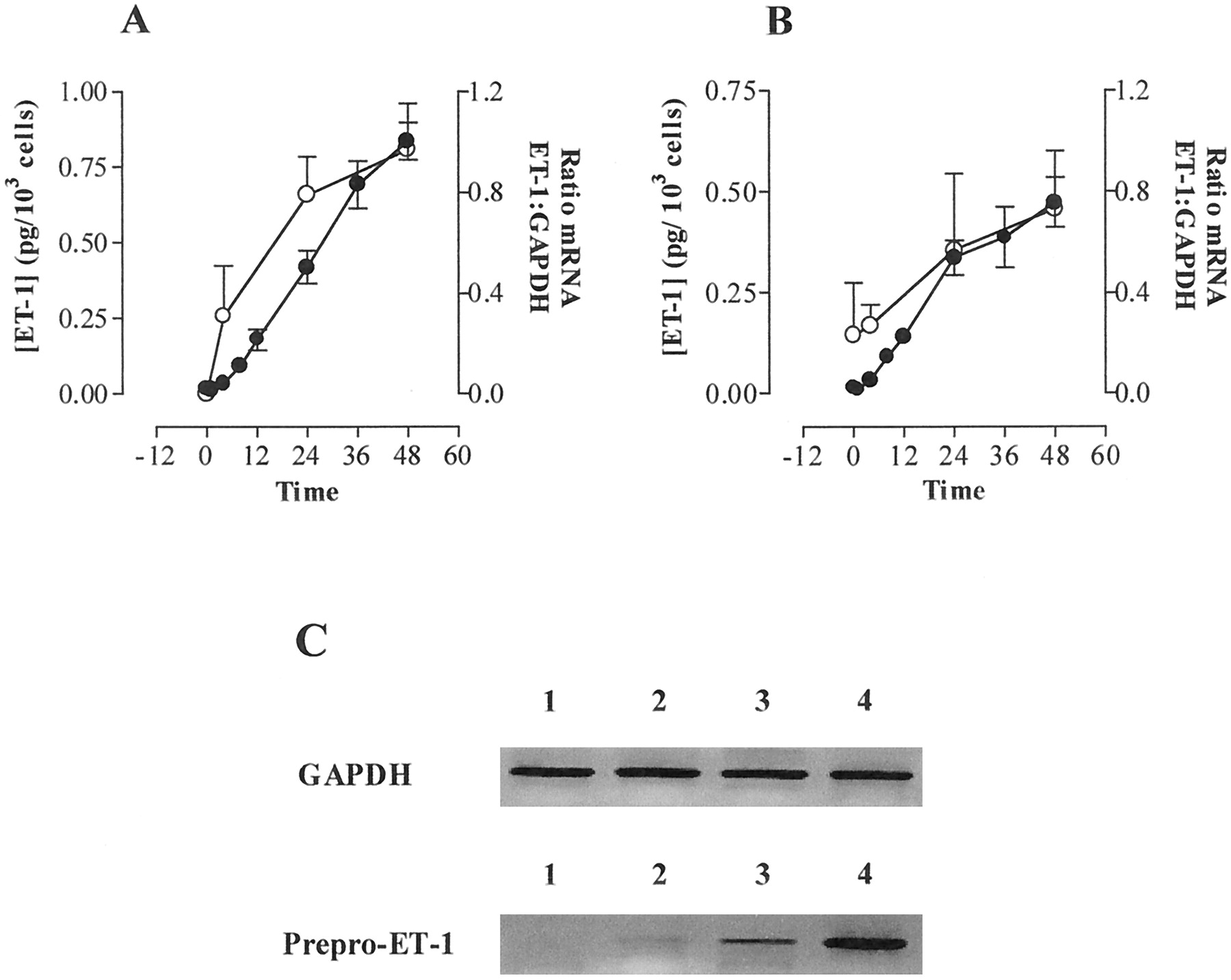
Effects of a combination of TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) on expression of prepro-ET-1 mRNA (○) and production of ET-1 peptide (●) over time in VSMCs cultured from human IMA (A) and SV (B). VSMCs were grown to confluence on 75-cm2 tissue culture flasks treated with a mixture of TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for times indicated between 4 and 48 h. RNA was extracted and RT-PCR carried out as outlined in Experimental Procedures. Relative amounts of prepro-ET-1 and GAPDH mRNA were quantified by densitometry. Accumulation of ET-1 in culture medium was measured by specific sandwich enzyme-linked immunoassay. Data represents mean ± S.E.M. for cells from three patients each assayed in triplicate. C, typical gel of time-dependent effect of TNF-α and IFN-γ on expression of prepro-ET-1 mRNA in cultured human SV VSMCs cells over time. Lane 1, unstimulated cells; lanes 2, 3, and 4, VSMCs exposed to TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for 4, 24, and 48 h, respectively. Similiar responses were seen in VSMCs derived from two other explant cultures.
