Article Text

Abstract
Background/Aims We studied the role of lysyl oxidase-like 2 (LOXL2) in collagen crosslinking and hepatic progenitor cell (HPC) differentiation, and the therapeutic efficacy of a LOXL2-blocking monoclonal antibody on liver fibrosis progression/reversal in mice.
Methods Anti-LOXL2 antibody, control antilysyl oxidase antibody or placebo was administered during thioacetamide (TAA)-induced fibrosis progression or during recovery. Therapeutic efficacy in biliary fibrosis was tested in BALB/c.Mdr2−/− and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)-fed mice. Collagen crosslinking, fibrosis progression and reversal were assessed histologically and biochemically. HPC differentiation was studied in primary EpCAM(+) liver cells in vitro.
Results LOXL2 was virtually absent from healthy but strongly induced in fibrotic liver, with predominant localisation within fibrotic septa. Delayed anti-LOXL2 treatment of active TAA fibrosis significantly reduced collagen crosslinking and histological signs of bridging fibrosis, with a 53% reduction in morphometric collagen deposition. In established TAA fibrosis, LOXL2 inhibition promoted fibrosis reversal, with enhanced splitting and thinning of fibrotic septa, and a 45% decrease in collagen area at 4 weeks of recovery. In the Mdr2−/− and DDC-induced models of biliary fibrosis, anti-LOXL2 antibody similarly achieved significant antifibrotic efficacy and suppressed the ductular reaction, while hepatocyte replication increased. Blocking LOXL2 had a profound direct effect on primary EpCAM(+) HPC behaviour in vitro, promoting their differentiation towards hepatocytes, while inhibiting ductal cell lineage commitment.
Conclusions LOXL2 mediates collagen crosslinking and fibrotic matrix stabilisation during liver fibrosis, and independently promotes fibrogenic HPC differentiation. By blocking these two convergent profibrotic pathways, therapeutic LOXL2 inhibition attenuates both parenchymal and biliary fibrosis and promotes fibrosis reversal.
- CIRRHOSIS
- HEPATITIS
- HEPATIC FIBROSIS
- PRIMARY BILIARY CIRRHOSIS
- PRIMARY SCLEROSING CHOLANGITIS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The lysyl oxidase (LOX) family of five proteins (LOX, LOX-like (LOXL)1–4) mediates collagen crosslinking and stabilisation, promotes liver fibrosis and limits its resolution. The relative contribution of individual family members, including LOXL2, to this process of collagen crosslinking was unknown.
LOXL2 is upregulated in fibrotic diseases, and early treatment with anti-LOXL2 antibody AB0023 inhibits fibrosis in a mouse model of mild liver fibrosis (Barry-Hamilton et al9). It was not clear whether delayed LOXL2 neutralisation will be effective in the setting of pre-established biliary and non-biliary fibrosis, or whether it would induce fibrosis reversal.
LOXL2 controls epithelial differentiation in certain tissues and cancers; the effect on epithelial homeostasis in the liver remained unclear.
What are the new findings?
A major contribution of LOXL2 to collagen crosslinking and stabilisation in vivo is directly demonstrated in hepatic fibrosis.
Delayed treatment with anti-LOXL2 antibody inhibits advanced, pre-established biliary and non-biliary fibrosis, and promotes reversal of advanced parenchymal liver fibrosis in mice.
Autocrine/paracrine LOXL2 controls the lineage commitment of hepatic progenitor cells (HPC) independently of collagen crosslinking. LOXL2 inhibition promotes HPC differentiation towards hepatocytes and attenuates ductular reaction.
How might it impact on clinical practice in the foreseeable future?
Our findings directly relate to novel therapies targeting LOXL2: the antibody simtuzumab that is currently undergoing broad clinical phase II evaluation for liver diseases, and several small molecule inhibitors in preclinical/phase I stages. Our data (1) suggest that targeting LOXL2 might slow down fibrosis progression in advanced stages of biliary and non-biliary liver diseases; (2) support the rationale of anti-LOXL2 treatment to reverse established fibrosis/cirrhosis (eg, after achieving sustained viral response (SVR) in HCV); (3) new mechanistic insights into the role of LOXL2 in regulation of HPC biology suggest potential advantages for cell-permeable small molecule LOXL2 inhibitors in diseases associated with ductular reaction.
Introduction
Liver fibrosis, characterised by excessive deposition of extracellular matrix, results from chronic liver injury of different aetiologies and represents a major worldwide health problem.1 The progression of liver fibrosis to cirrhosis gives rise to severe complications including portal hypertension, liver failure and hepatocellular carcinoma (HCC), and incurs a high liver-related mortality.2 Even in the era of highly effective antiviral therapy, curative treatment is not available for the majority of patients with chronic liver diseases, with liver transplantation remaining the only effective treatment for decompensated cirrhosis or HCC. Thus, the development of effective antifibrotic drugs to halt progression to cirrhosis, or even reverse advanced fibrosis, is urgently needed.3
Collagen crosslinking is an essential process for fibrotic matrix stabilisation, which contributes to fibrosis progression and limits reversibility of liver fibrosis.4 Thus, inhibition of collagen crosslinking is considered to be a promising therapeutic strategy in fibrotic diseases. At least two types of crosslinking enzymes, tissue transglutaminase (TG2) and the lysyl oxidase (LOX) family, are overexpressed in hepatic fibrosis. However, TG2-deleted mice display normal collagen crosslinking, are not protected from liver fibrosis development and do not show improved fibrosis reversal, casting doubt on the functional significance of TG2 in fibrotic matrix stabilisation.5 In contrast, our recent data suggest that LOX activity is a major contributor to collagen crosslinking and fibrotic matrix stabilisation in liver fibrosis, and functionally regulates its reversibility.6
LOX family enzymes are secreted, copper-dependent amine oxidases that oxidise and deamidate the side chain of peptidyl lysine, which produces α-aminoadipic-δ-semialdehyde residues that react with the amino group of peptidyl lysine on a second collagen (or elastin) chain to form a covalent interchain crosslink.7 The LOX family is comprised of five isoforms, LOX and the LOX-like enzymes LOXL1–4, with overlapping but distinct functions and expression patterns in normal and diseased tissues.8 ,9 Among them, LOX and LOXL2 have been reported to be overexpressed in Wilson's disease10 and murine liver fibrosis.9 Only recently, proof-of-concept experiments using the non-selective LOX inhibitor b-aminopropionitrile (BAPN) confirmed that overall LOX activity functionally contributes to fibrotic matrix crosslinking and stabilisation, and retards reversal of CCl4-induced liver fibrosis.6 However, it is currently unclear which specific enzyme(s) within the LOX family are most relevant for collagen stabilisation, and whether functions other than collagen crosslinking mediate their putative profibrotic effects. While an important role in fibrosis has been assigned to LOXL2,9 and clinical studies with function-blocking monoclonal antibody (mAB) GS-6624 (simtuzumab) have been initiated, mechanistic studies are lacking that examine its therapeutic potential in clinically relevant scenarios of pre-established liver fibrosis progression and its reversal, and elucidate its effects on collagen crosslinking and cells in fibrosis.
