Article Text

Abstract
Background: Particulate air pollution affects cardiovascular and pulmonary disease and mortality. A main hypothesis about the mechanisms involved is that particles induce inflammation in lower airways, systemic inflammation and oxidative stress.
Objectives: To examine whether short-term exposure to wood smoke in healthy subjects affects markers of pulmonary inflammation and oxidative stress.
Methods: 13 subjects were exposed first to clean air and then to wood smoke in a chamber during 4-hour sessions, 1 week apart. The mass concentrations of fine particles at wood smoke exposure were 240–280 μg/m3, and number concentrations were 95 000–180 000/cm3, about half of the particles being ultrafine (<100 nm). Blood and breath samples were taken before and at various intervals after exposure to wood smoke and clean air and examined for exhaled nitric oxide and Clara cell protein in serum and urine, and malondialdehyde in exhaled breath condensate.
Results: Exposure to wood smoke increased alveolar nitric oxide 3 hours post-exposure while malondialdehyde levels in breath condensate were higher both immediately after and 20 hours after exposure. Serum Clara cell protein was increased 20 hours after exposure.
Conclusions: Wood smoke at levels that can be found in smoky indoor environments caused an inflammatory response and signs of increased oxidative stress in the respiratory tract, especially in the lower airways.
Statistics from Altmetric.com
Exposure to ambient air pollution has been shown to increase mortality and morbidity in pulmonary and cardiovascular disease, but the mechanisms underlying these effects are still unclear. One main hypothesis is that inhaled particles induce inflammatory changes in the lower airways which in turn affect the balance between pro-thrombotic and anti-thrombotic factors in favour of increased clotting.1 2
Previous experimental studies in humans and animals have mainly focused on the effects of particulate matter (PM) from diesel exhaust or concentrated ambient particles (CAPs). Diesel exhaust has been shown to result in enhanced gene transcription and expression of cytokines in the bronchial epithelial cells, as well as upregulation of adhesion molecules responsible for migration of leucocytes to bronchial tissue.3 4 The inflammatory changes may be initiated by oxidative stress.5 Experimental studies have also been performed in healthy humans, using CAPs or ultrafine particles.6–10 In two CAP studies, fibrinogen levels in plasma increased after CAP exposure.6 7
In some studies, the fraction of exhaled NO (FENO) was reported to increase with high levels of air pollution.11 12 We have previously examined the effects of diesel exposure on FENO at a flow of 50 ml/s (FENO50) without detecting any significant changes.13 We found, however, only one previous study using modelling of NO production in the distal airways to examine the effects of exposure to particles.10
Animal studies have shown an increase in thiobarbituric acid-reacting substances, a marker of malondialdehyde (MDA) formation which in turn reflects lipid peroxidation, after wood smoke exposure.14
Clara cell protein (CC16) is an anti-inflammatory protein secreted by the Clara cells located in the bronchioli, and is normally found in the epithelial lining fluid.15 16 There is a normal transfer of lung secretory proteins to serum, with higher serum levels when the permeability of the lung epithelial barrier increases.
In the present study we report the results of experimental exposure of healthy humans to wood smoke; another source of particulate air pollution.17–19 The hypothesis tested was that short term exposure to wood smoke induces signs of pulmonary inflammation and oxidative stress. Other results from this study (systemic inflammatory biomarkers and coagulation), as well as details on the exposure, have been reported elsewhere.20 21
METHODS
Subjects
We recruited 13 healthy subjects from our department, six men and seven women aged 20–56 (mean 34) years. All were never-smokers with normal spirometry. The subjects had been free from known infections or airways symptoms for at least 1 week before the experiments, and had taken no drugs on the 2 days before the exposure sessions. The study protocol was approved by the institutional review board of Goteborg University, and written consent was obtained from the subjects.
Study design
Subjects were exposed first to filtered indoor air, and then to wood smoke 1 week later. The filtered air and wood smoke sessions were identical except for the air quality. The experiment was carried out in two rounds, with seven and six subjects, respectively. Sessions started with urine sampling, followed by FENO measurements, blood sampling and collection of exhaled breath condensate (EBC). Then the subject entered the exposure chamber. Consequently, for most of the time there were six or seven subjects in the chamber. There were two 25-minute periods of light exercise. For scheduling of post-exposure samples see table 1.
