Article Text

Abstract
Objective To clarify the clinical characteristics and epidemiology of idiopathic pulmonary arterial hypertension (IPAH) in childhood, a rare condition with a bad prognosis, poorly documented in children. Also, to describe the long-term outcome.
Design A retrospective study of 7 years' experience.
Setting UK Service for Pulmonary Hypertension in Children based at a tertiary referral centre.
Patients 64 children.
Interventions Patients were initially treated with prostanoids (n=15), bosentan (n=23), sildenafil (n=9), combination therapy (n=11) or calcium channel antagonists (n=6).
Main outcome measures WHO functional class, distance walked in 6 minutes, escalation of therapy, survival, transplant-free survival.
Results Incidence of IPAH was 0.48 cases per million children per year and the prevalence was 2.1 cases per million. 31% presented with syncope. Oedema was rare. During the first year of follow-up WHO functional class and 6-minute walk distance improved significantly. Survival at 1, 3 and 5 years was 89%, 84% and 75%, respectively; while transplant-free survival was 89% 76% and 57%, respectively. Factors predicting worse survival were WHO functional class (HR 2.4, p=0.04) and poor height and weight z-score (p<0.05 for both) at presentation.
Conclusions We showed, for the first time, that the incidence of IPAH is lower in children than adults and that the clinical features can be different. Most children present with clinical evidence of advanced disease and clinical status at presentation is predictive of outcome. This 7-year experience confirms the significant improvement in survival over historical controls.
- Hypertension, pulmonary
- paediatrics, epidemiology
- survival
- paediatric cardiology
- pulmonary arterial hypertension (PAH)
Statistics from Altmetric.com
- Hypertension, pulmonary
- paediatrics, epidemiology
- survival
- paediatric cardiology
- pulmonary arterial hypertension (PAH)
Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is a debilitating progressive disease characterised by increased pulmonary vascular resistance (PVR), right ventricular failure and death. The median survival in untreated children is less than 1 year,1 2 shorter than in adults. Little is known about this condition in childhood2–6 and there are no reliable estimates of the incidence in childhood IPAH.7 In 2001 the United Kingdom Service for Pulmonary Hypertension in Children (UKSPHC) was established to care for all children with IPAH in the country and to be the single referral centre for the investigation and treatment of children suspected or known to have this disease.6 We have previously described our treatment strategy and how it changed as new medicines became available and have detailed the survival data in both idiopathic and associated pulmonary hypertension.6 Two years later, in this paper we take advantage of the rare opportunity afforded by having a single UK centre to study in depth the clinical picture and epidemiology of children with IPAH in addition to assessing survival in the era of modern therapy in a larger cohort over a longer period of time.
Methods
This was a retrospective cohort study of the 64 children with IPAH who presented to the UKSPHC below 16 years of age and were treated between January 2001 and October 2007, with follow-up extended to January 2008. The study was approved by the institution ethics committee.
Patient groups
Twenty of the 64 children were diagnosed between 1986 and 2000, before the establishment of the UKSPHC in January 2001 and entered the service after its inception (prevalent group). The median time from diagnosis to entry into the UKSPHC was 45 months (IQR 25–98). The remaining 44 children were diagnosed after January 2001 (incident group), and entered the service a median of one month (IQR 0–7) after diagnosis.
Investigations
The initial investigation in all patients included an electrocardiogram (ECG), transthoracic echocardiogram and chest radiograph. Cardiac catheterisation under general anaesthesia with acute vasodilator testing using inhaled nitric oxide (NO) was performed where possible. Pulmonary blood flow was calculated based on measured oxygen consumption in the majority of cases. Computed chest tomography with pulmonary angiography (CTPA) was performed to exclude chronic thrombo-embolic and lung disease associated PAH. From 2003 a 6-minute walk test was performed on children over the age of 5 years who had the cognitive ability to perform the test. After the initial assessment the patients were followed up at approximately three-monthly intervals.
