


Abstract
Corticosteroids partially suppress cytokine production by chronic obstructive pulmonary disease (COPD) alveolar macrophages. p38 mitogen-activated protein kinase (MAPK) inhibitors are a novel class of anti-inflammatory drug. We have studied the effects of combined treatment with a corticosteroid and a p38 MAPK inhibitor on cytokine production by COPD alveolar macrophages, with the aim of investigating dose-sparing and efficacy-enhancing effects. Alveolar macrophages from 10 patients with COPD, six smokers, and six nonsmokers were stimulated with lipopolysaccharide (LPS) after preincubation with five concentrations of dexamethasone alone, five concentrations of the p38 MAPK inhibitor 1-(5-tert-butyl-2-p-tolyl-2H-pyrazol-3-yl)-3(4-(2-morpholin-4-yl-ethoxy)naphthalen-1-yl)urea (BIRB-796) alone, and all combinations of these concentrations. After 24 h, the supernatants were analyzed for interleukin (IL)-8, IL-6, tumor necrosis factor α (TNFα), granulocyte macrophage–colony-stimulating factor (GM-CSF), IL-1α, IL-1β, IL-1ra, IL-10, monocyte chemoattractant protein 3, macrophage-derived chemokine (MDC), and regulated on activation normal T cell expressed and secreted (RANTES). The effect of dexamethasone on p38 MAPK activation was analyzed by Western blotting. Dexamethasone and BIRB-796 both reduced LPS-induced cytokine production in a dose-dependent manner in all subject groups, with no differences between groups. Increasing the concentration of BIRB-796 in combination with dexamethasone produced progressively greater inhibition of cytokine production than dexamethasone alone. There were significant efficacy-enhancing benefits and synergistic dose-sparing effects (p < 0.05) for the combination treatment for IL-8, IL-6, TNFα, GM-CSF, IL-1ra, IL-10, MDC, and RANTES in one or more subject groups. Dexamethasone had no effect on LPS-induced p38 MAPK activation. We conclude that p38 MAPK activation in alveolar macrophages is corticosteroid-insensitive. Combining a p38 MAPK inhibitor with a corticosteroid synergistically enhances the anti-inflammatory effects on LPS-mediated cytokine production by alveolar macrophages from patients with COPD and controls.
Introduction
Corticosteroids are the most widely used anti-inflammatory therapy for patients with chronic obstructive pulmonary disease (COPD). Corticosteroids exert a wide range of anti-inflammatory effects by interfering with the ability of transcription factors such as nuclear factor-κB (NF-κB) to bind to the promoter regions of inflammatory genes (Adcock et al., 2004). However, many patients with COPD remain symptomatic despite high-dose corticosteroid therapy, and this is associated with persistent airway inflammation (Keatings et al., 1996; Bourbeau et al., 2007; Soriano et al., 2007).
Cigarette smoking increases the number of alveolar macrophages (Di Stefano et al., 1998; Hogg et al., 2004). These cells can release a variety of proinflammatory cytokines and chemokines as well as proteases (Barnes, 2004). Corticosteroids have a limited effect on the secretion of inflammatory mediators by COPD alveolar macrophages cultured ex vivo; there is only partial suppression of a range of cytokines and chemokines secreted after stimulation even in the presence of high drug concentrations (Culpitt et al., 2003; Cosio et al., 2004; Armstrong et al., 2009). It is noteworthy that we have also observed this profile of incomplete corticosteroid efficacy in alveolar macrophages from healthy subjects (Armstrong et al., 2009). This indicates that the inability of corticosteroids to fully suppress the inflammatory repertoire of alveolar macrophages is a “normal” phenomenon, which is also observed in patients with COPD. This idea is supported by the findings of Ogawa et al. (2005) who studied healthy mouse macrophages and showed that a large number of lipopolysaccharide (LPS)-responsive genes were insensitive to corticosteroids.