We performed an in-depth study on the role of LOXL2 in chronic liver disease using selective LOXL2 inhibition with a blocking AB0023 mAb in well-characterised and standardised mouse models of biliary and non-biliary fibrosis, and in vitro cell culture systems. We show that LOXL2 neutralisation efficiently inhibits collagen crosslinking, suppresses advanced fibrosis progression and accelerates its reversal. We also demonstrate that both crosslinking-dependent and crosslinking-independent mechanisms are responsible for profibrogenic LOXL2 activity.
Materials and methods
All mouse experiments were approved by the IACUC of the Beth Israel Deaconess Medical Center (BIDMC, protocols 004-2012, 010-2015).
Thioacetamide-induced model of liver fibrosis progression and reversal
Progressive pan-lobular liver fibrosis was induced according to an optimised dose-escalating protocol of chronic thioacetamide (TAA) administration established as described.5
BALB/c.Mdr2−/− mouse model of biliary liver fibrosis
Mdr2(abcb4)−/− mice on the fibrosis-susceptible BALB/c background, which spontaneously develop accelerated biliary fibrosis, early-onset portal hypertension and liver cancer, were generated as reported previously.11
DDC (3,5-diethoxycarbonyl-1,4-dihydrocollidine)-induced model of biliary fibrosis
DDC feeding induces progressive cholangitis, a pronounced ductular reaction and bridging fibrosis, recapitulating clinical features of human biliary fibrosis. C57Bl/6J male mice, 8-week-old, were fed DCC-supplemented diet (0.1%) for 4 weeks to induce advanced biliary fibrosis as described.12
Therapeutic anti-LOXL2 and anti-LOX mAb
LOXL2-specific mAb (AB0023) with high affinity (KD=0.37 nM) for mouse and rat LOXL2 was developed by Arresto and Gilead Sciences.13 IC50 for inhibition of enzymatic activity of LOXL2 by AB0023 is 46 nM. The LOX-specific mAb (M64, Gilead Sciences) for mouse and rat LOX and isotype control IgG1 (mouse mAb with irrelevant specificity) were used as controls. AB0023 (30 mg/kg), M64 (30 mg/kg) or vehicle (PBS) as placebo were administered to mice by intraperitoneal injection two times a week.
Cell lines and conditioned media
The spontaneously immortalised rat hepatic stellate cell (HSC)/myofibroblast line HSC-X was established from primary HSC isolated from male Wistar rats (Charles River, Sulzfeld Germany) as described.14 The immature murine ductal cell line 603B15 was a kind gift from Dr Ueno (University of Sendai, Japan). Conditioned medium (CM) was prepared by incubation of subconfluent 603B or HSC-X cells in serum-free DMEM for 24 hours and supplemented with 10% FBS.
Morphometry of fibrotic septa,6 measurements of fibrotic matrix stability and hepatic collagen content,5 ,16 immunostaining and immunoblotting (see online supplementary table S1 for primary antibody information), quantitative reverse transcriptional PCR (see online supplementary table S2 for primer/probe sequences),5 ,17 ,18 isolation and culture of primary HSCs19 and EpCAM(+) cells from the livers of Mdr2−/− mice,20 ,21 collagen gel contraction assay and chromogenic in situ hybridisation were performed as described previously. For details, see online supplementary material.
supplementary data
Statistical analysis
Data are expressed as mean±SE of mean. Comparisons between two groups were done by Student's t-test, and between three or more groups by analysis of variance with Dunnett's post-test. Statistical significance was defined as p<0.05. All statistical analyses were performed with GraphPad Prism V.5.00 (GraphPad Software, San Diego, California, USA).
Results
Delayed treatment with anti-LOXL2 antibody inhibits collagen crosslinking and attenuates advanced liver fibrosis
Progressive pan-lobular liver fibrosis was induced in C57Bl6 mice by repeated TAA injections for up to 12 weeks.5 TAA administration led to robust and progressive scarring, characterised histologically and biochemically as significant pericentral fibrosis with occasional bridging at week 6, and advanced pan-lobular, bridging fibrosis/precirrhosis at week 12. Immunohistochemistry revealed that LOXL2, hardly detectable in healthy non-fibrotic control livers, was strongly upregulated after 6 and 12 weeks of TAA (figure 1A). LOXL2 immunoreactivity was observed along the fibrotic septa in a pattern similar to the distribution of collagen type I and α-smooth muscle actin (α-SMA)-positive activated HSC (figure 1A).

Anti-LOXL2 antibody attenuates the progression of advanced pan-lobular thioacetamide (TAA)-induced fibrosis. (A) Immunohistochemistry for lysyl oxidase-like 2 (LOXL2), collagen I and α-smooth muscle actin (α-SMA) (200×). (B) Liver fibrosis was induced in C57Bl/6J mice with repeated TAA injections, and treatment with anti-LOXL2 monoclonal antibody (mAb) (AB0023, 30 mg/kg), vehicle or anti-LOX mAb (M64, 30 mg/kg) performed two times a week from week 7 to 12. (C) AB0023 inhibits bridging fibrosis compared with vehicle and M64 treatment (Sirius Red staining, 50΄). (D) Quantitative morphometry of collagen area (Sirius Red, n=5–8 for fibrotic groups, n=3 in healthy controls). Dotted bar indicates start and end of treatment. (E) Hepatic collagen content as determined biochemically via hydroxyproline (n=13–15). (F) Fibrotic matrix stability assessed ex vivo as described in M&M. AB0023 reduced the accumulation of highly crosslinked (insoluble) hepatic collagen at advanced liver fibrosis stages (n=4). Data are expressed as means±SEM. *p<0.05 (analysis of variance). CTRL, control.
To evaluate the antifibrotic efficacy of LOXL2 inhibition on advanced liver fibrosis, we administered anti-LOXL2 mAb AB0023 (30 mg/kg, intraperitoneally two times a week) concurrently with fibrosis induction, starting after 6 weeks on TAA for another 6 weeks (figure 1B). Vehicle (placebo control) and LOX-specific mAb M64 without cross-reactivity with LOXL2 were administered in parallel. Histologically, connective tissue staining revealed that liver scarring, and most notably bridging of septa, was reduced only in the AB0023-treated group (figure 1C), with a 53.4% decrease of collagen-stained area during the 6-week treatment period (figure 1D). Hepatic collagen levels (determined biochemically via hydroxyproline) confirmed a significant effect of AB0023, but not of M64, on overall collagen deposition, which was partly reduced by 21.3% (p=0.0198) when compared with the ‘start of treatment’ controls (figure 1E and see online supplementary table S3).