Effect of Phosphoramidon on Release of ET-1 and Big ET-1 from Cytokine-Treated Human IMA and SV VSMCs.
The release of ET-1 that followed exposure of the human VSMCs to TNF-α and IFN-γ was inhibited by phosphoramidon in a concentration-dependent manner (log IC50 values: IMA, −3.9 ± 0.05; SV, −4.1 ± 0.09) with accompanying reciprocal increases in the release of big ET-1 (log EC50 values: IMA, −4.4 ± 0.1; SV, −4.5 ± 0.09) (Fig.3).

Effect of phosphoramidon on accumulation over 48 h of ET-1 (○) and big ET-1 (●) in medium of IMA (A) and SV (B) VSMCs treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml). Data represents mean ± S.E.M. for cells from five patients each assayed in triplicate.
Effects of CGS 26303, CGS 26393, and CGS 24592 on Release of ET-1 and Big ET-1 from Cytokine-Treated Human IMA and SV VSMCs.
CGS 26303 caused a concentration-dependent inhibition of the cytokine-induced release of ET-1 from IMA and SV VSMCs with log IC50 values of −3.5 ± 0.09 and −3.6 ± 0.08, respectively (Fig. 4). Similarly, CGS 26393 inhibited endogenous ET-1 release with a log IC50 value of −4.4 ± 0.09 in IMA VSMCs and of −3.8 ± 0.08 in SV VSMCs (Fig.5). The inhibition of endogenous ET-1 production caused by both CGS 26303 and CGS 26393 was accompanied by matched increases in the accumulation of big ET-1 (Figs. 4 and 5). The structurally related compound CGS 24592, a NEP inhibitor lacking in ECE inhibitory activity, had no effect on cytokine-stimulated ET-1 release from IMA or SV VSMCs (data not shown).

Effect of CGS 26303 on accumulation over 48 h of ET-1 (■) and big ET-1 (▪) in medium of IMA (A) and SV (B) VSMCs treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml). Data represents mean ± S.E.M. for cells from five patients each assayed in triplicate.

Effect of CGS 26393 on accumulation over 48 h of ET-1 (▵) and big ET-1 (▴) in medium of IMA (A) and SV (B) VSMCs treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml). Data represents mean ± S.E.M. for cells from five patients each assayed in triplicate.
Incubation of VSMCs from both IMA and SV with either phosphoramidon, CGS 26303, CGS 26393, or CGS 24592 for 48 h did not affect cell viability.
Effect of Phosphoramidon and CGS 26303 on Conversion of Exogenous Big ET-1 by Human IMA and SV VSMCs.
Phosphoramidon inhibited the conversion of exogenous big ET-1 more potently than the endogenous production of ET-1 in both human IMA VSMCs (log IC50 values −4.8 ± 0.06 versus −3.9 ± 0.05, p < .0001) and SV VSMCs (log IC50 values −5.1 ± 0.08 versus −4.1 ± 0.09, p < .0001; Fig.6). Similarly, CGS 26303 inhibited the conversion of exogenous big ET-1 about 10 times more potently than the endogenous production of ET-1 (log IC50 values: IMA VSMCs, −4.3 ± 0.13 versus –3.5 ± 0.09,p < .001; SV VSMCs, −4.8 ± 0.15 versus −3.6 ± 0.08, p < .0001; Fig.7).
