Article Text

Abstract
Background: The prevalence of portopulmonary hypertension (PPHTN) in patients with cirrhosis and refractory ascites is unknown. Its presence may preclude patients from receiving a transjugular intrahepatic portosystemic shunt or liver transplantation as a definitive treatment for their end stage cirrhosis.
Purpose: To determine the prevalence, possible aetiological factors, and predictive factors for the development of PPHTN in these patients.
Methods: Sixty two patients (53 males, nine females; mean age 54.5 (1.4) years) with biopsy proven cirrhosis and refractory ascites underwent angiographic measurements of pulmonary and splanchnic haemodynamics. Endothelin 1 levels were measured from the pulmonary artery. Forty nine patients underwent radionuclide angiography for measurements of central blood volume, pulmonary vascular, and cardiac chamber volumes. Forty seven patients also underwent two dimensional echocardiography for measurements of cardiac structural and functional parameters. Cardiac output, and systemic and pulmonary vascular resistance were calculated.
Results: Ten patients (16.1%) fulfilled the criteria for PPHTN (mean pulmonary artery pressure ≥25 mm Hg and pulmonary vascular resistance ≥120 dyn×s/cm5), with significantly higher mean right atrial (15.4 (1.2) v 7.9 (0.5) mm Hg; p<0.001), and right ventricular pressures (24.7 (1.5) v 14.7 (0.6) mm Hg; p<0.001), and endothelin 1 levels (3.04 (0.40) v 1.98 (0.12) pg/ml; p=0.02). No significant differences in any of the other parameters measured were detected between the two groups. A right atrial pressure of ≥14 mm Hg had a 83% positive predictive value for the presence of PPHTN.
Conclusions: Portopulmonary hypertension is common in cirrhosis with refractory ascites, possibly due to excess endothelin 1 in the pulmonary circulation. An elevated right atrial pressure ≥14 mm Hg predicts the presence of PPHTN, which may be helpful in deciding management options in these patients.
- cirrhosis
- refractory ascites
- porto-pulmonary hypertension
- endothelin 1
- atrial natriuretic peptide
- PPHTN, portopulmonary hypertension
- TIPS, transjugular intrahepatic portosystemic shunt
- ET-1, endothelin 1
- RNA, radionuclide angiography
- PVR, pulmonary vascular resistance
- CO, cardiac output
- SV, stroke volume
- mRAP, mean right atrial pressure
Statistics from Altmetric.com
- PPHTN, portopulmonary hypertension
- TIPS, transjugular intrahepatic portosystemic shunt
- ET-1, endothelin 1
- RNA, radionuclide angiography
- PVR, pulmonary vascular resistance
- CO, cardiac output
- SV, stroke volume
- mRAP, mean right atrial pressure
Portopulmonary hypertension (PPHTN) is an uncommon complication of cirrhosis, affecting only 0.25–4% of these patients.1–3 PPHTN is defined as an increased mean pulmonary artery pressure of >25 mm Hg, increased pulmonary vascular resistance of >120 dyn×s/cm5, and pulmonary capillary wedged pressure of <15 mm Hg in the presence of portal hypertension.4 The pathogenesis is complex and involves interplay between haemodynamic and local factors. The mechanisms proposed for the development of PPHTN include excess local production of vasoconstrictor substances,5–7 increased pulmonary blood flow leading to endothelial damage and vascular remodelling,8 excess pulmonary vascular volume,9 and in situ microthrombosis.10,11 Portal hypertension and cirrhosis are thought to contribute to the pathogenesis of pulmonary hypertension by shunting hypertension inducing substances, not metabolised by the liver, into the pulmonary circulation. However, studies to date have not found a correlation between the extent of PPHTN and the height of portal hypertension, or the severity of liver disease, as assessed by the Child-Pugh score, or the degree of shunting.3,12 Furthermore, the recent recognition of cirrhotic cardiomyopathy with myocardial thickening and diastolic dysfunction as a complication of decompensated cirrhosis13 has added another potential aetiological factor in the development of PPHTN. Therefore, the pathogenesis of PPHTN remains incompletely understood.
Of all the vasoconstrictive factors implicated in the pathogenesis of pulmonary hypertension, endothelin 1 (ET-1) is the most potent vasoconstrictor.14,15 Indeed, systemic ET-1 levels have been shown to be increased in patients with primary pulmonary hypertension.16 Previous studies have also demonstrated higher systemic ET-1 levels in cirrhotic patients compared with controls.17,18 Furthermore, systemic ET-1 levels are even higher in cirrhotic patients with ascites compared with those without ascites.18 If ET-1 was implicated in the pathogenesis of PPHTN in cirrhosis, one could speculate that the prevalence of PPHTN should be higher in ascitic cirrhotic patients.