The p38 mitogen-activated protein kinase (MAPK) intracellular signaling pathway is activated by a range of inflammatory stimuli including cytokines and Toll-like receptor agonists (Zarubin and Han, 2005). Activated p38 MAPK enhances the function of transcription factors such as NF-κB (Saccani et al., 2002) and increases the post-transcriptional stability of some mRNAs (Winzen et al., 1999). There is evidence of p38 MAPK overactivation in COPD lung macrophages (Renda et al., 2008). p38 MAPK inhibitors reduce cytokine production from alveolar macrophages (Birrell et al., 2006; Smith et al., 2006; Kent et al., 2009) and are in clinical development for the treatment of COPD (Singh et al., 2010).
Corticosteroids do not suppress p38 MAPK activation in peripheral blood mononuclear cells (PBMCs) from patients with severe asthma (Bhavsar et al., 2010). This implicates p38 MAPK signaling as a cause of the failure of corticosteroids to suppress inflammation. There is evidence from studies with alveolar macrophages from patients with asthma and COPD that combining a p38 MAPK inhibitor with a corticosteroid can increase the suppression of cytokine production (Kent et al., 2009; Bhavsar et al., 2010). Because those previous combination studies used only one concentration of each drug it was not possible to determine whether the increased effectiveness of this combination approach was caused by an additive effect of each of the monotherapies or was the result of a mechanistic interaction leading to a synergistic effect.
We report an investigation of the effects of combined treatment with a corticosteroid and p38 MAPK inhibitor on LPS-stimulated alveolar macrophage cytokine production from patients with COPD, smokers without COPD, and healthy nonsmokers (HNS). We evaluated full dose-response curves for these drugs in combination to evaluate additive and synergistic interactions. The primary analysis was performed on secreted TNFα, IL-8, and IL-6 levels measured by specific immunoassays, with supporting analysis for a range of other cytokines and chemokines measured by multiplex. In addition, we studied whether p38 MAPK activation in alveolar macrophages is corticosteroid-insensitive, because this has previously been studied in PBMCs (Bhavsar et al., 2010), which may not be reflective of alveolar macrophages.
Materials and Methods
Study Subjects.
Eighteen patients with COPD diagnosed in accordance with current guidelines, six smokers, and six HNS were recruited. Ten of the patients with COPD and all of the smokers and HNS provided sufficient samples for experiments; the demography of these subjects is shown in Table 1. For Western blot experiments, three patients with COPD and three smokers undergoing lung surgery resection for suspected or confirmed lung cancer were recruited (Table 2). All subjects gave written informed consent. The study was approved by the Greater Manchester East and South Manchester research ethics committees.
Subject demography
Data are presented as mean (S.D.), mean (range) for age.
Lung subject demography
Data are presented as median (range).
Bronchoscopy.
Bronchoscopies were performed as described previously (Armstrong et al., 2009) with a maximum instilled volume of 480 ml. Bronchoalveolar lavage (BAL) fluid was placed on ice.
Cell Culture.
Alveolar macrophages were isolated from BAL as described previously (Armstrong et al., 2009) and cultured with or without dexamethasone or the p38 MAPK inhibitor 1-(5-tert-butyl-2-p-tolyl-2H-pyrazol-3-yl)-3(4-(2-morpholin-4-yl-ethoxy)naphthalen-1-yl)urea (BIRB-796) (reconstituted with dimethyl sulfoxide and diluted in supplemented RPMI medium at 1, 10, 100, 300, and 1000 nM) for 2 h followed by LPS (1 μg/ml) stimulation (serotype O26:B6) for 24 h. Each dexamethasone concentration was also used in combination with each BIRB-796 concentration. A baseline control was included by culturing cells with vehicle (dimethyl sulfoxide) for 2 h followed by an additional 24 h with no LPS. Supernatants were stored at −80°C.
Alveolar macrophages were isolated from resected lung tissue as described previously (Armstrong et al., 2009) and incubated with and without dexamethasone (1000 nM) and/ or LPS (1 μg/ml) for 5, 10, 20, 40, and 60 min. Cells were lysed for Western blot analysis.
Cytokine and Chemokine Assays.