To assess whether LOXL2 inhibition had a direct effect on collagen crosslinking and fibrotic matrix stabilisation, we performed stepwise collagen extraction assay.5 As expected, the highly crosslinked (insoluble) collagen fraction increased significantly during fibrosis progression, representing 13.3% and 19.8% of total collagen at 6 and 12 weeks of TAA, respectively, compared with 5.7% in healthy controls. Anti-LOXL2 antibody treatment significantly inhibited the increase in crosslinked collagens, with a 45% reduction in insoluble fraction between 6 and 12 weeks of TAA and a 20.8% reduction of overall crosslinked collagens, compared with placebo (figure 1F). M64 had no effect on collagen crosslinking. Interestingly, early intervention with AB0023 (from 3 to 8 weeks of TAA) was not effective at suppressing liver fibrosis (see online supplementary figure S1 and table S4), suggesting that LOXL2 plays a more prominent role in late stages of liver fibrosis.
LOXL2 inhibition accelerates the reversal of TAA-induced hepatic fibrosis
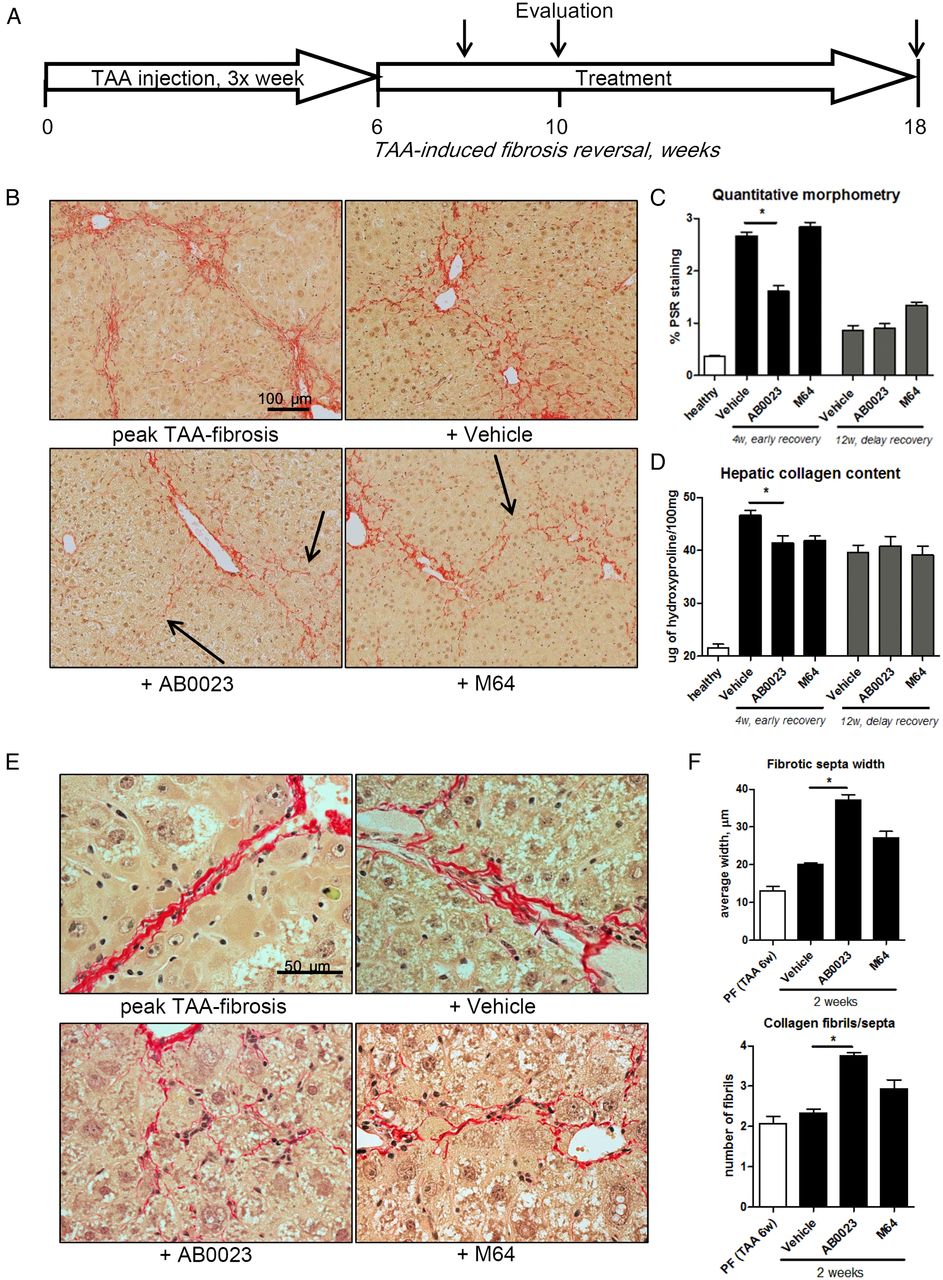
Because collagen crosslinking and fibrotic matrix stabilisation limit reversal of established fibrosis, we sought to test whether LOXL2 inhibition may promote fibrosis reversal. We induced liver fibrosis in mice with 6 weeks of TAA induction, after which TAA was discontinued and mice were allowed to recover for up to 12 weeks, with or without treatment with anti-LOXL2 mAb AB0023 or control M64 mAb. Fibrosis reversal was evaluated at early (4 weeks) and late (12 weeks) time-points of recovery (figure 2A). While the other groups at the early time-point of recovery demonstrated widened but uninterrupted septa, mice with anti-LOXL2 treatment displayed disruption of bridging and splitting of fibrotic septa (figure 2B). Quantitative morphometry showed a significant 45.7% reduction in connective tissue square at early recovery (4 weeks) in mice treated with AB0023, but not M64 (figure 2C). This was accompanied by a 20.9% and 19.1% reduction in hepatic collagen content (hydroxyproline) in AB0023-treated and M64-treated groups, respectively compared with placebo (figure 2D and see online supplementary table S5). Architectural changes in fibrotic matrix, such as widening of septa and splitting into thinner and less-organised fibrils (septal remodelling), often without a significant reduction of collagen, are a hallmark of fibrosis reversal.5 Septal remodelling signs were detected morphometrically in AB0023-treated mice: septal width increased to 183.8% and the number of thinned fibrils forming the septa increased twofold (splitting), compared with minimal changes in the placebo or M64 groups (figure 2E, F). At the late time-point of recovery (12 weeks), the differences in histological changes and hepatic collagen content between groups were no longer detectable. These findings suggest that anti-LOXL2 antibody accelerates reversal of TAA-induced hepatic fibrosis.

Lysyl oxidase-like 2 inhibition accelerates the reversal of thioacetamide (TAA)-induced hepatic fibrosis. (A) C57BL/6 mice with pre-established liver fibrosis (TAA for 6 weeks) were allowed to recover for up to 12 weeks, while treated with AB0023 (30 mg/kg), M64 (30 mg/kg) or vehicle intraperitoneally two times per week. (B) Collagen staining and morphometry (C, n=10) show that AB0023 promotes early resolution of bridging fibrosis with widening and disruption of fibrotic septa (arrows) (4 weeks of recovery, Sirius Red, 200×). (D) Hepatic collagen content (via hydroxyproline, 4 weeks of recovery, n=15–16). (E and F) Septal morphology demonstrates widening of septa and splitting of collagen bundles into thinner fibrils (E, 630×) in AB0023-treated mice. (F) Septal thickness and number of fibrils assessed in 10 randomly selected fibrotic septa (Sirius Red, 630×, 2 weeks recovery, n=4). *p<0.05 vs vehicle controls (analysis of variance).