Therefore, the aims of this study were: (i) to determine the prevalence of PPHTN in cirrhotic patients with refractory ascites, (ii) to assess the roles of various aetiological factors including pulmonary vascular volume, cardiac dysfunction, and vasoconstrictive factor such as ET-1 in the pathogenesis of PPHTN; and (iii) to determine if there were predictive factors for its development in these patients.
PATIENTS AND METHODS
Subjects
From March 1993 to June 2001, 62 patients (53 men, nine women), with a mean age of 54.5 (SEM 1.4) years, with biopsy proven cirrhosis and refractory ascites, underwent haemodynamic measurements prior to transjugular intrahepatic portosystemic shunt (TIPS) insertion at the Toronto General Hospital. Refractory ascites was defined as unresponsiveness to 400 mg of spironolactone plus 160 mg of furosemide daily for two weeks, without significant side effects, while on a sodium restricted diet of 44 mmol/day.19 All patients were infection free with a normal chest x ray, negative urine culture, normal white blood cell count, and absence of spontaneous bacterial peritonitis, as indicated by a negative ascitic fluid cell count and negative ascitic fluid culture. All diuretics were discontinued and no paracentesis was performed for at least two weeks before undergoing TIPS insertion. As part of the pre-TIPS assessment, all patients underwent a pulmonary function test and none of the patients had evidence of chronic lung disease. Additional evidence for the absence of cardiac disease was indicated by a normal electrocardiograph, and confirmed clinically by a cardiologist. Evidence of portosystemic shunting was recorded either by gastroscopy (oesophageal/gastric varices) or by abdominal ultrasound (intra-abdominal varices). The presence of portal hypertension was confirmed at the time of TIPS insertion. The indication for TIPS insertion was refractory ascites in 59 (96.1%) patients, hepatorenal syndrome type II in two, and recurrent variceal bleeds in one patient complicating refractory ascites. The patient with variceal haemorrhage was haemodynamically stable for two days after the bleeding episode prior to TIPS insertion. Patient demographics are presented in table 1.
Individual mean pulmonary artery and right atrial pressure measurements, and individual calculated pulmonary vascular resistance in the two study groups
Baseline demographics of the two study groups
Study protocol
Patient hospital records were studied retrospectively and the following parameters recorded: (i) liver profile and serum creatinine, and the Child-Pugh score calculated20; (ii) pressure measurements obtained during angiography just prior to TIPS insertion (right atrium, right ventricle, pulmonary artery, pulmonary capillary wedged pressures, portal vein, free hepatic vein, wedged hepatic vein, and inferior vena cava pressures); (iii) thoracic volume measurements and systolic function, as assessed by radionuclide angiography (RNA)21 (total central blood volume, right and left pulmonary vascular volumes, left ventricular end diastolic and end systolic volumes, and left ventricular ejection fraction); and (iv) cardiac structural and functional parameters as measured by two dimensional echocardiography. Cardiac structure was assessed by measuring left ventricular systolic and diastolic chamber dimensions, septal and posterior wall relative thickness (the thickness of the septum and posterior wall relative to the intraventricular diameter), and left ventricular mass.22 Diastolic function was assessed by measuring the E/A ratio (E velocity=early maximal ventricular filling velocity, A velocity=late diastolic or atrial velocity, which is an assessment of the atrial contribution of the ventricular volume), isovolaemic relaxation time, and deceleration time. An increase in isovolaemic relaxation time and deceleration time, and a decreased E/A ratio suggest impedance to left ventricular filling, or increased ventricular stiffness.23 All patients had RNA and two dimensional echocardiography within one week of their TIPS insertion. Blood samples that were collected at the time of angiography from the pulmonary artery were available for analysis from 19 patients for ET-1. These were collected in chilled tubes containing ethylene diaminetetraacetic acid and aprotinin, then separated by refrigerated centrifugation, and stored at −70°C.