Supernatants were analyzed by ELISA according to the manufacturer's instructions (R&D Systems, Abingdon, UK) to quantitate TNFα, IL-8, and IL-6 levels. These cytokines were chosen as primary endpoints from our previous work that has characterized their induction by LPS and sensitivity to corticosteroids (Armstrong et al., 2009; Kent et al., 2009). GM-CSF, IL-1α, IL-1β, IL-1ra, IL-10, MCP-3, MDC, and RANTES were measured simultaneously using a LINCOplex kit (Millipore Corporation, Billerica, MA). For ELISAs and multiplex, values below the lower level of detection were assigned a value of half the lower limit of detection (Laan et al., 2002). The lower limits of quantification for the TNFα, IL-8, and IL-6 ELISAs were 15.6, 31.3, and 9.4 pg/ml, respectively. The lower limit of quantification for the LINCOplex multiplex assay was 3.2 pg/ml.
Western Blots.
Cells (5 × 106) were lysed in radioimmunoprecipitation assay buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% Nonidet P-40) containing phosphatase (Sigma Aldrich, Poole, Dorset, UK) and protease inhibitors (Calbiochem, San Diego, CA). Cell lysates diluted in sample buffer (62.5 mM Tris, 10% glycerol, 1% SDS, 1% β-mercaptoethanol, and 0.01% bromphenol blue, pH 6.8) were electrophoresed on SDS-polyacrylamide gels (10%) and transferred to Hybond ECL membranes (Whatman, Maidstone, UK). Membranes were incubated with blocking buffer (5% dried milk in Tris-buffered saline containing 0.1% Tween 20) for 4 h at room temperature and then incubated with primary antibodies (diluted in blocking buffer) at 4°C overnight. After washing in Tris-buffered saline containing 0.1% Tween 20, the membranes were incubated for 60 min with a peroxidase-conjugated secondary antibody (diluted in wash buffer) and washed again, and the antibody-labeled proteins were visualized by enhanced chemiluminescence (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). Densitometric analysis was performed by normalizing band density to that for β-actin using Quantity One version 4.6.1 software (Bio-Rad Laboratories, Hemel Hempstead, UK).
The following antibodies were used: rabbit anti-p38, rabbit antiphospo-p38, rabbit antiphospho-HSP27 (all Cell Signaling Technology, Hitchin, UK), rabbit anti-β actin (Abcam plc, Cambridge, UK), and horseradish peroxidase-conjugated goat anti-rabbit (Dako North America, Inc., Carpinteria, CA).
Data Analysis.
Normality was assessed using the Kolmogorov-Smirnov test. BAL cell counts were compared between all subject groups using analysis of variance (ANOVA). All ELISA and multiplex data were normally distributed. Data were compared between all subject groups using ANOVA followed by unpaired t tests. Data were compared within subject groups using paired t tests. P < 0.05 was considered significant. To calculate IC50 values, dose-response curves were constructed by assuming that a concentration 10-fold lower than the lowest used in the experiment would give 0% inhibition, thus effectively constraining the minimum effect to zero. The maximum effect was estimated from the curve fit. The group mean percentage inhibitions were plotted against the log of the drug concentrations, and a nonlinear curve fit was then applied (GraphPad Software Inc., San Diego, CA; http://www.graphpad.com); the drug concentration at which 50% inhibition occurred was the IC50.
The equivalence of response between subject groups was assessed by fitting dose-response curves with separate parameters for each subject group, fitting dose-response curves restricting the parameter of interest, either IC50 or maximal inhibition, to be common across the subject groups and comparing the goodness of fit using an F statistic. Isobologram plots were generated to provide a graphical representation of the combined effect of dexamethasone and BIRB-796. Based on the monotherapy responses for each drug and assuming additivity, isobolograms, contours of constant expected inhibitions, were plotted. The average observed inhibition at each combination dose was superimposed on the isobolograms, allowing comparison between the expected and observed response, giving an indication of whether the data were synergistic, additive, or antagonistic.
Two analyses were performed to assess whether a combination of dexamethasone and BIRB-796 exhibited synergy: a dose-sparing analysis to assess whether equivalent responses can be achieved at lower doses of compound than expected given the monotherapy response of the two compounds, and an efficacy-enhancing analysis to assess whether the combination results in a significantly greater maximal effect than either compound alone as monotherapies. The dose-sparing analysis calculates a combination index with confidence intervals using the method described by Harbron (2010). The efficacy-enhancing analysis fits Hill dose-response curves to the monotherapy and combination results using both common and separate parameters for maximal response and tests for the improvement in fit from allowing the parameter to vary by using an F test. Both analyses were performed assuming a slope parameter in the Hill dose-response equation equal to one. Robustness analyses were also performed estimating the slope parameters and found to give the same conclusions.