Anti-LOXL2 therapeutic antibody suppresses the progression of biliary fibrosis in Mdr2−/− mice
Next, we tested the effect of LOXL2 inhibition on the progression of periportal, biliary-type fibrosis in our new BALBc.Mdr2−/− mouse model.11 LOXL2 expression progressively increased between 4 and 8 weeks of age, when BALBc.Mdr2−/− mice show rapid progression,11 with immunoreactivity restricted to portal fibrotic lesions, similar to collagen I and α-SMA expression (figure 3A). Next, we administered AB0023 or M64 mAb in Mdr2−/− mice starting from 4 weeks of age and evaluated liver fibrosis assessed 4 weeks later (figure 3B). Connective tissue staining revealed diminished scarring in the AB0023-treated group, with reduced onion skinning and portal–portal bridging, compared with vehicle-treated or M64-treated animals (figure 3C). Connective tissue square was reduced by 77.7% in the AB0023-treated group compared with placebo, while M64 mAb did not have a significant effect (figure 3D). Hepatic collagen (hydroxyproline) levels were diminished by AB0023, but not by M64 (30.6% of reduction in collagen deposition; figure 3E and see online supplementary table S6). In the Mdr2−/− model, the fibrotic matrix was crosslinked to a much higher degree than in the TAA-induced fibrosis model, with the insoluble collagen fraction representing over 40% of total collagen at 8 weeks of age. However, we have not been able to detect significant changes in collagen crosslinking after 4 weeks of anti-LOXL2 therapy using stepwise collagen extraction method (figure 3F).

Targeting lysyl oxidase-like 2 (LOXL2) with a therapeutic antibody suppresses the progression of biliary fibrosis in BALB/c.Mdr2−/− mice. (A) Immunohistochemistry for LOXL2, collagen I and α-smooth muscle actin (α-SMA) in BALB/c.Mdr2−/− livers (40΄). (B) BALBc.Mdr2−/− mice were administered AB0023 or M64 (30 mg/kg) intraperitoneally two times per week from 4 to 8 weeks of age. (n=9–10) (C) Diminished fibrosis in AB0023-treated mice (representative Sirius Red staining, 50΄). (D) Quantitative collagen morphometry (n=4–6). Dotted bar indicates start and end of treatment. (E) Hepatic hydroxyproline content (n=8–15). (F) Fibrotic matrix stability as assessed ex vivo via serial collagen extraction (n=4 for each bar). Data are expressed as mean±SEM. *p<0.05 (analysis of variance). CTRL, control.
The ductular reaction and HSCs activation are diminished in BALBc.Mdr2−/− mice with LOXL2 inhibition
To unveil the mechanism by which LOXL2 inhibition suppressed the progression of biliary fibrosis, apparently unrelated to fibrotic matrix crosslinking, we evaluated the extent of the ductular reaction, the driving force of biliary fibrosis, using pan-cytokeratin immunohistochemistry in BALBc.Mdr2−/− mice with treatment. LOXL2 inhibition with AB0023 mAb (but not control M64 mAb) significantly decreased the ductular reaction (p=0.0228, figure 4A), associated with diminished replication of ductular cell (Ki-67 immunohistochemistry). Conversely, there was a significant increase in Ki-67 positive hepatocytes in AB0023 group (figure 4B). Quantitative morphometry confirmed a highly significant 3.5-fold reduction in Ki-67(+) ductal cells, paralleled by a 1.9-fold increase in Ki-67(+) hepatocytes (figure 4C). This was accompanied by diminished activation of HSC/myofibroblasts, as evidenced by 57.4% reduction of α-SMA protein levels (figure 4D) and downregulation of several profibrogenic messenger RNAs (mRNAs) (figure 4E).
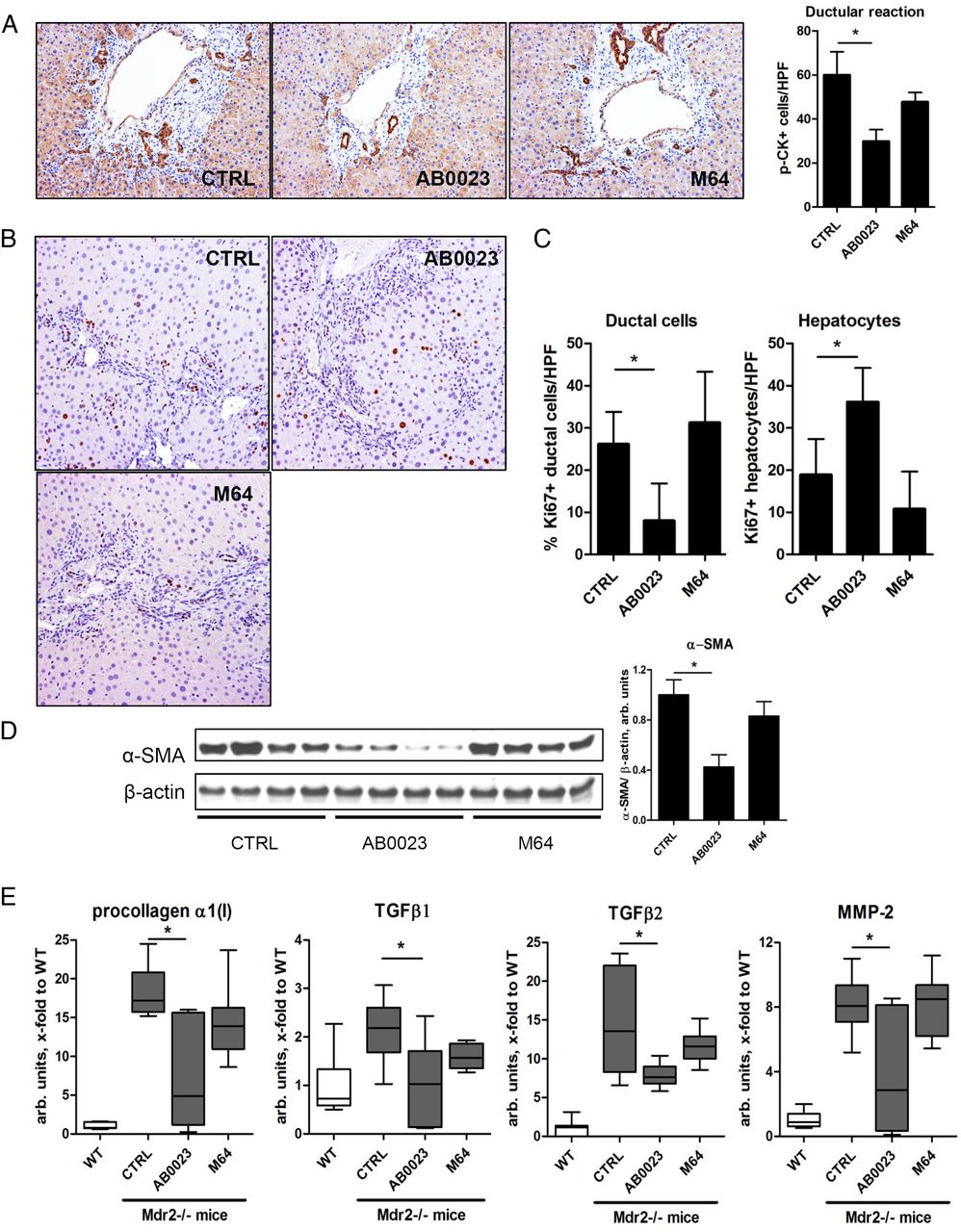
Profibrogenic ductular reaction is attenuated upon lysyl oxidase-like 2 (LOXL2) inhibition in BALBc.Mdr2−/− mice. (A) Representative pan-cytokeratin (p-CK) immunohistochemistry and p-CK+ cell numbers (>7 random portal fields, n=4, 200×). (B) Cell proliferation (Ki-67 immunohistochemistry). Note the increased proliferation in ductular structures in untreated controls, whereas labelled hepatocytes predominate in AB0023-treated Mdr2−/− mice. (C) Quantification of Ki-67+ hepatocytes and ductal cells (>7 HPF, n=4, 200΄). (D) α-smooth muscle actin (α-SMA) expression in liver lysates (representative western blot and densitometry, with β-actin as a loading control, n=8–9). (E) Hepatic messenger RNA expression of procollagen α1(I), transforming growth factor β (TGFβ)1, TGFβ2 and matrix metalloproteinase 2 (MMP2) (n=9–15). *p<0.05 (analysis of variance). CTRL, control; HPF, high-power field; WT, wild type.