Procedures
Pulmonary and splanchnic angiography:
Cardiac, pulmonary, and splanchnic pressure measurements were made during angiography prior to TIPS insertion. All pressure measurements were performed through a catheter via a Hewlett-Packard (Andover, Massachusetts, USA) transducer and monitor. To measure pulmonary and right sided cardiac pressures, a 7 French end hole Swan-Ganz catheter was advanced through a sheath in the right internal jugular vein under fluoroscopic control. The catheter balloon was inflated in the right atrium. Subsequently, the catheter was advanced through the right ventricle and pulmonary artery to a wedged position in the right pulmonary artery. The balloon was then deflated and the catheter withdrawn to the main pulmonary artery. Pressure measurements were taken in the right atrium and ventricle, pulmonary artery, and wedged pulmonary capillary positions. To measure hepatic and indirect portal venous pressures, an end hole or an occlusion balloon angiographic catheter was used. Either the end hole catheter was advanced to the “wedged” position in any peripheral hepatic vein and the pressure measured or, alternatively, the balloon of an occlusion balloon catheter was inflated centrally in a hepatic vein to create a “wedge” and the pressure measured. In either situation, the pressure measured was equivalent to the direct portal vein pressure, providing that there was no haemodynamically significant obstruction between the catheter tip and the intrahepatic portal veins. The end hole catheter was then withdrawn to a central hepatic vein or the balloon deflated, and the “free” hepatic vein pressure was measured. The difference between the “wedged” hepatic pressure and the “free” hepatic pressure gave the corrected sinusoidal pressure, a reflection of portal pressure. Portal pressure was measured directly during the TIPS procedure by direct puncture of the intrahepatic portion of the portal vein.
Radionuclide angiography
Radionuclide angiography was performed with the patient supine. A description of the technique of cardiac chamber, pulmonary vascular, and total thoracic vascular volume measurements was detailed previously.21 Briefly, red blood cells were labelled using Tc-99m pertechnetate. After cardiac volume imaging was completed, a large field of view image was taken of the entire thorax, including both lung fields, central cardiac, and vascular structures for five minutes onto a 256×256 matrix. Cardiac volumes were measured based on regional activity corrected for attenuation. Ejection fraction and cardiac volumes were analysed using semi automated software. Quality assurance studies in our Nuclear Cardiology Laboratory have established the standard error of the estimate of the left ventricular ejection fraction calculation to be less than 2% using semi automated techniques. The standard error of the estimated ventricular volume calculation is less than 5 ml.24
Two dimensional echocardiography
A complete transthoracic echocardiograph examination was performed using commercially available cardiac ultrasound machines (Hewlett Packard). Patients were placed on the left lateral decubitus position and standard parasternal, apical, and substernal views were obtained. Pulse and colour Dopplers were used to interrogate mitral venous inflows, and isovolaemic relaxation and deceleration times. All images were then recorded onto magnetic optical discs and VHS tapes for offline analysis.
Analytical assays
ET-1
Plasma concentrations of ET-1 were measured using an enzyme linked immunoassay (R&S Systems, Minneapolis, Minnesota, USA). The assay consists of a two step procedure: firstly, ET-1 is extracted from plasma and purified using reverse phase solid extraction. Following extraction, samples are assayed by a sandwich immunoassay technique which involves the simultaneous reaction of ET-1 present in the sample or standard with two antibodies directed at two different epitopes of the ET-1 molecule. One antibody is coated onto the surface of the wells of a microtitre plate and the other is conjugated to the enzyme horseradish peroxidase. Any ET-1 present forms a bridge between the two antibodies. After removal of unbound material by aspiration and washing, the amount of conjugate bound to the wells is detected by reaction with a substance specific for the enzyme which yields a coloured product proportional to the amount of conjugate and thus ET-1 in the sample. The coloured product is quantified photometrically at 450 nm. Values of unknowns are calculated directly from the standard curve of known concentrations.
Calculations
Pulmonary vascular resistance (PVR)
PVR (dyn×s/cm5)=(mean pulmonary artery pressure (mm Hg)−pulmonary capillary wedged pressure (mm Hg)) × 80/cardiac output (CO) (l/min). Patients who had both an increased pulmonary artery pressure of more than 25 mm Hg, a PVR of more than 120 dyn×s/cm5, and a pulmonary capillary wedged pressure of less than 15 mm Hg were considered as having pulmonary hypertension.
Systemic haemodynamics and vascular volumes
Central blood volume, and pulmonary vascular and left ventricular volumes were measured directly during RNA. Central cardiac and vascular blood volume, which represents the total vascular volume in the four cardiac chamber and the great vessels, was calculated as total central blood volume minus right and left pulmonary vascular volume.21 Stroke volume (SV) is the difference between end diastolic and end systolic volumes, CO is the product of heart rate and SV, and systemic vascular resistance (dyn×s/cm5) is calculated using the formula: mean arterial pressure (mm Hg) × 80/CO (l/min).25 All blood volumes and CO were corrected for body surface area using the subject’s height and weight.