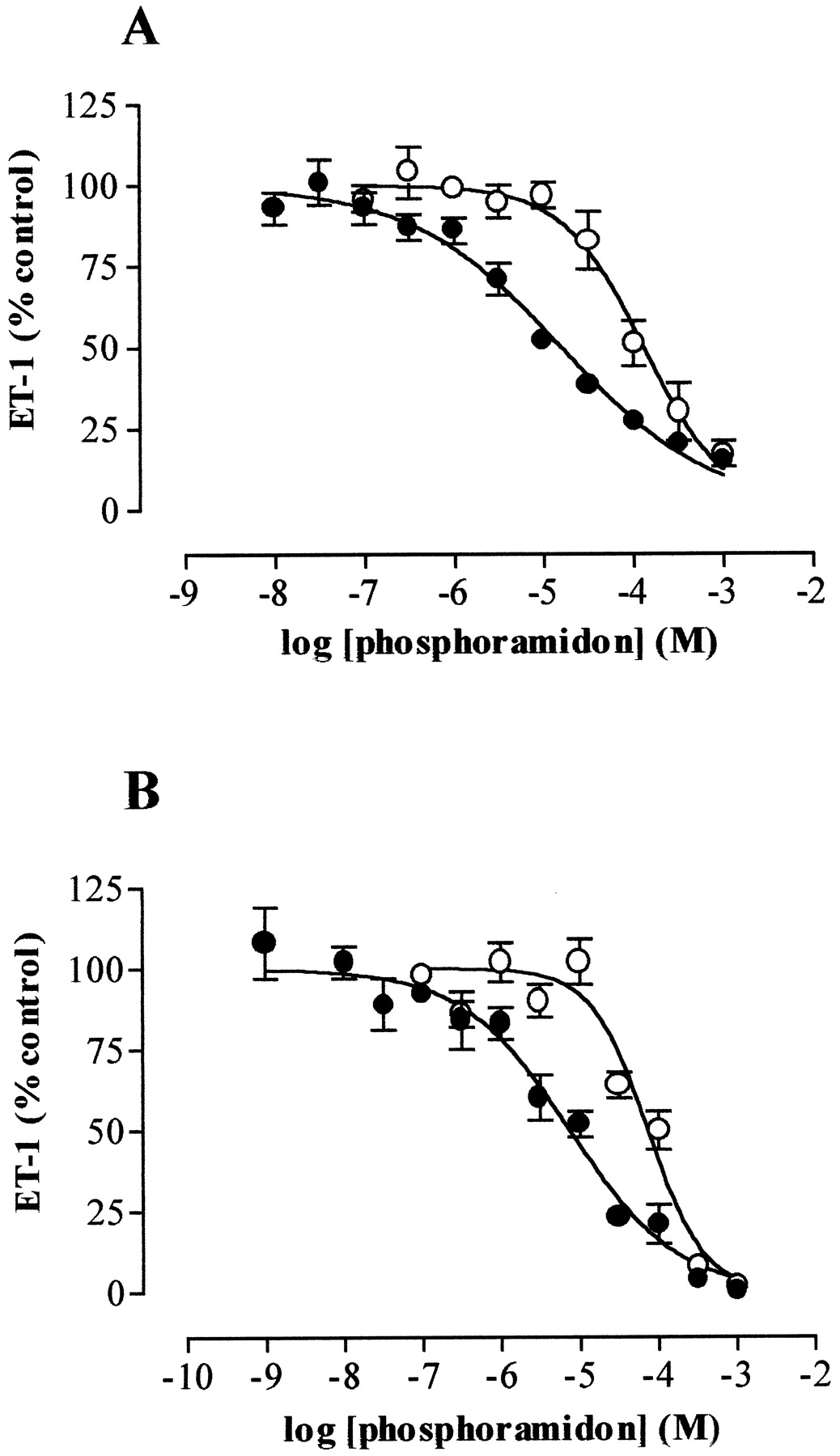
Effect of phosphoramidon on production over 48 h of endogenous ET-1 (○) and conversion over 2 h of exogenous big ET-1 (1 μM, ●) by IMA (A) and SV (B) VSMCs treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml). Data represents mean ± S.E.M. for cells from five patients each assayed in triplicate.