LOXL2 regulates differentiation of hepatic progenitor cells in vitro
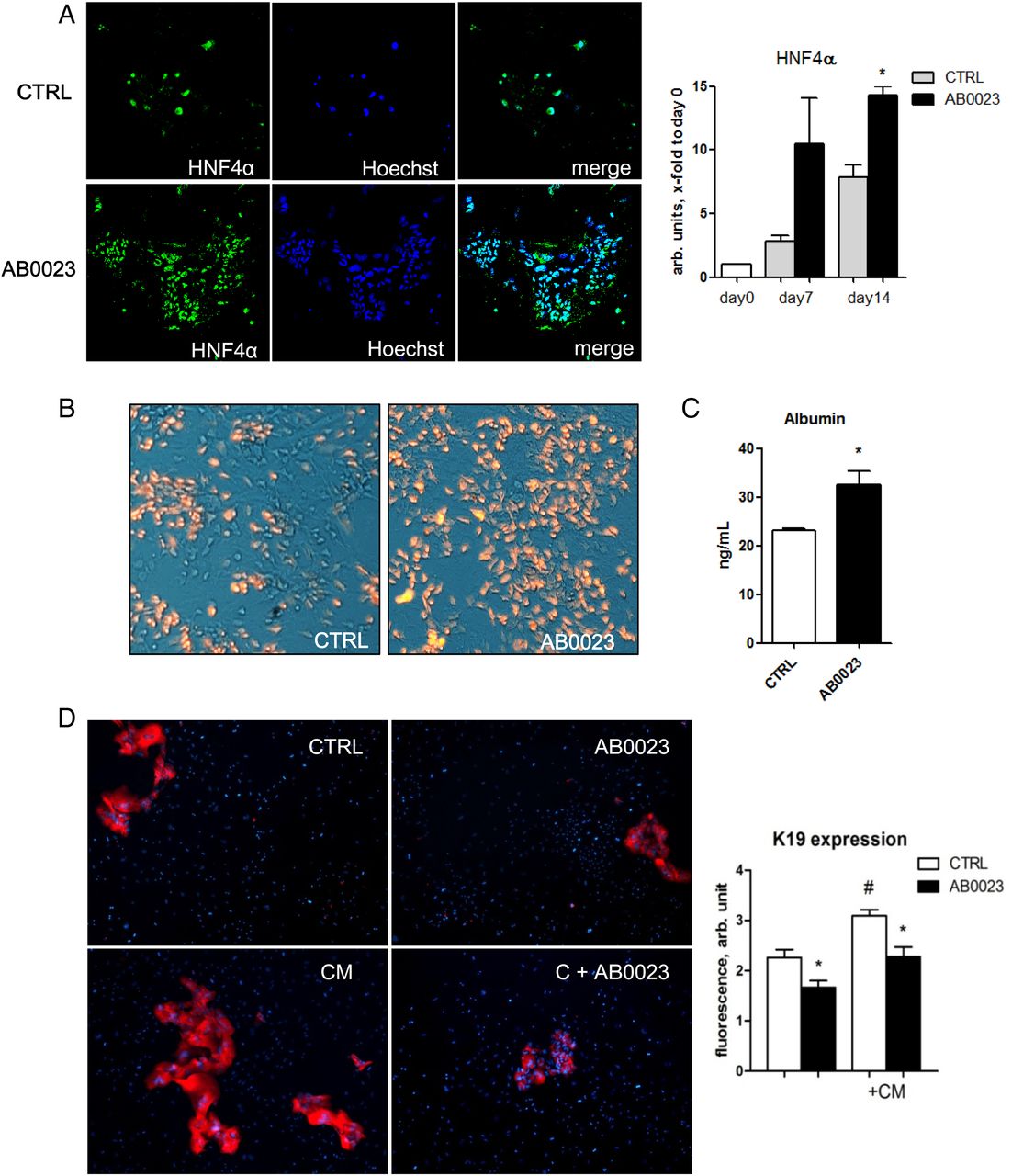
Our in vivo findings on a switch in cell proliferation from ductal cells to hepatocytes upon LOXL2 inhibition suggested a potential role in regulating lineage commitment and differentiation of bipotent hepatic progenitor cells (HPC). Double immunofluorescence for LOXL2 and the HPC marker EpCAM in Mdr2−/− livers revealed a strong LOXL2 signal immediately adjacent to EpCAM(+) cell clusters (figure 5A). However, since LOXL2 is a secreted protein, all immunopositivity signals were found extracellularly and could not be unequivocally assigned to specific cell type. To circumvent this issue, we performed in situ hybridisation for LOXL2 mRNA, which revealed LOXL2 expression by pseudoducts (HPC/reactive cholangiocytes) and spindle-shaped myofibroblast-like cells in fibrotic Mdr2-/- liver (figure 5B). To further test this hypothesis, we isolated primary HPC from 4–5 weeks old Mdr2−/− mice using EpCAM antibody-coated magnetic beads20 and studied their differentiation in vitro. As expected,20 when cultured in appropriate differentiation medium, EpCAM(+) HPC formed cell colonies from day 5 with morphological features of ductal cells and hepatocytes, and upregulation of both cholangiocyte (HNF1β, K19) and hepatocyte (HNF4α, albumin) differentiation markers between day 7 and 14 (not shown). LOXL2 mRNA was enriched 17-fold in freshly isolated EpCAM(+) cell fraction compared with total liver RNA, and was further upregulated during culture (figure 5C, D). When cultured in the presence of LOXL2-neutralising mAb, HPC-derived cell colonies demonstrated a marked expansion of HNF4α-positive cells and upregulated HNF4α mRNA expression between days 7 and 14 (figure 6A). Low-density lipoprotein (LDL) uptake and albumin secretion in HPC-derived cultures increased with LOXL2 neutralisation, suggesting that autocrine/paracrine LOXL2 suppresses differentiation of HPC into functional hepatocytes (figure 6B, C). Interestingly, while LOXL2 neutralisation increased the hepatocyte replication rate in diseased Mdr2−/− mice (figure 4C), DNA synthesis in mature and differentiated (non-HPC derived) hepatocytes freshly isolated from healthy mouse liver was not affected by LOXL2 inhibition, as assessed by BrdU incorporation (not shown), suggesting that LOXL2-dependent effects might be limited to HPC-derived hepatocyte lineages. On the other hand, proliferation of K19(+) ductal cells of cholangiocyte lineage within HPC-derived colonies was inhibited by anti-LOXL2 antibody (figure 6D). CM from HSC cultures further promoted growth of K19(+) cholangiocyte from EpCAM(+) HPC cells, and was likewise inhibited by AB0023 (figure 6D). Taken together, these data strongly suggest that autocrine/paracrine LOXL2 controls lineage commitment of HPC in vitro, inhibiting differentiation towards a hepatocyte lineage while favouring differentiation towards fibrogenic cholangiocytes.