Statistical analysis
All results are expressed as mean (SEM). Differences between patient groups with and without PPHTN were compared using the Student’s t test. A p value of <0.05 was considered statistically significant. Relationships between two variables were analysed by linear regression, with the same significance levels.
The positive predictive value was calculated as the number of patients with PPHTN (true positive) divided by the sum of the patients with PPHTN and patients with no PPHTN but a mean right atrial pressure (mRAP) of ≥14 mm Hg (true positive+false positive). The negative predictive value was calculated as the number of patients without PPHTN (true negative) divided by the sum of patients without PPHTN and patients with PPHTN and a mRAP of <14 mm Hg (true negative+false negative).
RESULTS
Table 1 describes the demographic data of the study patients. Fifteen of the 62 patients received a general anaesthetic for the procedure, 12 (26%) in the no PPHTN group and three (30%) in the PPHTN group, reflecting equal distribution of the use of general anaesthetic between the two subgroups of patients. There was no significant difference between patients with and without pulmonary hypertension in terms of sex, age, liver function, presence of collateral circulation, or aetiology of liver disease.
Systemic, pulmonary, and splanchnic haemodynamics
Systemic haemodynamics
CO and systemic vascular resistance were indirectly assessed using RNA. Thirteen of 62 study patients could not undergo RNA because either the presence of ascites interfered with accurate measurements of cardiac volumes from which CO was calculated or the TIPS procedure was performed on a semi urgent basis, thereby bypassing RNA. Of the remaining 49 patients, there was no significant difference in systemic haemodynamics between those patients with and without pulmonary hypertension (table 2).
Systemic and splanchnic haemodynamics in the two study groups
Pulmonary haemodynamics
Ten of 62 study patients (16.1%) fulfilled the criteria for PPHTN.4 Pulmonary haemodynamic parameters for the two study groups are shown in table 3 while mean pulmonary artery pressures and PVR of those patients with and without PPHTN are shown in fig 1. Patients with PPHTN had a significantly higher mRAP (15.1 (1.2) mm Hg) and a higher mean right ventricle pressure (24.7 (4.7) mm Hg) compared with patients with no PPHTN (7.9 (0.5) mm Hg (p<0.001) and 14.7 (0.6) mm Hg (p<0.001), respectively).
Pulmonary haemodynamics in the two study groups
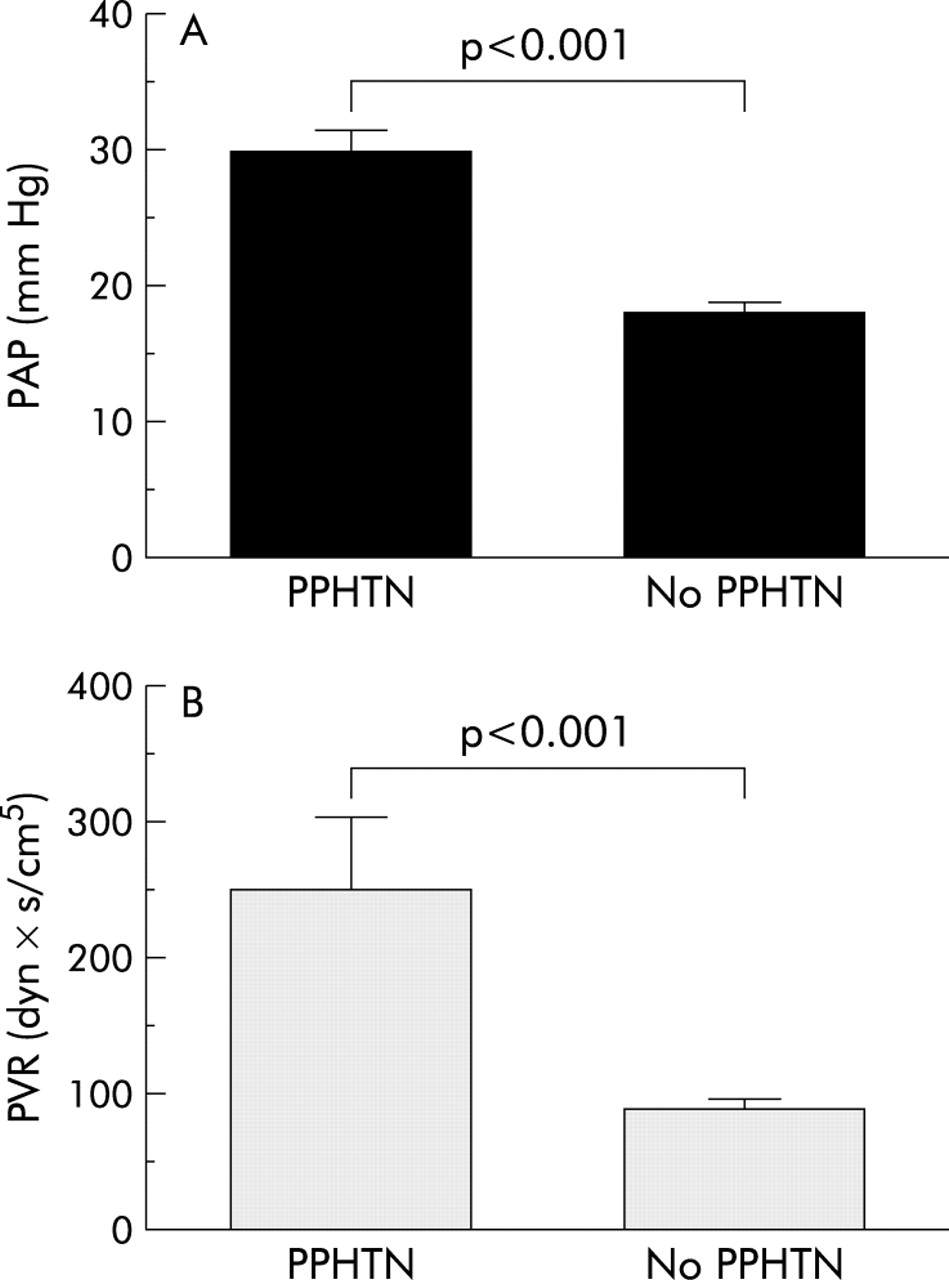
(A) Pulmonary artery pressure (PAP) and (B) pulmonary vascular resistance (PVR) in patients with and without portopulmonary hypertension (PPHTN).