Generation of wood smoke and characterisation of exposure
The exposure chamber was 44 m2×2.9 m, and the interior was covered with Teflon-impregnated fibreglass fabric. Wood smoke was generated in a cast iron stove placed just outside the chamber. A partial flow of the wood smoke was mixed with indoor air, filtered using a high-efficiency particulate air filter, to the target concentration. A mixture of hardwood/softwood was fired in the stove. After approximately 1 hour the mass concentration was stable, and the first subject entered the chamber. Details have been reported elsewhere.21
Personal and stationary measurements of particulate matter of diameter 2.5 μm (PM2.5) and particulate matter of diameter 1 μm (PM1) mass concentrations were performed in each session. Number concentrations and size distributions of particles (0.007–6.7 μm, with 12 size intervals), were measured in real time with an electric low pressure impactor. Nitrous oxides, CO2, CO, temperature, and relative humidity were measured with direct-reading instruments and loggers. Personal and stationary measurements of benzene and 1,3-butadiene were performed using SKC-Ultra diffusive samplers (SKC, USA) filled with Carbopack X (Supelco, USA) and active sampling of formaldehyde and acetaldehyde was performed using pumps and Sep-Pak 2,4-dinitrophenylhydrazine (DNPH)-impregnated silica cartridges (Waters, USA).21
Nearly all particles were <1 μm in size. Similar results for mass concentrations were obtained with both stationary and personal measurements. The median mass concentrations were 279 μg/m3 and 243 μg/m3, respectively, in the two wood smoke rounds. The PM number concentrations were 180 000/cm3 and 95 000/cm3, with 65% and 28% ultrafine particles (UFPs) (<100 nm). Particle size distributions have been reported elsewhere.21 In the clean air sessions, mass concentrations were low, many of them below the detection limit. For NO2, the mean concentration was 0.01 ppm in the clean air experiments and 0.08–0.09 ppm in the two wood smoke sessions. The median concentrations of formaldehyde and acetaldehyde were 114 μg/m3 and 75 μg/m3, respectively, in the first wood smoke session, and 64 μg/m3 and 40 μg/m3 in the second. In the clean air sessions levels of both substances were at or below 10 μg/m3. The median benzene and 1,3-butadiene concentrations were 30 μg/m3 and 6.3 μg/m3 in the first wood smoke session and 20 μg/m3 and 3.9 μg/m3 in the second. The levels in the clean air sessions were at least 10 times lower.21
Symptoms and biomarkers
Symptoms were registered using a self-administered questionnaire.22 Clara cell protein in serum (S-CC16) and urine (U-CC16) was analysed using ELISA techniques (Bio Vendor, Czech Republic). U-CC16 was assessed using excretion rate (μg/h) and creatinine-corrected concentration.23
A chemiluminescence analyser (NIOX system; Aerocrine AB, Sweden) was used to measure FENO at a 50 ml/s (FENO50), 100 ml/s and 270 ml/s (FENO270) flow rate. A plus or minus 10% deviation of the instant flow and plus or minus 5% of the mean flow during the plateau phase was accepted. All measurements were performed in duplicate according to the 2005 ATS/ERS recommendations after at least 1 hour of fasting. A two-compartment model was used to calculate alveolar NO concentration (ppb) and NO flux (nl/s).24
Exhaled breath condensate samples were collected using an RTube (Respiratory Research, Charlottesville, VA, USA). The exhaled volume was measured with an ECoVent (Jaeger, Germany). Subjects wore a nose clip, rinsed their mouth with distilled water for 30 seconds before sampling, and breathed tidally through the RTube until 80 litres had been exhaled. Samples were frozen (–20°C) and the MDA analyses were carried out as previously described.25
Statistics
The FENO, MDA and U-CC16 levels were not normally distributed. Intraindividual differences (differences in level before and after wood smoke, subtracting differences before and after clean air, that is, “net changes”) were calculated, and tested using Wilcoxon’s signed-rank test. Associations between biomarkers were assessed using Spearman’s rank correlation coefficient (rs). For S-CC16 (normally distributed), the changes at wood smoke exposure compared with clean air were analysed with repeated-measures analysis of variance using PROC MIXED in SAS. Exposure (wood smoke, yes/no) and time (before/after chamber) were fixed effects and subject level was a random factor. Separate analyses were performed for the potential effects directly after exposure, 3 hours after exposure and in the morning on the next day. For morning samples the analysis included three unexposed samples (before and after the clean air session and before wood smoke) and one exposed sample for each subject. If there was no significant effect of time in these comparisons, we used a model with exposure as the only fixed effect.