Assessing the data
The 6-minute walk distance was expressed as a percentage of predicted based on a published reference equation for children.8 Height and weight were expressed as z-scores for normal British children.9 Electrocardiograms were analysed for evidence of right ventricular hypertrophy (RVH), right ventricular strain and right atrial enlargement as previously described.10 On the echocardiograms, a subjective, semiquantitative assessment of right ventricular functional impairment, dilatation and hypertrophy were made using the following grades: 1=normal, 2=mild, 3=moderate and 4=severely affected.10 11 From the beginning of the study period, an acute vasodilator response was accepted as positive when the pulmonary vascular resistance fell to a near normal value (ie, <5 U.m2), with a normal or high cardiac output. In all our acute responders the pulmonary arterial pressure fell to less than 40 mm Hg on vasodilator testing.
Statistical analysis
Values are presented as median and IQR or mean and SD as appropriate. Comparisons between groups were made using the Student t test, the paired t test, Wilcoxon rank sum test or Fisher's exact test as appropriate. Incidence of IPAH was calculated for patients newly diagnosed between January 2001 and January 2007 with reference to the total UK childhood population (Office for National Statistics). Factors associated with syncope were assessed using logistic regression. In the analysis of change in WHO functional class, patients who died were assigned to functional class IV. Kaplan-Meier survival curves were constructed for the determination of median survival. Survival time was taken to be the time from presentation to our service to the time of death or the first censoring event. Patients were censored at time of transplantation or at the end of the study period. Factors associated with a poorer prognosis were assessed using a Cox proportional hazards model. The assumption of proportionality was tested using the Schoenfeld residuals. Analysis was performed using Stata version 10.0 (StataCorp). A two-sided p value <0.05 was considered statistically significant.
Results
Incidence, prevalence, ethnicity and demographics