Lysyl oxidase-like 2 (LOXL2) is expressed by hepatic progenitor cells (HPCs) in fibrotic livers. (A) Double immunofluorescence for LOXL2 (green) and EpCAM (red) in Mdr2−/− mice reveals prominent LOXL2 expression adjacent to EpCAM(+) cells (arrows). PV, portal vein. (B) In situ hybridisation reveals LOXL2 messenger RNA (mRNA)-specific signal (magenta) within HPC/ductal cells forming pseudoduct (closed arrows). Note spindle-shaped myofibroblast-like cells in periductular scar, also positive for LOXL2 transcripts (open arrows). Representative images from four Mdr2−/− mice shown (original magnification, ×40). (C) LOXL2 and LOX mRNA expression in freshly isolated EpCAM(+) cells compared with whole liver RNA from Mdr2−/− mice, and during prolonged culture in vitro (D). Data are means±SEM. *p<0.05 (Student's t-test).

Function-blocking experiments reveal that lysyl oxidase-like 2 (LOXL2) regulates lineage commitment of EpCAM+ hepatic progenitor cell (HPC) in vitro. EpCAM(+) progenitor cells were grown in differentiation medium for up to 14 days in the presence of AB0023 (30 μg/mL, added 48 hours after plating) or isotype control IgG (CTRL, 30 μg/mL). (A–C) In vitro LOXL2 neutralisation promotes HPC differentiation into functional hepatocytes. (A) Accumulation of HNF4α-expressing cells in EpCAM(+)-derived colonies (left, green HNF4α, blue nuclei) and HNF4α messenger RNA levels (right) in cell lysates. (B) Low-density lipoprotein (LDL) uptake analysed using Dil-AcLDL (red fluorescence overlayed on phase-contrast image). (C) Albumin levels in the EpCAM(+) cell supernatant after 12 days of culture with AB0023 or control IgG. (D) Proliferation of K19(+) ductular-like cell clusters (red) from EpCAM(+) progenitors is promoted by hepatic stellate cell-conditioned medium (CM), and inhibited by anti-LOXL2 AB0023 antibody. Quantification of K19 expression in EpCAM(+) HPC-derived colonies (ImageJ, n=3). Data are means±SEM. *p<0.05 (Student's t-test).
Contractile activity of HSC is regulated by LOXL2
Next, we investigated whether (HPC/cholangiocyte-derived) LOXL2 contributes to a profibrogenic epithelial–mesenchymal crosstalk between cholangiocytes and HSC. A collagen gel contraction assay was performed with rat HSC-X and primary murine HSC in the presence of anti-LOXL2 mAb. Addition of CM from the mouse cholangiocyte (immature ductular) cell line 603B15 markedly accelerated collagen gel contraction by HSC (figure 7A, B). Anti-LOXL2 antibody AB0023, but not M64 mAb, completely abrogated the stimulatory effect of 603B medium on collagen gel contraction by HSC-X and primary mouse HSC (figure 7C). In the absence of collagen, when HSC were simply cultured on plastic, LOXL2 neutralisation did not have an effect on their fibrogenic activity (as assessed by mRNA of COL1A1, etc., cell proliferation, not shown). These results suggest that ductal cell-derived LOXL2 promotes fibrogenic (contractile) activity of HSC, likely in an indirect (collagen crosslinking-dependent) manner.

Collagen gel contraction by hepatic stellate cells is inhibited by lysyl oxidase-like 2 (LOXL2) blocking antibody. (A–C) Collagen gel contraction assay using the activated rat hepatic stellate cell (HSC)-X cell line (A, representative gel images after 4 days) and primary murine HSC in the presence of AB0023, isotype control IgG or M64 (all 10 μg/mL). The per cent reduction in collagen gel surface area was analysed at days 4 in HSC-X cells (B) or 15 in primary HSC (C) and quantified using ImageJ software in triplicates. *p<0.05 (Student's t-test); CM, conditioned medium. (D–F) Anti-LOXL2 antibody achieves antifibrotic efficacy in a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)-feeding model. (D) Low-magnification representative images of collagen staining (50΄, upper row) show decrease in periportal and bridging fibrosis in AB0023-treated mice. Lower panel shows high magnification of selected portal areas (200×, asterisk marks portal vein). (E) Hepatic collagen deposition (via hydroxyproline content, n=9–10 in fibrotic groups, n=5 in healthy controls). (F) The ductular reaction is attenuated in mice with LOXL2 inhibition (K19 immunohistochemistry, left) with (G) quantification of K19+ cells (>7 random portal fields/liver, n=9–10, 200×). Data expressed as means±SEM. *p<0.05 (analysis of variance). CTRL, control.