RESULTS
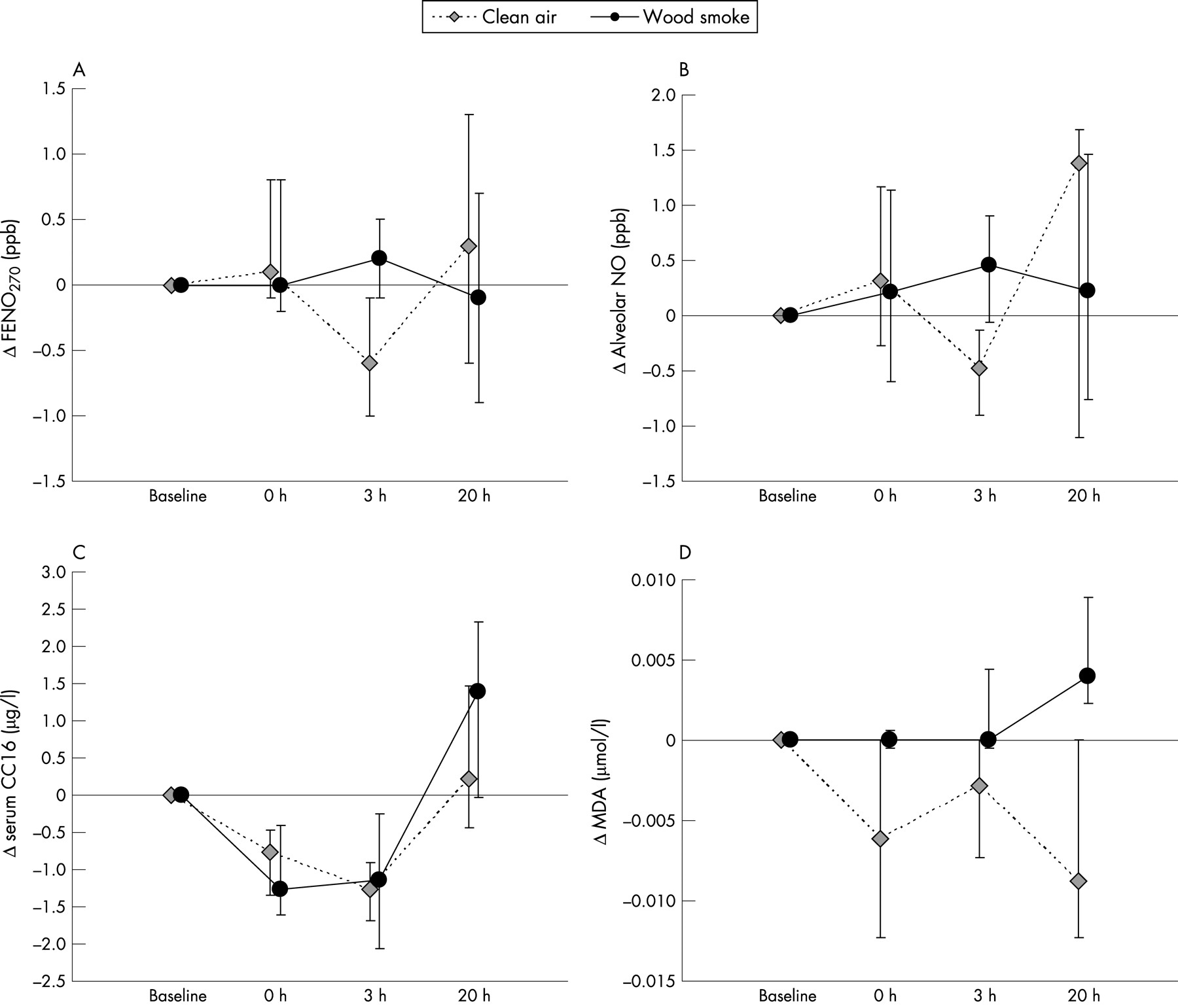
Subjective symptoms were weak, with a mild eye irritation after the wood smoke exposure. The medians and ranges of the various biomarkers of effects on airways are presented in table 2.