The estimated incidence of childhood IPAH was 0.48 (95% CI 0.31 to 0.66) per million children per year. Estimated regional incidence ranged from 0–1.09, but the differences were not statistically significant. The prevalence of childhood IPAH was 2.07 cases per million children. A family history of pulmonary arterial hypertension was present in five (7.8%) of the 64 patients. The ethnic breakdown was 83.9% white, 12.5% Asian, 3.6% mixed race and zero from black and other ethnic groups, compared with the expected ethnic breakdown for UK children of 88.1%, 5.7%, 2.2%, 2.9% and 1.1%, respectively. The small numbers preclude statistical analysis. The female to male ratio was 1.7:1. A fifth of patients were below the age of one year at the time of diagnosis (table 1).
Demographic, clinical and haemodynamic characteristics at presentation to the United Kingdom Service for Pulmonary Hypertension in Children
Baseline clinical and investigative findings
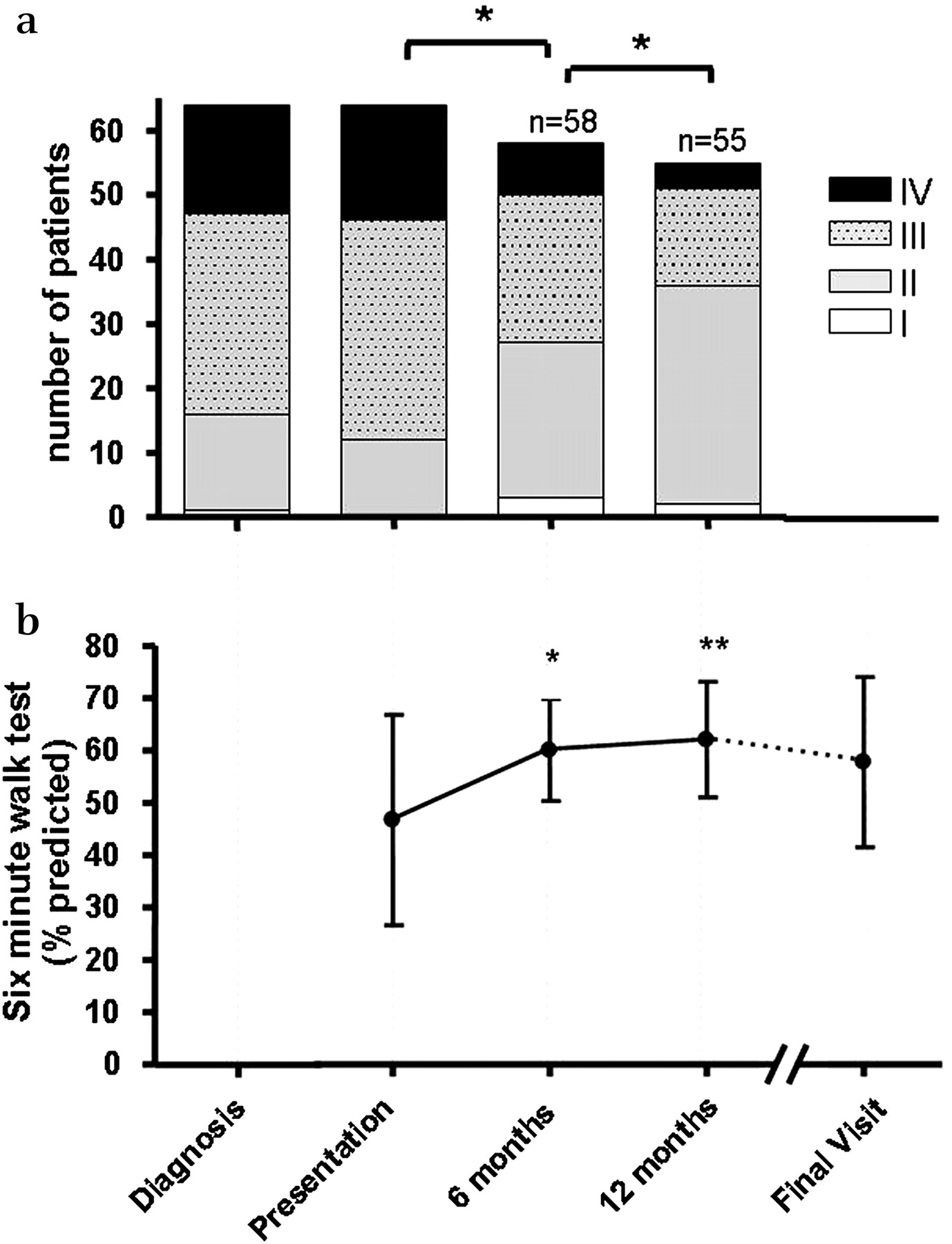
The frequency of symptoms at diagnosis was dyspnoea in 75%, exercise limitation in 31%, syncope in 31%. Eighty per cent of patients had had two or more of these symptoms. The children were significantly growth retarded, the z-score for both weight and height being significantly reduced, p<0.002 and p<0.001, respectively. Most were in WHO class III (table 1) (figure 1a) at diagnosis. Fifteen more children, all untreated, deteriorated to class III by the time they reached our service. An accentuated pulmonary component of the second heart sound was present in 95% of patients, a right ventricular heave in 80%, hepatomegaly in 23%, an elevated jugular venous pressure in 8%, likely under observed in the younger children and peripheral oedema in just one 15-year-old patient. Two or more physical signs were present in 83%. The median peripheral oxygen saturation by pulse oximetry was 95% (IQR 90–97.5). One child had Noonan's syndrome, one had triple X syndrome and another three had Down's syndrome.

(a) WHO functional class *p<0.01. (b) Percentage predicted 6-minute walk distance n=19, *p=0.04, **p<0.01 compared to baseline.
The electrocardiogram showed evidence of right ventricular hypertrophy in 85%, P pulmonale in 58% and right ventricular strain in 50% of patients. All had at least one of the following echocardiographic abnormalities: right ventricular dilatation, hypertrophy or dysfunction. The mean (±SD) score for each of these features was 3.0±0.8, 2.7±0.7 and 1.8±−0.8, respectively. Tricuspid regurgitation was present in 91% patients with a maximum jet velocity of 4.6±0.9 m/s. Pulmonary regurgitation was detectable in 34% of patients with an end diastolic velocity of 2.8±0.7 m/s. An inter-atrial communication was detected in 28 (44%) patients, a patent foramen ovale in 16 and an atrial septal defect in 12, all of whom had the same history and clinical findings as the children without an atrial communication.