Effect of CGS 26303 on production over 48 h of endogenous ET-1 (■) and conversion over 2 h of exogenous big ET-1 (Δ 1 μM, ▪) by IMA (A) and SV (B) VSMCs treated with TNF-α (10 ng/ml) and IFN-γ (1000 U/ml). Data represents mean ± S.E.M. for cells from five patients each assayed in triplicate.
Effect of ECE Inhibitors on Cytokine-Stimulated Expression of Prepro-ET-1 mRNA in Human VSMCs.
Phosphoramidon (300 μM), CGS 26303 (1 mM), or CGS 26393 (300 μM) had no effect on TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) stimulated expression of prepro-ET-1 mRNA over 48 h (Fig. 8).

Effect of TNF-α and IFN-γ on expression of mRNA for prepro-ET-1 in cultured human IMA VSMCs. VSMCs grown to confluence on 75-cm2 tissue culture flasks were exposed to TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for 48 h. Total RNA was extracted and RT-PCR was performed using specific primers as described inExperimental Procedures. Expression of mRNA for GAPDH was used as positive control. Lane 1, control or unstimulated VSMCs; lane 2, VSMCs exposed to TNF-α and IFN-γ alone; and lanes 3 to 5, VSMCs exposed to TNF-α and IFN-γ in the presence of phosphoramidon (300 μM), CGS 26393 (300 μM), and CGS 26303 (1 mM), respectively. Data is shown from a representative experiment.
Expression of mRNA for ECE in Human SV VSMCs.
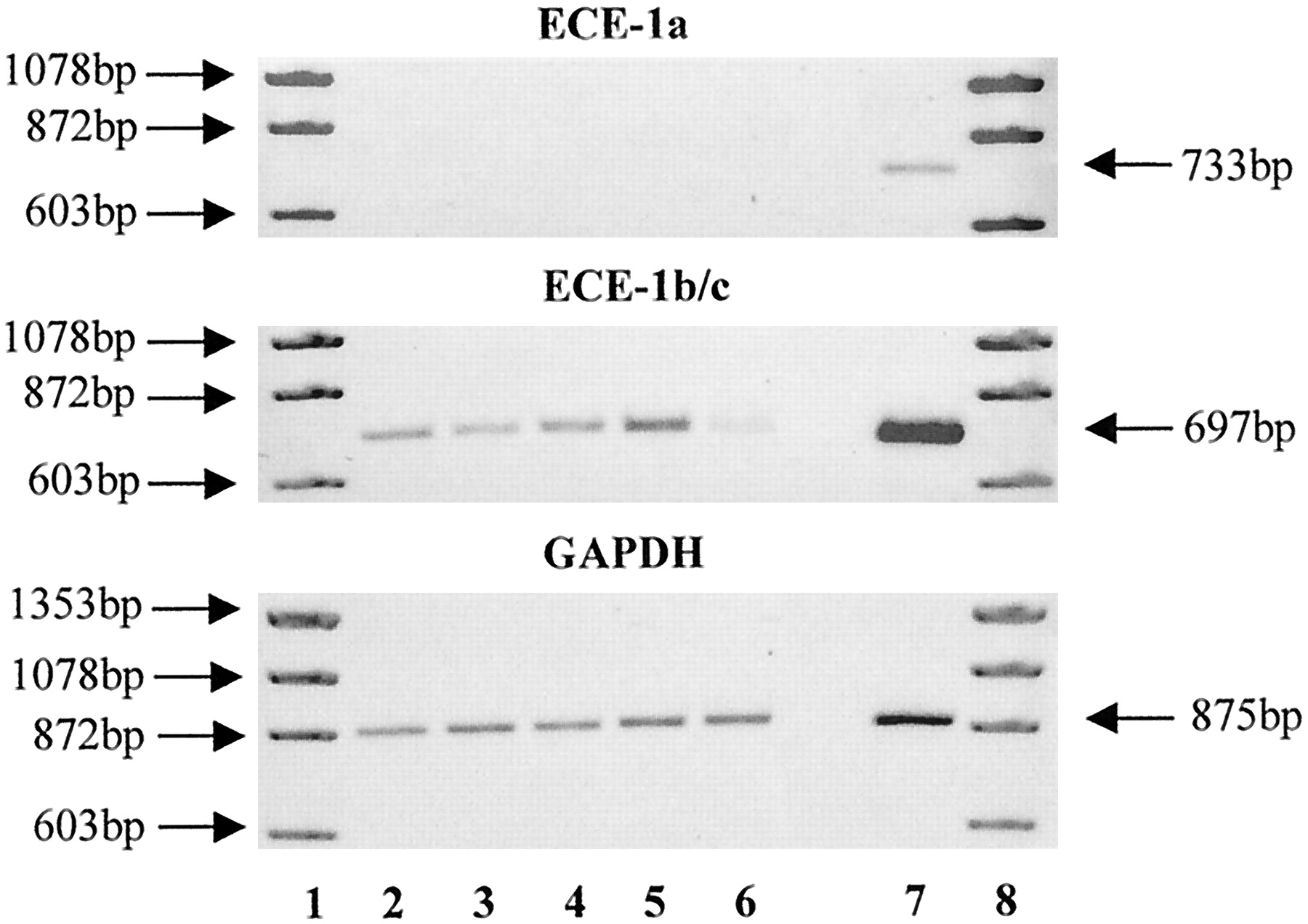
RT-PCR confirmed the presence of ECE-1b/c mRNA in SV VSMCs (Fig.9). No mRNA for ECE-1a was found in these cells, although it was readily detected in human umbilical vein endothelial cells used as a positive control. In contrast to the marked elevation in prepro-ET-1, expression of mRNA for ECE-1b/c showed little variation during exposure of SV VSMCs to TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for 48 h, although a reduction was apparent at 48 h.

Effect of TNF-α and IFN-γ on expression of mRNA for ECE in cultured human SV VSMCs over time. VSMCs grown to confluence on 75-cm2 tissue culture flasks were exposed to TNF-α (10 ng/ml) and IFN-γ (1000 U/ml) for 4, 12, 24, and 48 h. Total RNA was extracted and RT-PCR was performed using specific primers as described in Experimental Procedures. Expression of mRNA for GAPDH was used as positive control. Lane 1, marker; lane 2, control or unstimulated VSMCs; and lanes 3 to 6, VSMCs exposed to TNF-α and IFN-γ for 4, 12, 24, or 48 h, respectively. Lane 7, human umbilical vein endothelial cells that were used as a positive control for ECE-1a mRNA expression. Data is shown from a representative experiment with SV VSMCs.