When analysing the intraindividual changes in biomarkers after exposure, there was some variability over time, as shown in figure 1 and several statistically significant differences between wood smoke exposure and clean air. For FENO270 there was a net increase (adjusted for the change at exposure to clean air) 3 hours after exposure to wood smoke compared with clean air (p = 0.013) (fig 1A). This was true also for calculated alveolar NO (p = 0.033) (fig 1B). There was no difference in the effects of wood smoke versus clean air on FENO50 levels or NO flux.

Serum CC16 showed a significant net increase 20 hours after exposure (fig 1C). The point estimate of the fixed effect of wood smoke exposure was an increase in S-CC16 of 1.3 μg/l (17%; p = 0.004, fig 1C). The net increase in urinary CC16 was not statistically significant (p = 0.08).
The levels of MDA in EBC were significantly increased immediately after wood smoke compared with changes after clean air (p = 0.012). The difference was smaller at 3 hours (p = 0.07), but more pronounced 20 hours post-exposure (p = 0.0005) (fig 1D).
Since wood smoke exposure levels were slightly higher in round 1 than in round 2, separate analyses were performed, stratified for round. In round 1 the analysis showed a significant increase in S-CC16 levels in the morning after wood smoke exposure, while there was no significant effect in round 2. For the FENO and MDA measurements, the results of the two rounds were similar.
Also acute phase proteins (C-reactive protein (CRP), fibrinogen, and amyloid A (AA)) were measured.20 There were significant overall correlations between S-CC16 and plasma fibrinogen and CRP in serum (S-CRP), as well as between U-CC16 and serum AA and S-CRP. On separately analysing the sampling occasions we found statistically significant positive associations between S-CC16 and plasma fibrinogen (rs = 0.48–0.75 on the various occasions) and the morning excretion rate of CC16 (rs = 0.43–0.59). The net change in alveolar NO 3 hours after exposure to wood smoke was associated with the net changes in S-CRP (directly after exposure: rs = 0.71, p = 0.006; 3 hours after exposure: rs = 0.54, p = 0.06; 20 hours after exposure rs = 0.68, p = 0.01) (fig 2).

{kind=link}
{kind=link}
There was a clear diurnal variation in S-CC16 with lower levels during daytime, as previously reported, while the U-CC16 excretion rate showed the opposite pattern, being higher in the day than during the night.22
DISCUSSION
This study on wood smoke exposure in humans showed an increase of alveolar NO and FENO270 3 hours after exposure, compared with filtered air, indicating inflammation at the distal part of the airways. After 20 hours there was also an increase in S-CC16, probably reflecting an increased permeability of the air-blood barrier. Levels of MDA in EBC were also higher after wood smoke exposure, indicating an increased lipid peroxidation, possibly in the airways. This increase was seen already immediately after exposure, but it was more pronounced in the morning after exposure. These findings support the hypothesis that particulate air pollution causes oxidative stress and distal pulmonary inflammation. As reported elsewhere, exposure to wood smoke increased the levels of S-AA, a cardiovascular risk factor, as well as factor VIII in plasma and the factor VIII/von Willebrand factor (vWf) ratio, indicating a slight effect on the balance of coagulation factors.20 Moreover, there was increased urinary excretion of free 8-iso-prostaglandin F2α, a major F2-isoprostane and marker of free radical-mediated lipid peroxidation. Taken together, the findings strengthen the link between pulmonary inflammation and cardiovascular risk factors.
How to accurately model the production of nitric oxide in the distal airways is a field in progress and still under debate. In our study, the finding of an increase of alveolar NO is strengthened by a parallel increase of FENO270, which to a higher extent than FENO50 represents NO derived from the distal airways. Neither FENO50 nor maximal NO-flux from the airways was increased after wood smoke. This is in accordance with our findings in a previous experimental study on effects of diesel exposure, in which we found no influence on FENO50, indicating no significant inflammatory effect of the exposure on the conducting airways.13
Knowledge about the significance of increased alveolar NO is still limited. Modelled in a similar way as in this study, it has been shown to be increased among subjects with allergic alveolitis, and chronic obstructive pulmonary disease (COPD), supporting an association with distal airway inflammation.26 In our study, the increase in alveolar NO was significantly correlated with an increase in S-CRP, which supports the hypothesis that the increase in alveolar NO reflects the onset of an inflammatory process.