Nineteen children, with a mean age of 11.7±3.6 years, had a baseline walk test. The mean walk distance was 285±122 m (47±20% predicted) (figure 1b). Testing could not be carried out on one child who had behavioural problems and one who was extremely ill at presentation.
Fifty-eight patients underwent initial cardiac catheterisation (table 1). The six children who were not catheterised were extremely ill and three died within 90 days of presentation. The mean pulmonary artery pressure (mPAP) was severely elevated at 58±22 mm Hg, and in 43% it exceeded the systemic arterial pressure. Five of the 54 children successfully tested were considered as true acute responders in that the PVRI fell from 8.2±5.5 (range 6.4–19.3) U.m2 to 4.6±2.4 (range 3.0–5.1) U.m2 and the mean PAP fell from 40±9.6 mm Hg to 22±4.5 mm Hg. These children were treated with a calcium channel antagonist alone. In another five patients the PVRI fell, but remained at a higher level, falling from 18.8±5.3 (range 14.1–27.4) U.m2 to 7.0±2.0 (range 6.2–9.9) U.m2 and the mean PAP fell from 48±8.1 mm Hg to 27.4±3.0 mm Hg. These children were given sildenafil in addition to a calcium channel antagonist. None of the children died during catheterisation.
Clinical correlates at the time of presentation
Influence of age and gender
There was no correlation between age and mean PAP, PVRI or CI. The median age of acute responders was 8.0 years (IQR 7.1–12.8) compared with 4.6 years (IQR 2.4–9.5) for non-responders, p=0.25. There was no male:female difference in mean age (5.8 vs 5.4 years), functional class (2.7 vs 3.0), mean PAP (63 vs 55 mm Hg) or PVRI (22 vs 18 U.m2). On acute vasodilator testing, however, boys were less responsive than girls, and had a higher mean PAP and PVRI of 62 vs 45 mm Hg (p<0.01), and 19.6 vs 12 U.m2 (p=0.02), respectively.
Syncope
Twenty patients (31%) had experienced syncope by the time of diagnosis. The median time from first syncope to presumptive diagnosis by the referring physician was 9 months (IQR 1–19). Inter-atrial communications were associated with a lower incidence of syncope, unadjusted OR 0.13 (95% CI 0.03 to 0.50, p=0.004). On univariate analysis syncope was also associated with older age at diagnosis OR 1.14 (95% CI 1.1 to 1.23, p=0.03), worse WHO functional class OR 3.3 (95% CI 1.4 to 7.95), higher RV dilatation score on echocardiography OR 2.1 (95% CI 1.0 to 4.6), and a higher weight z-score OR 1.7 (95% CI 1.1 to 2.6). There was no significant relation between syncope and gender, right atrial pressure, baseline PVRI or response to acute vasodilator testing. On multivariate testing, only WHO functional class and RV dilatation score remained significantly associated with an increased risk of syncope, adjusted ORs 4.6 (95% CI 1.3 to 16.5, p=0.02) and 3.0 (95% CI 1.1 to 8.7, p=0.04), respectively.
Treatment
Considering both prevalent and incident groups, at presentation to the service 41 patients had never received treatment (treatment naive), and 23 were already receiving treatment, the median duration of previous treatment being 23 months. Eight children were being given calcium channel antagonists. Fifteen were on a prostanoid, or an endothelin receptor antagonist or sildenafil, six of these having deteriorated on a calcium channel antagonist. Four of the children who were referred on sildenafil and one child who was referred on a calcium channel antagonist died before they could be fully investigated and appropriately treated.