For the 10 patients who had PPHTN, there was no evidence of structural cardiac or pulmonary disease. Their chest x ray showed normal cardiac size and no evidence of pulmonary parenchymal disease. Only one patient had a prominent left hilum. Two patients had a pleural effusion in addition to their tense ascites. The ECGs of two of the 10 patients showed tall p waves in leads II, III, V1, and V2, and were otherwise normal. Pulmonary function tests on two of the 10 patients showed a mildly reduced total lung capacity of 74% of predicted. Forced expiratory volume in one second (FEV1) and FEV1/vital capacity ratios were within the normal range.
Splanchnic haemodynamics
Portal hypertension was present in all study patients. There was no significant difference in inferior vena caval, free and wedged hepatic venous, portal, and corrected sinusoidal pressures in patients with and without PPHTN (table 2).
Central blood volume and cardiac volumes
Forty nine patients had measurements of blood volumes by RNA. End diastolic, end systolic, and stroke volumes were similar between patients with and without pulmonary hypertension. In addition, blood volumes in the pulmonary vascular bed and other compartments of the central chest cavity were also not significantly different between the two groups (table 4).
Central blood volume, and pulmonary vascular and cardiac chamber volumes in the two study groups
Cardiac structure and function
Forty seven patients had measurements of cardiac structure and systolic and diastolic function by two dimensional echocardiography. Fifteen patients did not have assessments of the various cardiac parameters either secondary to body habitus interfering with accurate measurement of the various parameters or because TIPS was performed as a semi urgent procedure, thereby bypassing two dimensional echocardiography. There was no significant difference between patients with and without PPHTN in terms of cardiac structure and left ventricle systolic and diastolic function (table 5).
Cardiac structure and function in the two groups of patients
Hormone profile
Pulmonary artery blood samples were available in 19 patients for measurement of ET-1 (five with PHTN and 14 without PHTN).
Endothelin-1
ET-1 levels in all study patients as a whole (2.31 (0.19) pg/ml) were significantly higher than levels in normal controls (0.30–0.90 pg/ml). There was a statistically significant difference in ET-1 levels between patients with (3.04 (0.40) pg/ml) and patients without (1.98 (0.12) pg/ml) PPHTN (p=0.02).
Correlations
A positive significant correlation (p<0.001) was found between mean pulmonary artery pressure and mRAP and mean right ventricle pressure (fig 2). There was no correlation between pulmonary artery pressure and any other haemodynamic parameter. A mRAP of ≥14 mm Hg had a positive predictive value of 83% for the presence of PHTN while a mRAP of ≤14 mm Hg had a negative predictive value of 91% (fig 3).