LOXL2 inhibition suppresses the ductular reaction and fibrosis progression induced by DDC feeding
To further validate the antifibrotic efficacy of AB0023 mAb, we employed the murine DDC-feeding model as a second, mechanistically different model of biliary fibrosis. Immunohistochemistry revealed that LOXL2 is also overexpressed in DDC model in the pattern similar to that in TAA or Mdr2−/− models. Notably, LOXL2 signal around proliferating bile ducts and (pseudo)-ducts was again observed (see online supplementary figure S2), confirming our observations in Mdr2−/− livers (figure 5A). Vehicles as placebo, AB0023 or M64 mAb were administered for 4 weeks intraperitoneally concurrently with DDC feeding. Histologically, periportal and bridging fibrosis was significantly attenuated only in AB0023-treated mice (figure 7D), accompanied by a 38.6% reduction in biochemical collagen deposition compared with the placebo group (p=0.0453, figure 7E and see online supplementary table S7). Similarly, the ductular reaction, as assessed by K19 staining and quantification of K19+ cells was significantly reduced by the anti-LOXL2 AB0023 mAb, but not by M64 mAb (p=0.0092, figure 7F, G). Interestingly, administration of LOX-specific control antibody M64 also seem to have an impact on average collagen content (figure 7E), although this did not reach statistical significance when compared with the placebo group.
Discussion
The process of fibrotic matrix stabilisation through collagen crosslinking contributes to scar tissue accumulation and limits reversibility of fibrosis.4 Early descriptive studies have implicated TG2 in collagen crosslinking. However, collagen crosslinking, liver fibrosis or its reversibility are not altered in TG2−/− mice.5 Instead, the process of collagen crosslinking and stabilisation depends largely on LOX enzymatic activity.6 Here, we identify LOXL2, a member of the LOX family induced during liver fibrosis, as a key contributor to fibrotic matrix stabilisation. Direct analysis of the liver extracellular matrix (ECM) via collagen fractionation revealed that selective LOXL2 inhibition reduced the deposition of highly crosslinked collagen in progressive TAA-induced liver fibrosis (figure 1F) and accelerated fibrosis resolution after cessation of liver injury (figure 2F). This is the first direct evidence that shows that LOXL2 directly and significantly contributes to hepatic collagen stabilisation and retards fibrosis reversal. Moreover, the extent to which LOXL2 neutralisation inhibits stable, crosslinked collagen deposition in TAA-induced fibrosis (by approximately 45%) is comparable with unselective LOX inhibition with BAPN, as we reported in a similar CCl4-induced fibrosis model (40%–74% stable collagen reduction6). Although we did not compare anti-LOXL2 antibody in a side-by-side experiment with non-selective inhibitor BAPN, these results may suggest that LOXL2 is a major contributor to hepatic collagen crosslinking/stabilisation within the LOX family. Interestingly, 6 weeks of delayed anti-LOXL2 antibody therapy during progression (weeks 7–12 of TAA) partly inhibited further progression of the already advanced liver fibrosis (figure 1D), whereas the same treatment was ineffective when administered in earlier stages of progressive fibrosis (during weeks 3–8 of TAA; see online supplementary figure S1). These results are in agreement with our recent data showing that overall LOX-mediated collagen crosslinking is more prominent and pathophysiologically significant in late fibrosis stages. This is clinically relevant, since most patients in need of antifibrotic therapy present with already advanced fibrosis. Moreover, anti-LOXL2 antibody promoted fibrosis reversal, resulting in favourable septal remodelling and collagen removal (figure 2), when treatment started during recovery. Interestingly, control M64 antibody with specificity to LOX appeared to reduce hepatic collagen levels, as well as septal remodelling, although to a lesser degree than anti-LOXL2 mAb (figure 2). This suggests that LOXL2-mediated, and to some degree LOX-mediated, collagen crosslinking occurs even after cessation of chronic liver injury, thus retarding collagen degradation and fibrosis reversal. This is important even in the era of the novel, highly efficient direct-acting antivirals against HCV,22 since patients with advanced liver fibrosis23 and cirrhosis24 remain at a high risk for HCC development after achieving SVR.
Recent studies suggest that ECM stiffness due to collagen crosslinking in fibrosis is necessary and sufficient to trigger and maintain the activation of HSC and portal fibroblasts, as demonstrated in vitro by culturing cells on artificial polyacrylamide supports of increasing rigidity.25 Moreover, an increase in liver stiffness precedes HSC activation and at least partly depends on LOX activity, since chemical LOX inhibition in vivo attenuates increases in liver stiffness in experimental fibrosis.26 This represents a potential mechanism of perpetuation of fibrotic responses and HSC activation by progressive collagen crosslinking, and is consistent with our findings that both general LOX inhibition6 and selective anti-LOXL2 treatment are more effective at advanced compared with early stages of fibrogenesis (figure 1, and see online supplementary figure S1).
Progression of biliary fibrosis is associated with a pronounced ‘ductular reaction’27 characterised by proliferation of adult bipotent hepatic progenitors and their progeny (reactive cholangiocytes and intermediate hepatocytes). Reactive cholangiocytes are important targets for antifibrotic therapies, since they drive HSC/myofibroblast activation through secretion of multiple proinflammatory and profibrogenic factors.3 ,28 Interestingly, LOXL2 neutralisation potently suppressed the ductular reaction in our two biliary fibrosis models (figures 4 and 7), both of which are fuelled by the proliferation of HPC/reactive cholangiocytes. Notably, suppression of the ductular proliferation by LOXL2 inhibition relieved the proliferative block on hepatocyte proliferation (figure 4C), consistent with redirection of (bipotent) HPC differentiation from a (fibrogenic) biliary towards a (regenerative) hepatocyte lineage. This was accompanied by decreased HSC activation (figure 4D) and downregulation of profibrogenic genes including Col1A1 and transforming growth factor β1 (figure 4E), suggesting that the profibrogenic ductular reaction is driven by LOXL2 activity in vivo. To address the presumed direct role of LOXL2 in regulating HPC biology, we isolated EpCAM(+) HPCs and studied their differentiation in vitro in the presence of LOXL2-blocking antibody. LOXL2 neutralisation leads to the expansion of cells expressing the hepatocyte-specific transcription factor HNF4α and promoted their differentiation into functional hepatocytes, as evidenced by increased albumin secretion and LDL uptake (figure 6A–C). At the same time, basal and HSC CM stimulated the emergence of ductal K19+ cells from HPC which can be suppressed by the LOXL2-blocking mAb (figure 6D), implicating HSC-derived LOXL2 as an additional paracrine factor for HPC. Taken together, delayed LOXL2 targeting effectively suppressed advanced, pre-established biliary fibrosis in the Mdr2−/− model (figures 3 and 4), apparently independent of its crosslinking function (figure 3F), that is, via direct regulation of HPC fate and inhibition of the profibrogenic ductular reaction. Importantly, while we could not directly assess delayed treatment modalities also in the DDC model due to its short duration, anti-LOXL2 mAb administered in parallel with fibrosis progression clearly diminished both fibrosis and the ductular reaction (figure 7), thus supporting our findings in the central Mdr2−/− model by using a second, mechanistically different biliary fibrosis model. Although we could not detect statistically significant differences, in DDC model, anti-LOX antibody M64 administration seem to reduce mean values of hepatic collagen levels compared with placebo. In another, more robust Mdr2−/− model11 of biliary fibrosis, M64-treated group also demonstrated a modest trend to lower means of connective tissue squares (figure 3D) and ductular reaction (figure 4A) compared with placebo, although neither change in DDC nor Mdr2−/− was statistically significant. However, these observations suggest that the potential role of LOX in fibrotic response, as implicated in prior reports,6 ,26 cannot be completely ruled out, and may merit specific investigation in follow-up studies.