Initial targeted therapy following assessment by the UK service
Choice of initial therapy was guided by clinical and haemodynamic status as previously described.6 The significant association between functional class and PVRI and treatment choice are shown in figure 2. The five acute responders were treated with calcium channel antagonists alone. A further five children were treated with sildenafil with or without calcium channel antagonist. Thirty-one patients were given bosentan (combined with sildenafil in eight), and 18 were treated with intravenous prostacyclin (combined with sildenafil in three). Five patients had undergone atrial septostomy before referral and a further 17 children had an atrial septostomy at the time of initial presentation to the UKPHS. The majority of children were anticoagulated with warfarin but the youngest were given aspirin. Supplemental oxygen and diuretics were given when clinically indicated.

Relation of PVRI and WHO functional class with treatment choice. (a) Pulmonary vascular resistance (PVRI) at baseline (upper circles) and during vasodilator testing (lower circles). The mean fall in PVRI for the three treatment groups was 42±29%, 23±23% and 17±14%, respectively (ANOVA p=0.03). (b) Mean WHO functional class for the three treatment groups; 2.7±0.7, 3.0±0.7 and 3.4±0.6 (ANOVA p=0.01). CCB, calcium channel antagonist.
Clinical course on follow-up
WHO functional class improved during the first 6 months (p<0.01). During the second 6 months another 18 patients improved, five worsened and one died (p<0.01) (figure 1a). The children continued to grow but there was no catch-up growth, the z-scores for weight and height being −0.87 and −0.83, respectively, at the last assessment. The mean 6-minute walk distance increased significantly from 47% (285 metres) of predicted to 60% (385 metres) at 6 months (p=0.04) and remained stable until the final visit (33±10 months) (figure 1b).
Of the 44 patients who had not experienced syncope by the time of initial diagnosis a further seven children developed syncope by the time they reached the service. During follow-up a further nine patients developed syncope after a median time of 6.2 years.
Right ventricular strain, evident in the electrocardiograms of 32 patients at presentation, developed in a further nine (14%) patients after a median of 5.6 years. Twenty-one patients had at least one repeat cardiac catheterisation study. In the non-responders (n=17) there was no significant change in basal PVRI, PVRI on vasodilator testing or in mean PAP. Five acute responders being treated solely with a CCB were re-catheterised and all had maintained their acute vasodilator status.
Modification of treatment regimen
Twenty-seven of the 47 patients who were initially treated with monotherapy progressed to dual therapy after a median of 23 months, prostacyclin with sildenafil (2), prostacyclin with bosentan (17) and bosentan with sildenafil (8). Seven of these progressed to triple therapy. Atrial septostomy had been performed in 22 children initially and was carried out in a further nine during follow-up, the primary indication being recurrent syncope. One child died during a salvage procedure for recurrent syncope despite being on triple therapy, 15 months after the initial diagnostic catheterisation.
Seven children had a bilateral lung transplant and one a heart-lung transplant at a median age of 7.1 years. There were five boys and three girls. The median (range) time on the waiting list was 2.9 (0.5–9.9) months. Two patients died awaiting transplantation, 2.7 and 6.7 months after listing.