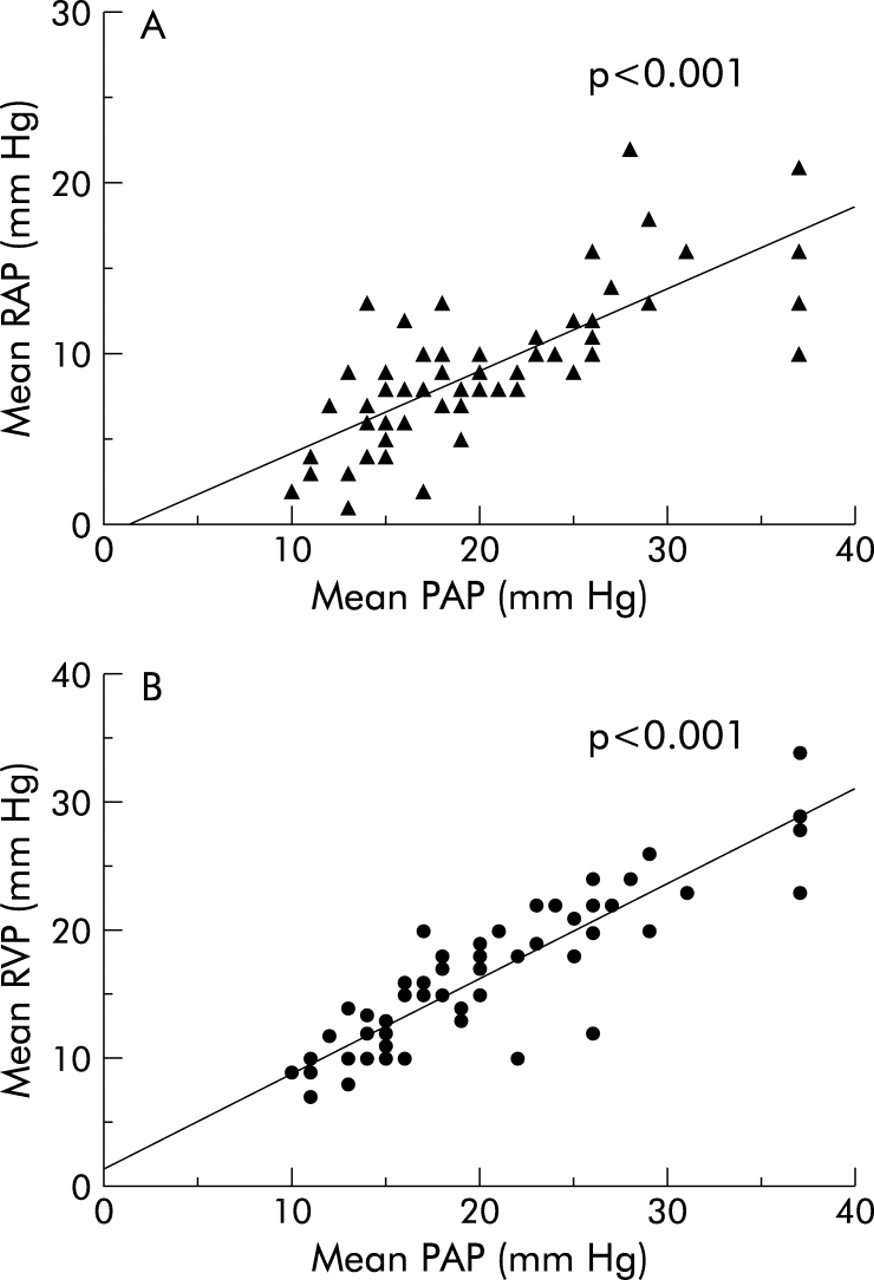
Correlation between (A) right atrial pressure (RAP) and pulmonary artery pressure (PAP) and (B) right ventricular pressure (RVP) and PAP in patients with and without portopulmonary hypertension.

{kind=link}
{kind=link}
{kind=link}
Positive and negative predictive values of right atrial pressure (RAP) for the presence of pulmonary hypertension in cirrhosis with refractory ascites in patients with and without portopulmonary hypertension (PPHTN).