Results
In eight patients with COPD, the number of viable alveolar macrophages recovered was less than 5 million, which was insufficient to perform all of the experiments required. The data presented are therefore from the remaining 10 patients with COPD. The percentage recovery of BAL fluid was lower in these patients with COPD compared with smokers and HNS (p = 0.0038 and 0.0006, respectively) (Table 3). The cellularity expressed as number of macrophages per milliliter of BAL was greatest in patients with COPD (p = 0.049 for COPD versus HNS).
Cell counts in bronchoalveolar lavage
Data are presented as mean (S.D.) or median (range). Data were compared between all subject groups using ANOVA, with P values shown.
LPS-Stimulated Protein Production
The secretion of inflammatory proteins from unstimulated alveolar macrophages measured by ELISA and multiplex is shown in Table 4; there was no difference between groups for any proteins. Table 4 also shows that LPS increased the secretion of all of these inflammatory proteins, with no difference between groups for any inflammatory mediator except IL-6, which was higher in smokers compared with patients with COPD (p = 0.01).
Lipopolysaccharaide-stimulated inflammatory mediator production from alveolar macrophages
Data are presented as mean (SD).
Effects of Dexamethasone
ELISA Results.
Dexamethasone significantly reduced LPS-stimulated IL-8, IL-6, and TNFα production measured by ELISA in a concentration-dependent manner in all subject groups (Fig. 1 and Supplemental Fig. 1, A, C, and E) with the top of the dose-response curve observed by 100 nM. There was no difference between patients with COPD and controls for the effects of dexamethasone at any concentration on these cytokines (ANOVA, p > 0.05 at each concentration). Table 5 shows no difference for the maximal effect of dexamethasone observed at the 1 μM concentration between patients with COPD and controls.



Combination effect of dexamethasone and BIRB-796 on inhibition of LPS-induced IL-8 (A), IL-6 (B), and TNFα (C) in alveolar macrophages. Data shown are mean ± S.E.M. percentage inhibition for IL-8, IL-6, and TNFα measured by ELISA for HNS (n = 6), smokers (S) (n = 6), and COPD (n = 10).
IC50 Values and maximal inhibition
Maximum percentage inhibition is defined as inhibition observed for 1000 nM of dexamethasone or BIRB-796. Maximum percentage inhibition data are presented as mean (S.D.).
Table 5 shows that IL-8 was less sensitive to inhibition by dexamethasone than IL-6 and TNFα, because a higher concentration of dexamethasone was required to inhibit IL-8 production by 50% (IC50) than for IL-6 and TNFα in all subject groups. In addition, the maximal inhibition of IL-8 achieved by dexamethasone (at 1 μM) was significantly lower than for IL-6 and TNFα in all subject groups (p < 0.01 for all comparisons).
Multiplex Results.
There was no difference in the effects of dexamethasone on multiplex measurements between patients with COPD and controls (Table 5), because there were no statistical differences in the maximal effect (at 1 μM dexamethasone) between groups (p > 0.05 for all proteins). Furthermore, IC50 values were similar between groups. The effects of dexamethasone varied between proteins, with the least suppression being observed for IL-1α, IL-1β, and MDC where less than 50% inhibition was achieved.
Effects of p38 Inhibitor BIRB-796
ELISA Results.
BIRB-796 significantly reduced LPS-stimulated IL-8, IL-6, and TNFα production in a concentration-dependent manner in all three subject groups (Fig. 1 and Supplemental Fig. 1, B, D, and F). The dose response for IL-8 was less clear, but statistically significant inhibition (p < 0.05) of IL-8 production was observed in all three subject groups only at the highest concentrations (300 and 1000 nM) of BIRB-796. There was no difference between patients with COPD and controls for the effects of BIRB-796 on measurements of IL-8, IL-6, and TNFα by ELISA (ANOVA, p > 0.05 at each concentration).
Multiplex Results.