In agreement with our results, Jansen et al reported increased levels of exhaled FENO350 after using an off-line method in seven subjects with asthma, on days with higher exposure to PM10 and PM2.5.12 However, Pietropauli et al found no increase in distal NO production after exposure to ultrafine carbon particles.10 Wood smoke particles have a more complex chemistry, which may cause a more pronounced inflammatory response.
The levels of FENO270 and alveolar NO were higher 3 hours after wood smoke than in the morning, but the effects of wood smoke were statistically significant only when the FENO270 or alveolar NO results were adjusted for changes after clean air. Although this is the logical way of analysing data, it raises the question of whether a decrease in FENO270 and alveolar NO should be expected in the afternoon (fig 1A and B). FENO70 has been shown to exhibit a slight diurnal variation, with levels about 15% lower at 16.00 than at 10.00.27 In our study, the median intraindividual decrease in FENO270 between 09.00 and 16.00 was 12% in the clean air session, while that for alveolar NO was 32%. In a separate subsequent study, we measured alveolar NO in 16 healthy individuals from our staff in the morning and afternoon (unpublished data). Alveolar NO was again 35%, and FENO270 16% lower in the afternoon than in the morning (median difference −1.1 pbb; and −1.1 ppb respectively, both p<0.001). Therefore we believe that the decrease in the afternoon after clean air in the present study is not a chance finding.
CC16 in the epithelial lining fluid is thought to protect the respiratory tract from inflammatory reactions, and has an inhibitory potential on several cytokines, including interleukin 6. Chronic airways disease may damage the Clara cells and decrease excretion of CC16 in the airways. However, serum CC16 levels will be higher when the permeability of the lung epithelial barrier is increased.28 For CC16 the normal transfer to serum probably occurs via pores in the terminal non-ciliated bronchioli, and possibly also at alveolar level.15 16 Our findings of increased levels of serum CC16 after exposure to wood smoke are consistent with similar findings after exposure to smoke from fires.29 30 The diurnal variability in S-CC16 in our study was very similar to that described by others.22
There was also increased urinary excretion of CC16 in the morning after exposure to wood smoke, which would be the result of the increased S-CC16 levels, although the effect, based on 10 subjects only, was not statistically significant. The normal intraindividual and interindividual variability is higher for U-CC16 than for S-CC16, which results in lower power to detect changes in lung permeability.23 Nevertheless, Timonen et al reported a study from Helsinki showing an increase in U-CC16 levels with ambient PM2.5, which they interpreted as an increased permeability of the epithelial barrier from particulate air pollution.31
Previous experimental studies using diesel exhaust showed signs of inflammatory changes in airways in bronchoalveolar lavage (BAL) and bronchial biopsies, but rarely in peripheral blood. No increase in S-CC16 was found after exposure to diesel exhaust (Blomberg A, personal communication), while such an effect has been shown after ozone exposure.22 The timing of sampling may be important. We found increased levels of S-CC16 only after 20 hours, while blood sampling in the diesel studies was performed 6 hours after exposure. In the CAP studies S-CC16 was not measured.
Malondialdehyde is one of the major products of lipid peroxidation, and has been found to be elevated in EBC in children and women with asthma.25 However, collection of EBC is still a method under development and is currently associated with methodological difficulties including the problem of oral contamination of the exhaled air. We tried to reduce the influence of contamination from constituents of saliva by mouth washing in each subject before measurements. We also standardised the collection by sampling the exact volume of 80 litres exhaled air in each subject, irrespective of sampling time. This was done based on the assumption of a fast steady state between MDA and exhaled air, and in order to minimise the influence of variations in exhaled volumes. However, many of the MDA determinations showed results below the detection limit. As shown in figure 1D, the morning results for MDA in EBC varied. The net increase in MDA the morning after wood smoke exposure was affected by the decrease after clean air. Although our results should be interpreted with caution, the increase in MDA in the morning after wood smoke was statistically significant also without “correction” for results after clean air exposure.
In the present study, the PM mass concentrations were similar to those of the first diesel study but higher than those of a second diesel study and the CAP studies.4 6–8 32 We exposed subjects to PM from wood smoke for 4 hours, in order to increase the dose, while in the diesel and the CAP studies an exposure time of 1–2 hours was generally used. The inhaled dose of PM mass in the present study should be two to four times higher than that in the other studies.