Survival
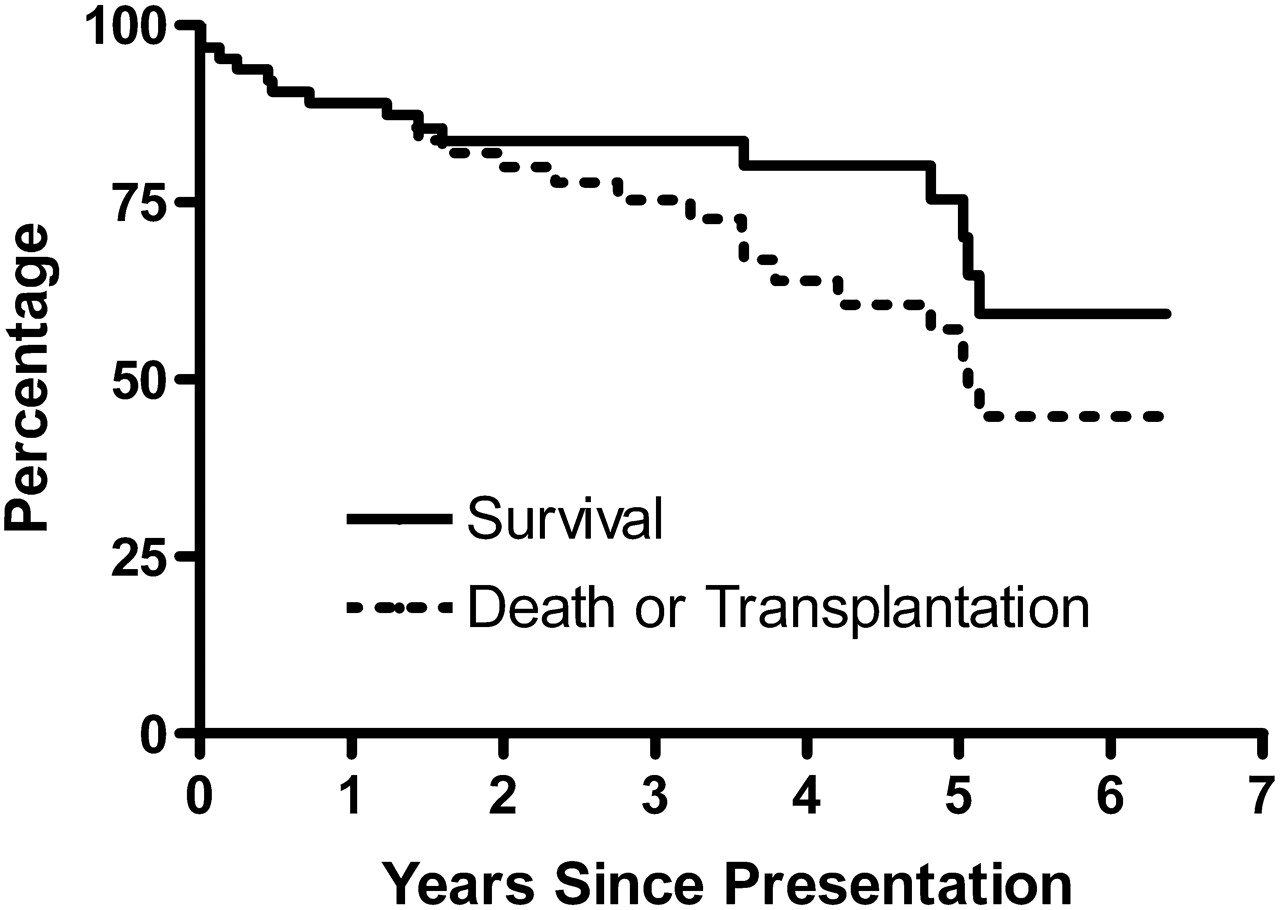
The 1, 3 and 5-year survival rates were 89% (78–95%), 84% (72–91%) and 75% (58–86%) for the entire group (figure 3), giving an annual mortality rate of 7.6% (CI 4.6% to 12.3%). Freedom from death or transplantation at 1, 3 and 5 years was 89% (78–95%), 76% (62–85%) and 57% (40–71%) for the entire group. There was no significant difference in either survival or freedom from death or transplantation between incident and prevalent groups. There were no deaths among the five acute responders. Of the other five patients whose mean PAP also fell to less than 40 mm Hg on acute vasodilator testing but whose PVRI was higher, 7.0±2.0 U.m2, and therefore were initially treated with oral PH specific therapies, one needed transplantation, one was in WHO functional class III and three remained in class II (median follow-up 13, range 7–24 months).
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier curves for survival and freedom from death or transplantation for the entire cohort.
Considering all baseline data as a predictor of outcome (table 2), univariate survival analysis for the group as a whole demonstrated that worse survival was associated with poorer WHO functional class (HR 2.35, p=0.04), and lower weight and height z-scores (HR 1.5, p=0.008 and HR 1.4 p=0.04), respectively. Worse survival was also associated with female gender (HR 8.9, p=0.03) but the effect disappeared when assessing freedom from death or transplantation. A higher proportion of males underwent transplantation. Age was not a prognostic factor for death or transplantation.