Discussion
In this study we addressed two questions. First, can cytokines and/or endotoxin induce human VSMCs to produce ET-1, and second, how is endogenously produced big ET-1 processed?
As outlined in the Introduction, ET-1 appears to be involved in many disease states associated with elevations in the plasma concentrations of proinflammatory cytokines. Because proinflammatory cytokines induce expression of other endothelial- derived mediators, such as NO and prostaglandins in smooth muscle, we were interested in determining whether cytokines may also induce expression of ET-1. We therefore examined the effects of interleukin-1β, interleukin-6, TNF-α, and IFN-γ, as well as bacterial endotoxin, on the production of ET-1 by human VSMCs. Combinations of TNF-α and IFN-γ caused the greatest increases in ET-1 production, with the other agents, either alone or in combination, having no additional effect. Most notably, in experiments examining the time course of the ET-1 response to TNF-α and IFN-γ, a very dramatic increase in ET-1 formation was evident (Fig. 2). These experiments used large numbers of cells, permitting a more reliable and precise sample analysis. The particular effects of TNF-α and IFN-γ in the human cells contrast with the rat in vivo, in which we also found that interleukin-1β produces marked increases in ET-1 production, albeit in a TNF-α-dependent manner (Klemm et al., 1995). This draws attention to the variation in cytokine responsiveness of vascular smooth muscle derived from different species, as has been previously noted for the expression of iNOS (Beasley and McGuiggin, 1994). It is also important to note that the amount of ET-1 produced by the human VSMCs after stimulation with cytokines (approximately 1 pg per 103 cells in 48 h) is of a similar magnitude to that of normal human aortic endothelial cells in culture (our unpublished data, 1998). As in many blood vessels, VSMCs form a much larger population than the single layer of endothelial cells, production of ET-1 by the smooth muscle could clearly equal or even surpass that by the endothelium.