The PM number concentrations in the present study were much lower than in the initial diesel study but much higher than in the CAP studies.4 6–8 The increase in CC16 was most pronounced in round 1. The number concentration was higher in round 1, while the PM mass concentration was similar to that in round 2. Therefore one could speculate that the number concentration was more important than mass concentration for the increase of serum CC16. There is some toxicological evidence that UFPs have a more pronounced capacity to cause oxidative stress and pro-inflammatory effects than have the somewhat larger particles.1 33 However, the chemical composition of the particles may also be important.
The subjects were not exposed to PM alone, but also to gaseous components of the smoke, some of which (NO2 and aldehydes) are airway irritants. The levels of NO2 and aldehydes in the present study were similar to those in the CAP studies, while both NO2 and aldehydes were much higher in the diesel chamber studies.3 We consider it unlikely that the relatively moderate levels of NO2 or aldehydes caused the changes in airway biomarkers.
In residential areas where wood burning is common, it may be the major source of PM2.5. Comparing a few epidemiological studies where outdoor air pollution from wood smoke was common with studies in which it was not, Boman et al came to the conclusion that PM from wood smoke seems to be at least as harmful as PM derived from traffic exhaust.18 In Sweden, we found typical contributions to personal exposure and indoor PM2.5 levels of 1–10 μg/m3 (24-hour averages) in subjects who used wood for space heating.19 Levels may be high during shorter periods, especially when operating non-airtight stoves. The wood smoke levels used in the present study (PM of about 250 μg/m3) are therefore not unrealistic. Several hundred micrograms per m3 or in the order of milligrams per m3 of PM have been reported for households using wood firing in India and Latin America.17 In developing countries, smoke from burning of solid fuels indoors is a serious threat to health of women and children. In the Global Burden of Disease study, such indoor smoke was among the 10 leading risk factors for death and disease, and was estimated to cause nearly two million deaths per year, mainly in low-income countries.34 Recently, wood smoke exposure in childhood and adolescence was reported to increase the risk of COPD later in life in Spain.35
All subjects in the present study were also exposed to clean air during an identical chamber session in order to control for diet, exercise, transport and unknown factors that could possibly induce changes in post-exposure versus pre-exposure levels of the biomarkers. The sequence of the sessions was, however, not randomised in the present study. This weakness of the study was the result of time constraints, since if wood smoke (but not clean air) induces long-lasting inflammatory effects, these could have affected a control session 1 week after wood smoke exposure. It seems, however, very unlikely that exposure to clean air could affect the response to wood smoke 1 week later.
For S-CC16, and MDA in EBC the effects were most clearly shown in the next morning, 20 hours after exposure. No further sampling was performed, and consequently the duration of the changes remains unclear.
In conclusion, the present study indicates that wood smoke exposure affects the distal airways, as shown by the changes in alveolar NO, S-CC16, and MDA in exhaled breath condensate. Together with the findings of increased levels of cardiovascular risk factors like acute phase proteins, factor VIII and urinary 8-iso-prostaglandin F2α, it indicates that wood smoke at levels that can be found in smoky indoor environments may affect cardiopulmonary morbidity and mortality.20 This set-up with exposure to wood smoke may be a suitable experimental model for further examining the relations between acute phase proteins and airways biomarkers, and their duration. By varying the exposure conditions, and examining the chemical character of the aerosol, information could be gathered with regard to which properties of PM are crucial for the observed effects.
Main message
This experimental study in healthy humans indicates that wood smoke exposure affects the respiratory tract, especially the lower airways, as shown by changes in exhaled alveolar NO, Clara cell protein in serum, and malondialdehyde in exhaled breath condensate.
Policy implication
Wood smoke seems to be as least as powerful as diesel exhaust in terms of proinflammatory potential. Efforts should be made to replace old stoves with modern ones with low emissions of particles.
Acknowledgments
Linda Johansson, Annika Claesson, Sandra Johannesson, and Gunnel Garsell are acknowledged for skilful technical assistance and Kjell Torén for valuable comments on a previous version of this manuscript.
REFERENCES
Footnotes
Funding: This study was part of the Swedish National Air Pollution and Health Effects programme (SNAP) funded by the Swedish Environmental Protection Agency.
Competing interests: None.