Likewise, cytokine measurements by multiplex showed no difference in the effects of BIRB-796 between groups (Table 5), because there were similar IC50 values and no statistical difference in the maximal effect (p > 0.05 for all proteins). The maximal inhibition of cytokine production achieved with BIRB-796 was lower than dexamethasone, because 50% inhibition was not reached for the majority of cytokines. BIRB-796 had little or no effect on the production of MCP-3 and MDC.
Combination Inhibition of LPS-Stimulated Inflammatory Mediator Production
Fig. 1 shows the effects of combining dexamethasone and BIRB-796 on IL-8, IL-6, and TNFα levels measured by ELISA. The lowest concentration of BIRB-796 (1 nM) added to dexamethasone caused greater inhibition than dexamethasone alone. Increasing the concentration of BIRB-796 in combination with this corticosteroid produced progressively greater inhibition of cytokine production.
Figure 2 compares the observed responses for each combination dose with the expected isobolograms that assume additivity for IL-6, IL-8, and TNFα. Comparison of the observed percentage inhibition (numerical values in black) with the expected percentage inhibitions (isobolograms in color) gives an indication of whether the response is additive (observed percentage inhibitions lie along the expected response contours) or synergistic (observed percentage inhibitions are greater than the expected response contours). For the majority of cases, the observed inhibition was greater than the corresponding isobolograms, suggesting synergy.
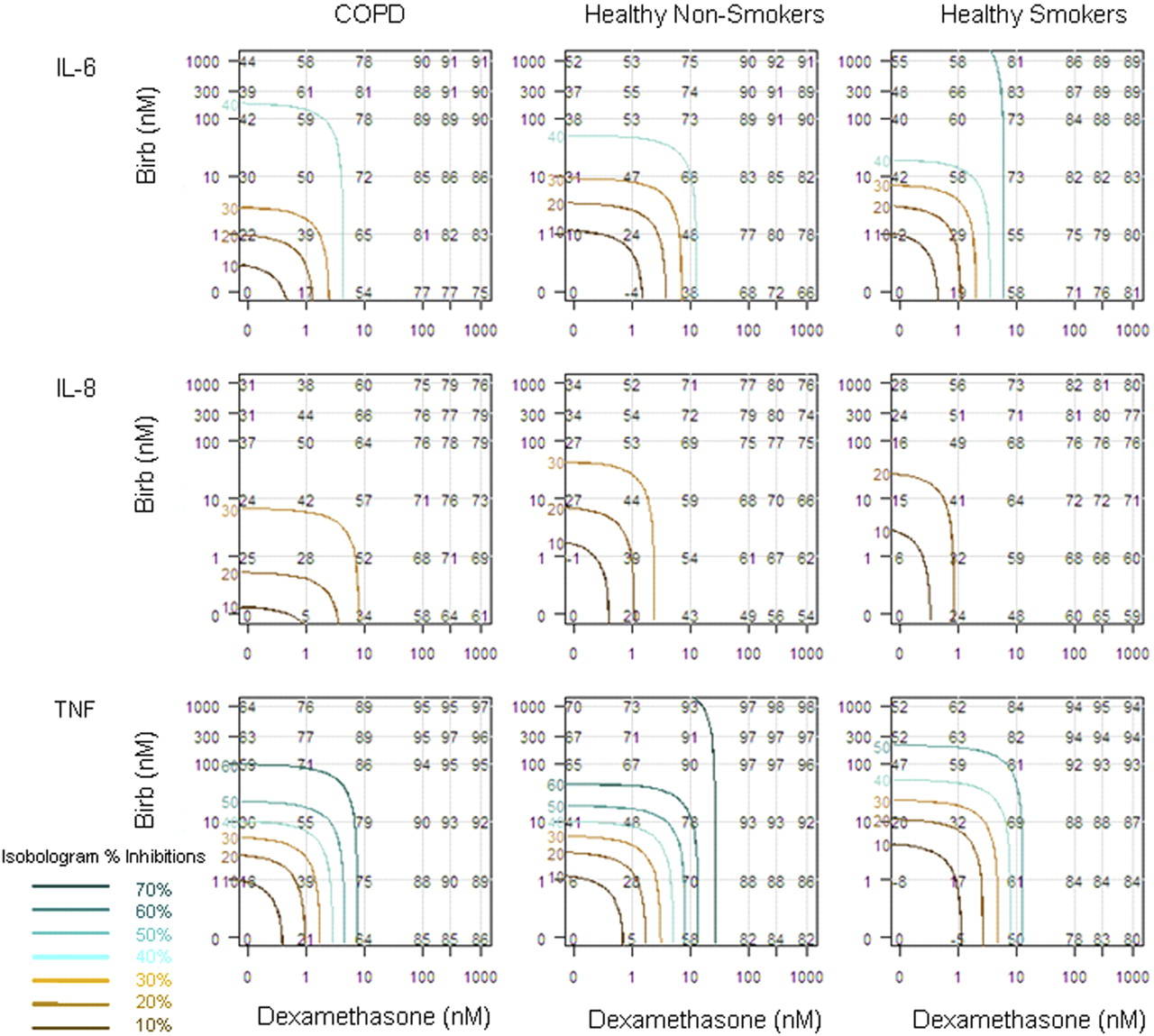
Isobolograms of the combination effect of dexamethasone and BIRB-796. Observed and expected inhibitions of LPS induced IL-6, IL-8, and TNFα in alveolar macrophages from each of the subject groups are shown. The black numerical values within the plots show the observed mean percentage inhibition at each combination of dexamethasone (x-axis) and BIRB-796 (y-axis). Isobolograms are plotted in color for different expected levels of response assuming additivity. The percentage expected inhibitions for these isobolograms are stated in corresponding color on the y-axis and in the key, 70 to 40% inhibition are shades of green and 30 to 10% inhibition are shades of brown. Comparison of the observed percentage inhibition (numerical values) with the expected percentage inhibitions (isobolograms) gives an indication whether the response is additive (observed percentage inhibitions lie along the expected response contours) or synergistic (observed percentage inhibitions are greater than the expected response contours).