After our experiments addressing the stimuli regulating ET-1 production by VSMCs, we investigated the effects of known inhibitors of ECE. Although ECE exists in two principal forms, ECE-1 and ECE-2, it is ECE-1 that appears to be the enzyme mainly responsible for the conversion of big ET-1 into ET-1 (Schmidt et al., 1994; Shimada et al., 1994; Xu et al., 1994). It has been well documented that ECE-1 is a zinc metalloprotease that is inhibited by phosphoramidon, and phosphoramidon has been shown to inhibit the conversion of exogenous big ET-1, both within the circulation and in isolated vascular preparations (Ikegawa et al., 1991; Matsumura et al., 1991; McMahon et al., 1991; Sawamura et al., 1991; Warner et al., 1992). Notably, however, phosphoramidon is often more potent as an inhibitor of the conversion of exogenous big ET-1 than as a suppresser of the endogenous production of ET-1 by endothelial cells (McMahon et al., 1991; Sawamura et al., 1991; Warner et al., 1992; Corder et al., 1993, 1995; Plumpton et al., 1996). Similarly, we found that in the human VSMCs phosphoramidon was 10 times more potent at inhibiting the conversion of exogenous big ET-1 than the endogenous production of ET-1. This difference was not confined to phosphoramidon, but was also seen with CGS 26303, which displayed a similar 10-fold difference in potency (De Lombaert et al., 1994; Trapani et al., 1995). Two pieces of evidence suggest that the effects of phosphoramidon and CGS 26303 were mediated at the level of ECE-1. First, and most important, the inhibition of ET-1 production was accompanied by a reciprocal increase in the accumulation of big ET-1 within the culture medium. Second, the inhibitors of ET-1 production were not associated with reductions in the expression of mRNA for prepro-ET-1. Third, CGS 24592, a NEP inhibitor with little activity on ECE-1, had no effect on the production of ET-1.
It is of particular interest that the ECE inhibitors acted with 10-fold different potencies against the endogenous and exogenous conversion of big ET-1 to ET-1. We can consider two likely reasons. First, that the contrasting potencies are explained by the presence within the VSMCs of multiple ECE enzymes differently responsible for the conversion of endogenous and exogenous big ET-1, and second, that the conversion of exogenous and endogenous big ET-1 may take place at different sites. To address the first possibility, we used RT-PCR to assess the presence within the VSMCs of ECE-1. There are three isoforms of ECE-1, ECE-1a, ECE-1b, and ECE-1c that have distinct subcellular localizations (Shimada et al., 1995; Valdenaire et al., 1995; Schweizer et al., 1997). ECE-1a and ECE-1c are present at the cell surface, whereas ECE-1b appears to be predominantly intracellular. We found no evidence that the VSMCs express ECE-1a mRNA, although we could detect it in control human umbilical vein endothelial cells, as previously reported (Schweizer et al., 1997). Conversely, RT-PCR revealed the presence of ECE-1b/c in human IMA and SV VSMCs. These data indicate that the VSMCs express known ECE-1 isoforms. Interestingly, we found no evidence that the expression of the ECE-1b/c was increased following exposure to cytokines, although small changes may well not have been detected by the methodology we employed. This is in comparison with the clear increase in expression of prepro-ET-1 that was detected in matched experiments. It therefore appears that the second possibility considered above is the most likely, i.e., VSMCs contain ECE-1b/c at both intra- and extra-cellular sites. The intracellular ECE, possibly ECE-1b, is responsible for processing endogenously produced big ET-1 and has a similar sensitivity to inhibitors of ECE, as does the intracellular ECE-1b found in endothelial cells (Turner, 1993;Schweizer et al., 1997). The resistance of this intracellular ECE-1b to inhibition may be explained by inability of compounds to penetrate to the inside of the cells. Phosphoramidon, for example, is a phosphorylated sugar derivative and penetrates cells poorly (Turner, 1993; Corder et al., 1995). Earlier reports that vascular smooth muscle converts big ET-1 to ET-1 in a readily inhibitable manner leading to the conclusion that VSMCs cells express extracellular ECE, probably ECE-1c, is therefore too simple a conclusion (Balwierczak et al., 1993;Plumpton et al., 1996; Maguire et al., 1997). In fact, VSMCs appear to have an inducible synthetic pathway for ET-1 that is similar to that present constitutively within endothelial cells, and similarly sensitive as endothelial cell ECE to enzyme inhibitors.