Recently, novel and distinct roles of LOXL2 beyond ECM crosslinking have been reported in the context of epithelial cell biology. Intracellular LOXL2 acts as a transcription factor in cancer cells and downregulates cadherin-1 expression by deaminating lysine 4 within histone H3, altering epigenetic controls and inducing epithelial–mesenchymal transition.29 ,30 Moreover, both endogenous and exogenous LOXL2 inhibited differentiation of keratinocytes through its fourth ‘scavenger receptor cysteine-rich domain’ and independently of the crosslinking catalytic domain via yet unidentified receptor.31 In our study, and consistent with the previously reported preponderance of HPC to differentiate to cholangiocytes in vitro,32 isolated EpCAM(+) cells readily differentiated into K19+ ductal cells, which was further promoted by CM from HSC. Both basal and CM-stimulated ductal expansions were inhibited by LOXL2 mAb addition (figure 6D). Since cells do not internalise the AB0023 antibody from the extracellular space, this suggests that secreted, extracellular LOXL2 controls HPC fate and the ductular reaction in an autocrine and paracrine manner. While mesenchymal cells (HSC and portal fibroblasts33) were described as the significant source of LOX and LOXL2 in the liver, we found that HPC/ductal cells express LOXL2 as well. Thus, purified EpCAM(+) cells contain significant levels of LOXL2 mRNA, upregulate LOXL2 when cultured in vitro (figure 5C, D), and LOXL2 protein can be readily detected in immediate proximity to EpCAM+ HPC clusters (figure 5A). Moreover, in situ hybridisation provided the direct evidence of a LOXL2 mRNA within both ductular cells (reactive cholangiocytes/HPC) and myofibroblasts cellular source (figure 5B). These data together are strongly suggestive of HPCs lineage being a plausible source of biologically active LOXL2 in the fibrotic liver. Thus, antibody neutralisation of LOXL2 in purified primary EpCAM(+) HPC cultures profoundly affected their lineage commitment in vitro (figure 6). However, LOXL2-dependent effects were also observed with the addition of HSC-CM to HPC (figure 6D), and conversely, by addition of immature ductal cell 603B-CM on HSC (figure 7A–C), strongly suggesting a paracrine LOXL2-mediated HPC–HSC crosstalk. The identification of a putative LOXL2 receptor on the surface of epithelial cells31 and the precise molecular mechanism by which LOXL2 regulates HPC lineage commitment are important questions that clearly need to be addressed in future studies.
Taken together, our data in biliary and non-biliary fibrosis models indicate at least two distinct and independent mechanisms by which LOXL2 promotes liver fibrosis: via direct enzymatic collagen crosslinking/stabilisation and via promoting the differentiation of HPC towards a profibrogenic ductular/cholangiocyte lineage. In pan-lobular TAA-induced fibrosis, the effect of LOXL2 inhibition on collagen crosslinking and matrix stabilisation was prominent, and we were able to readily detect a significant effect of AB0023 on matrix stabilisation (figure 1F). However, in biliary fibrosis model, Mdr2−/− (figure 3F), collagen stability does not appear to be altered by LOXL2 inhibition; instead, the direct action on fibrogenic HPC differentiation is prominent (figure 4). It is possible that we simply failed to detect the effect on collagen crosslinking due to considerably shorter duration of treatment (4 vs 6 weeks in the TAA model) and the much higher degree of crosslinking in Mdr2−/− (40% of insoluble collagen compared with 20% in TAA-induced fibrosis). However, it is apparent that in biliary fibrosis, direct effect of LOXL2 inhibition on HPC/ductular reaction and fibrosis at least precedes (and thus unlikely to result from) the effect on collagen crosslinking. HPC activation/ductular reaction is most prominent in biliary disease and also occurs in advanced stage pan-lobular human liver fibrosis, correlating with the progression to cirrhosis, including non-biliary liver diseases such as chronic hepatitis C34 and non-alcoholic steatohepatitis.35 The important role of reactive cholangiocytes/HPC in driving fibrosis progression has been demonstrated in rodent models of biliary fibrosis,18 ,36 and in settings of advanced pan-lobular fibrosis, for example, during fibrotic liver regeneration after partial hepatectomy.37
In summary, we report that LOXL2 is overexpressed in liver fibrosis and promotes collagen matrix stabilisation, fibrosis progression and irreversibility, as well as directs unfavourable HPC differentiation towards fibrogenic cholangiocytes. Blocking of LOXL2 activity in the extracellular space (1) inhibits collagen crosslinking, fibrosis progression and accelerates its reversal; (2) redirects HPC differentiation from fibrogenic cholangiocytes (ductular reaction) in favour of hepatocytes, thereby promoting pro-resolution and regeneration pathways (figure 8). LOXL2 is a promising therapeutic target to treat biliary and non-biliary fibrosis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme of the proposed role of lysyl oxidase-like 2 (LOXL2) in promoting hepatic fibrogenesis. (1) LOXL2-mediated crosslinking stabilises collagen into a more rigid and degradation-resistant ECM. The stiff crosslinked matrix microenvironment further promotes hepatic stellate cell activation via mechanosensing. (2) Progenitor cell responses are controlled by autocrine/paracrine LOXL2, which promotes progenitor commitment towards a (profibrogenic) cholangiocyte lineage, while suppressing differentiation into (proregeneration) hepatocytes. (3) Blocking of LOXL2 activity in the extracellular space with AB0023 monoclonal antibody (mAb) (a) attenuates collagen crosslinking and (b) redirects hepatic progenitor cell (HPC) differentiation from profibrogenic cholangiocytes (ductular reaction) to hepatocytes, promoting fibrosis resolution and liver regeneration. HCC, hepatocellular carcinoma; TGF, transforming growth factor.
References
Footnotes
Contributors NI performed most experiments, analysed data and wrote the manuscript. Z-WP and SY performed oval cell characterisation and some animal experiments. SBL and DYS performed mouse breeding and genotyping, assisted with animal experiments and assays. AMV performed immunohistochemistry and connective tissue square morphometry. DS and VS provided experimental tools and critically revised the manuscript. YVP designed the experiments, analysed data, supervised the study and wrote the manuscript.
Funding This study was supported in part by a research grant from Gilead Sciences, and an institutional grant from Department of Medicine (BIDMC) to YVP. NI was supported by a fellowship award from the Uehara Memorial Foundation and Z-WP was a recipient of career development awards from The First Affiliated Hospital of Sun Yat-sen University (Guangzhou, China), National Natural Science Foundation of China (81301842) and Pearl River S&T Nova Program (2014J2200087).
Competing interests AM-V and VS are employees of Gilead Sciences which develops anti-LOXL2 antibody and holds related patents. YVP received research funding from Gilead Sciences.
Provenance and peer review Not commissioned; externally peer reviewed.