DISCUSSION
In the present study, the prevalence of PPHTN in cirrhotic patients with refractory ascites was found to be 16.1%, associated with significantly higher pulmonary arterial ET-1 levels compared with those without PPHTN, although cirrhotic patients with refractory ascites as a group had significantly higher than normal ET-1 levels. A significant positive correlation was found between pulmonary artery pressure and right atrial and ventricular pressures. A right atrial pressure of ≥14 mm Hg with cirrhosis and refractory ascites had a positive predictive value of 83% for the presence of PPHTN.
Although the study population was small and the cohort consisted of a very selected group of patients, namely cirrhotic patients with refractory ascites, the finding of a significantly higher prevalence of pulmonary hypertension of 16.1% compared with 0.25–4% in cirrhosis generally reported in the literature1–3 is of great clinical importance. In contrast with previous studies which assessed heterogeneous groups of patients at various stages of cirrhosis, our population was homogenous and consisted only of patients with decompensated cirrhosis and refractory ascites. This would suggest that the development of PPHTN is somehow related to the presence of advanced cirrhosis. In agreement with previous studies,3,12 there was no correlation between the extent of PPHTN and degree of liver dysfunction, as measured by the Child-Pugh score, or portal pressure. Therefore, it appears that it is not the severity of liver dysfunction or portal hypertension per se that directly contributes to the development of PPHTN. Rather, it is some aspects of advanced cirrhosis that are implicated in the pathogenesis of PPHTN. The factors involved include the hyperdynamic circulation of cirrhosis, increased pulmonary vascular volume, and excess vasoconstrictors causing pulmonary vasoconstriction.26 These in turn all contribute to the obliterative vascular changes which further promote pulmonary hypertension.
Unlike patients with hepatopulmonary syndrome who have hypoxaemia at rest and a widened alveolar-arterial oxygen gradient, patients with PPHTN can be asymptomatic. When symptoms develop, patients complain of dyspnoea on exertion, fatigue, and peripheral oedema.27 Patients with hepatopulmonary syndrome have vasodilatation affecting the pulmonary pre-capillary and capillary vessels whereas patients with PPHTN usually have pulmonary vasoconstriction. Several pathogenetic factors for the development of PPHTN were considered in this study, and these included the haemodynamic changes of advanced cirrhosis and increased pulmonary vascular volume. Severe sodium and fluid retention and volume expansion are cardinal features of cirrhosis with refractory ascites. Some of this excess circulatory volume is redistributed to the central chest cavity21 and hence to the pulmonary circulation. However, there was no difference in pulmonary vascular volume between those patients with and without PPHTN. Furthermore, there was no correlation between pulmonary vascular volume and pulmonary arterial pressure (data not shown). Therefore, it is unlikely that an increased pulmonary vascular volume contributed significantly to the development of PPHTN in these patients. An increase in inflow into the pulmonary circulation as part of the hyperdynamic circulation was also not responsible for the development of PPHTN in patients with refractory ascites, as there was no difference in CO or systemic vascular resistance between the two groups of patients with or without PPHTN. Cirrhotic cardiomyopathy, a newly recognised complication of cirrhosis,13,28 characterised by subtle myocardial hypertrophy diastolic dysfunction23,29 and systolic incompetence under conditions of stress,23,30 was also considered. The hypertrophied and therefore stiff myocardium cannot only impede ventricular filling but can also impair active relaxation. This potentially could offer increased resistance to the pulmonary circulation. In our patients, cardiac investigations excluded the presence of heart failure as a cause of pulmonary hypertension. Furthermore, there was no evidence of any difference in cardiac structural or functional parameters between patients with or without PPHTN, thereby discounting cirrhotic cardiomyopathy as a significant cause for PPHTN in these patients.
Recently, increased levels of vasoconstrictors such as serotonin, ET-1, norepinephrine, and angiotensin II, produced in the splanchnic circulation and reaching the pulmonary circulation via portosystemic shunts, have been proposed as the mechanism responsible for the development of PPHTN in cirrhosis.31,32 This coupled with a reduced production of vasodilators such as nitric oxide and prostacyclin has been attributed as the mechanism involved in the increased smooth muscle contractile and proliferative response in the pulmonary circulation. ET-1 is one of the most potent vasoconstrictors which has been implicated in the pathogenesis of pulmonary vasoconstriction in primary pulmonary hypertension.14,16,33 In patients with primary pulmonary hypertension or pulmonary hypertension secondary to heart disease, high levels of ET-1 have been found in lung tissues.33,34 High expression of ET-1 mRNA35 has also been documented in damaged pulmonary vessel walls of these patients. These findings suggest that elevated pulmonary levels of ET-1 may be responsible for pulmonary vasoconstriction, leading to pulmonary hypertension.