In conclusion we show that cytokines can greatly increase the expression and release of ET-1 by human VSMCs. Previous investigators have reported that VSMCs in culture can produce low amounts of ET-1, and that this is increased following exposure to steroids, oxyhemoglobin, or cyclosporine (Kanse et al., 1991; Kasuya et al., 1993; Takeda et al., 1993; Yu and Davenport, 1995; Schweizer et al.,1997; Morin et al., 1998). It has also been found that VSMCs cultured from coronary plaques produce more ET-1 than those cultured from normal coronary artery (Haug et al., 1996). There has not, however, been an analysis of the effects of proinflammatory cytokines known to be involved in vascular disease states. Furthermore, there has been no data presented as to the synthetic pathway via which ET-1 could be produced. Generally, it has appeared that vascular smooth muscle is a passive responder to ET-1, which expresses extracellular ECE to convert any big ET-1 released from endothelial or other cell types. Our data indicates that in fact the vascular smooth muscle can actively produce ET-1 to a similar extent as the endothelium. The important conclusion that we can draw from this is that the vascular smooth muscle, traditionally thought to be an “ET-1 responder” can, under the influence of cytokines, become an “ET-1 producer”. This means that we should add ET-1 to the list of vasoactive endothelial mediators that can be induced in vascular smooth muscle: e.g., NO, PGI2, PGE2, and now ET-1. It may well be that this is part of an adaptive response in which the vascular smooth muscle takes on some endothelial characteristics to compensate for endothelial dysfunction. Under such conditions, however, the normal checks and balances present between mediators derived from the endothelium, e.g., between NO and ET, may no longer be present (Warner, 1996). This is indeed the case for NO, which can be produced by iNOS in a largely unregulated manner (Wong and Billiar, 1995). Unregulated production of ET-1 by the vascular smooth muscle following loss or dysfunction of the endothelium may similarly underlie the causative role of ET-1 in a number of disease states (Haynes and Webb, 1993; Warner et al., 1996; Parris and Webb, 1997).
Footnotes
-
Send reprint requests to: Dr. Timothy D. Warner, Vascular Inflammation, The William Harvey Research Institute, St. Bartholomew’s and the Royal London School of Medicine and Dentistry, Charterhouse Square, London, EC1M 6BQ, United Kingdom. E-mail:t.d.warner{at}mds.qmw.ac.uk
-
M. W. was in receipt of a European Commission Training and Mobility Fellowship, N. R. W. holds an Aelwyn Bursary from the Joint Research Board of St. Bartholomew’s and the Royal London School of Medicine and Dentistry, J. A. M. is a Wellcome Foundation Career Development fellow, and T. D. W. is a British Heart Foundation Lecturer (BS/95003). Citation of meeting abstracts in which this work was previously presented: Woods M, Bishop-Bailey D, Pepper JR, Evans TW, Mitchell JA and Warner TD (1997) Cytokine and lipopolysaccharide stimulation of endothelin-1 release from internal mammary artery and saphenous vein smooth muscle cells.Br J Pharmacol122:18P; and Woods M, Wood EG, Mitchell JA and Warner TD (1998) Evidence for intracellular endothelin converting enzyme in human vascular smooth muscle cells.FASEB J12:A384.
- Abbreviations:
- Big ET-1
- big endothelin-1
- ECE
- endothelin-converting enzyme
- ET-1
- endothelin-1
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- iNOS
- inducible nitric oxide synthase
- IFN-γ
- interferon-γ
- IMA
- internal mammary artery
- NEP
- neutral endopeptidase
- NO
- nitric oxide
- PGI2/E2
- prostaglandin I2/E2
- RT-PCR
- reverse transcription-polymerase chain reaction
- SV
- saphenous vein
- TNF-α
- tumor necrosis factor-α
- VSMCs
- vascular smooth muscle cells
- Received November 4, 1998.
- Accepted February 18, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}