Univariate survival analysis
Survival free from transplantation was again worse in children with a poorer functional class (HR 2.8, p=0.003) and a lower weight z-score (HR 1.37, p=0.02). On baseline echocardiography, the presence of an inter-atrial communication (HR 2.3, p=0.05), increased tricuspid regurgitation velocity (HR 1.8, p=0.02) and right ventricular dilatation (HR 1.7, p=0.04) were all associated with a worse outcome. Indicators of a poor prognosis at initial cardiac catheterisation included the PVRI, both basal and on vasodilator testing (HR 1.04, p=0.03 and HR 1.06, p=0.01, respectively), and mean PAP during vasodilator testing (HR 1.02, p=0.05). Non-responders were significantly more likely to die or require transplantation (p=0.05).
On multivariate analysis, restricted to variables statistically significant on univariate analysis, low weight and a worse WHO functional class were predictors of a poor survival (HR 2.5, p=0.003 and HR 2.5, p=0.05, respectively).
Discussion
IPAH is a rare disease. Establishing a national clinical network for the entire UK provided a unique opportunity to study the incidence and prevalence of IPAH in children for the first time. We found that the incidence of IPAH in children was 0.48 per million children per year, lower than that in adults, estimated at 1–2 per million per year in the Western world.12–14 While it is unlikely that children with IPAH were not referred to the UK service it is possible that IPAH remains undiagnosed in some children. IPAH did not however, feature as a cause of sudden, unexplained death in a large UK study of unexplained death in children and adolescents published in 2000.15 A family history was present in 7.8% of patients, a figure similar to that previously reported in adults16–18 and in a Mexican paediatric study.4 We found a female preponderance, as in previous studies.2 4 14 16 This was true for infants as well as older children, suggesting a female predominance throughout life. The ethnic profile indicated a higher than expected proportion of Asian children, 12.5% compared with 5.7% in the general population, but the small numbers involved preclude statistical analysis.
Dyspnoea was the commonest presenting feature, as in adults. The most striking differences in presentation between children and adults were the greater frequency of syncope, 31% compared with 8%, and the absence of oedema in young children.14 16 The absence of oedema was consistent with there being a lower mean right atrial pressure in our paediatric cohort, 7.1 mm Hg, compared to the 9 mm Hg reported in adults.14 16 An inter-atrial communication appeared to protect against syncope, as expected, but there were no haemodynamic associations. Importantly, syncope appeared to be a feature of greater clinical deterioration at the time of presentation. Lack of an atrial communication was, however, associated with earlier presentation. Considering the entire cohort, the physical findings and investigations frequently indicated the presence of advanced disease at the time of presentation. The majority (75%) were in functional classes III and IV, similar to reported studies on both adults18 and children,3 and had electrocardiographic, echocardiographic and haemodynamic features of severe pulmonary hypertension. They had also failed to thrive. In five children IPAH was associated with a syndromal abnormality. Other investigators have made similar observations but the association is not understood.19
The main determinant of treatment was, and remains, the response to vasodilator testing, nowadays with nitric oxide. Until recently the definition of an acute responder had been a fall in pulmonary vascular resistance and arterial pressure of 20% with no reduction in cardiac output, but despite such a fall the resistance and pressure may still be high. We have always used a stricter definition of a positive response, looking for a fall in resistance to a near normal level. In 2004 it was thought that long-term CCB responders could be identified by a drop of mean PAP of >10 mm Hg, leading to a mean PAP <40 mm Hg and a normal cardiac output.20 We had 10 patients showing such a response. There were five patients in whom the resistance fell to a mean of 4.6 U.m2 and all remain well and stable on a calcium channel antagonist, while of the five whose mean resistance fell to 7 U.m2 and who were treated initially with sildenafil as well as a calcium channel antagonist, one has been transplanted, and one remains in WHO functional class III, despite escalating therapy. This demonstrates the fallibility of any management guideline, but also raises the possibility that different criteria may apply to children. In 2001 when the UKSPHC was started there was no treatment algorithm and no endothelin receptor antagonists or phosphodiesterase inhibitors for the treatment of pulmonary hypertension and all this changed during the study period. Despite this, retrospective analysis showed that there was a relation between the clinical assessment of the child and the treatment given. The patient characteristics (WHO functional class and PVRI) did correlate with the choice of therapy (figure 2). The worse the functional class and the higher the PVRI the more intensive was the treatment given.