In cirrhosis, ET-1 levels have been shown to be elevated compared with controls.17 Furthermore, levels of ET-1 increase as the patient progresses through the natural history of cirrhosis.18,36–38 Therefore, it is feasible that the higher pulmonary levels of ET-1 in these refractory ascitic cirrhotic patients are responsible for the overall higher prevalence of PPHTN. The fact that refractory ascitic patients with PPHTN had significantly higher concentrations of pulmonary ET-1 than those without PPHTN would support this contention. The pulmonary circulation is an important site for both the production and clearance of ET-1.39 In normal individuals, there is a normal physiological balance of ET-1 across the pulmonary circulation. In patients with pulmonary hypertension, endothelin may contribute to the pathogenesis of pulmonary hypertension by either reduced pulmonary clearance or increased production of this peptide. Although our single measurement of ET-1 in the pulmonary artery does not reflect the net result of ET-1 metabolism in the pulmonary circulation, it does represent the amount of ET-1 that is contributed by the splanchnic circulation and supplied to the pulmonary circulation. Cirrhotic patients have been reported to have increased production of ET-1 in the liver,17,38,40 gut,17,37,41 and spleen.41 Martinet and colleagues37 measured ET-1 levels in the portal vein in cirrhotic patients with refractory ascites and found them to be significantly elevated. Gerbes and colleagues17 reported a concentration gradient of ET-1 levels between the hepatic artery and vein in cirrhotic patients, suggesting hepatic production of ET-1. The stimulus for the increased splanchnic production of ET-1 could be endotoxaemia,42–44 increased angiotensin II,45,46 and epinephrine46 levels. Therefore, activated neurohumoral systems and endotoxaemia, common features of decompensated cirrhosis, acting via increased production of ET-1, may provide the explanation for the increased prevalence of PPHTN in cirrhotic patients with refractory ascites. The fact that patients with PPHTN have a further increase in their pulmonary arterial ET-1 levels would suggest that additional ET-1 could be produced locally in the pulmonary artery. However, the stimulus for a further increase in pulmonary arterial production of ET-1 in refractory ascitic patients is still unclear. Irrespective of the source of the increased pulmonary ET-1 levels, excess ET-1 levels most likely have disrupted the balance between vasoconstrictors and vasodilators in the pulmonary circulation, favouring vasoconstrictors, leading to pulmonary vasoconstriction and endothelial proliferation.47
Recognition of PPHTN in cirrhosis has important clinical implications in the management of these patients, especially in cirrhosis complicated by refractory ascites. For example, the presence of PPHTN may exaggerate the “splanchnic steal” phenomenon of advanced cirrhosis by favouring retention of the intravascular volume on the right side of the circulation, compromising left ventricular cardiac output, as evidenced by reduced central blood volume and cardiac volumes in patients with refractory ascites and PPHTN, compared with patients with refractory ascites but no PPHTN and ascites patients in general.21 Unrecognised PPHTN can also lead to devastating clinical outcomes in procedures designed to manage refractory ascites, such as post-transplantation or post-TIPS right ventricular failure and death, despite a successful procedure.48,49 This study suggests that the finding of increased ET-1 levels may indicate the presence of PPHTN. The additional finding of a right atrial pressure of greater than14 mm Hg mandates further investigations to diagnose PPHTN, especially prior to any procedure that places significant haemodynamic stress on the cardiovascular system such as TIPS insertion and liver transplantation. Until now, PPHTN has been considered a contraindication for liver transplantation50 based on reports of up to 80% perioperative mortality.51,52 However, patients with mild to moderate pulmonary hypertension appear to have no increased cardiopulmonary mortality after liver transplantation.52 The recent development of ET-1 receptor antagonists makes treatment of PPHTN a possibility.53,54 Therefore, it is imperative to diagnose PPHTN early in these patients, thereby improving their likelihood for selection for liver transplantation.
In conclusion, the prevalence of PPHTN in cirrhotic patients with refractory ascites is significantly higher than the prevalence in the general population of cirrhotic patients. This is related to increased levels of ET-1 in the pulmonary circulation, causing pulmonary vasoconstriction. The strong positive and negative predictive values of the right atrial pressure make it a simple way to identify cirrhotic patients with PPHTN.
Appendix
Table A1 shows individual mean pulmonary artery and right atrial pressure measurements, and individual calculated pulmonary vascular resistance in the two study groups. Thirteen patients could not have their pulmonary vascular resistance calculated as there were no cardiac output measurements because patients did not undergo radionuclide angiography.