Balloon atrial septostomy was carried out primarily to treat syncope and was an early rather than an end stage therapy. Considering all therapies, the early response to treatment was encouraging in that the children showed an improvement in WHO functional class and the 6-minute walk distance. However, the majority who were not acute responders showed evidence of disease progression. There was no catch-up growth and new syncopal events occurred at the rate of 7.7% per year. The majority of children (83%) were initially treated with monotherapy but by the end of the study period 64% were receiving combination therapy. Whether or not all children who are not positive responders should be started on combination therapy at diagnosis is yet to be decided, but would be a logical approach given the natural history of the disease. The necessity of using complicated drug delivery systems in some children emphasises the need for prompt referral to a specialised centre.
The overall survival rate was 89% at 1 year, 84% at 3 years and 75% at 5 years, figures comparable with American and Japanese paediatric cohorts3 5 and with adult studies.14 18 Patients in the prevalent group did not fare worse than those in the incident group, perhaps indicating that the prevalent group included self-selected longer-term survivors. The clinical and haemodynamic features at presentation were predictive of poor transplant free survival—namely, poor WHO functional class, an elevated PVRI at baseline and an elevated PVRI and PAP during acute vasodilator testing, as others have noted in both adult and paediatric studies.2 18 21 Younger age did not confer benefit and the haemodynamic findings were at least as severe in young as in older children, as Sandoval et al4 also found. In 1999 Barst2 found that survival was better the younger the child, possibly owing to the higher proportion of acute responders among the younger children in their cohort. In future it will be important to find out whether a specific genetic mutation is predictive of survival in children.
A novel finding in the present study was the prevalence of growth retardation and the important effect this has on prognosis. Low weight z-score on presentation was associated with increased mortality on multivariable analysis. While low BMI is known to be associated with poor outcome in adult patients with heart failure22 this association has not previously been described in either adult or paediatric patients with pulmonary hypertension. There was no significant change in either height or weight z-score between first presentation and final follow-up. If further study shows that in individual children changes in growth parameters correlate with survival then growth would provide a much needed cheap, simple and non-invasive tool for assessing treatment response.
Conclusion
The organisation of healthcare delivery to children with IPAH in the UK, around one reference centre to which all children suspected of the disease should be referred has provided a unique opportunity to study the clinical characteristics and epidemiology of this rare condition. This is the first study to estimate the incidence of IPAH in children in an audited national cohort and shows that the incidence is significantly lower than in adults. We found that most children present with signs of advanced disease and that clinical status at presentation is predictive of survival. We confirm the improvement in survival over historical controls.
Acknowledgments
We should like to thank all the physicians who referred the children to the service and helped take care of them.
References
Footnotes
Linked articles 195248.
Funding SM is supported by a grant from the Pulmonary Hypertension Association UK, PHA-UK. AAH is partially funded by Actelion Ltd. Gewerbestrasse 16CH-4123 Allschwil Switzerland Unit 3A Enterprise Court Farfield ParkManversRotherhamSouth Yorkshire S63 5DB, UK.
Competing interests HF and AAH declare no conflicts, SM has received an honorarium from Actelion Ltd. ISN and SGH have acted as a consultant and received unrestricted educational grants from Actelion Ltd, Encysive pharmaceuticals, GlaxoSmithKline and Pfizer.
Ethics approval This work was approved by the ethics committee of Great Ormond Street Hospital and UCL Institute of Child Health.
Provenance and peer review Not commissioned; externally peer reviewed.