Figures 3 shows the combination index with 95% confidence intervals calculated for each of the cytokines. Where the entirety of the 95% confidence interval lies below the value of one, this is a significant synergistic effect. IL-8, IL-6, TNFα, IL-10, and RANTES all showed a significant dose-sparing effect in all subject groups. IL-8 had the largest effect with a combination index less than 0.1, corresponding to a 10-fold dose sparing for the COPD and HNS groups. GM-CSF, IL-1β, IL-1ra, and MDC all showed significant synergistic dose sparing in one or more of the subject groups.

Dose-sparing effect of dexamethasone in combination with BIRB-796. Estimates of the combination index are plotted with 95% confidence intervals calculated for each cytokine and subject group. A combination index of one corresponds to additivity; values less than one represent synergy. Endpoints where the 95% confidence interval lies completely below one show statistically significant synergy. Missing lines indicate it was not possible to fit the model.
Figure 4 shows the efficacy-enhancing benefit with 95% confidence intervals calculated for each of the cytokines. Where the entirety of the 95% confidence interval lies above the value of zero, this is a significant synergistic effect. IL-8, IL-6, and MDC all showed a significant efficacy-enhancing benefit in all three subject groups with approximately 20% additional maximal inhibition achieved compared with dexamethasone alone. GM-CSF, IL-10, IL-1ra, TNFα, and RANTES all showed significant efficacy-enhancing benefit in one or more of the subject groups.

Efficacy enhancing benefit of dexamethasone in combination with BIRB-796. Estimates of the efficacy enhancing benefit are plotted with 95% confidence intervals calculated for each cytokine and subject group. An efficacy enhancing benefit of zero corresponds to additivity; values greater than zero represent synergy. Endpoints where the 95% confidence interval lies completely above zero show statistically significant synergy.
Effects of Dexamethasone on Phospho-p38 MAPK and Phospho-HSP27
LPS increased the expression of both phospho-p38 and phospho-HSP27 in macrophages from patients with COPD (n = 3) and smokers (n = 3); representative Western blots are shown in Fig. 5. The maximum expression of phospho-p38 and phospho-HSP27 were observed at 20 to 60 min in both patient groups. In both patients with COPD and smokers, dexamethasone did not change the expression of these p38 MAPK activation markers. Because there was no visual difference between patients with COPD and smokers, densitometry analysis was performed by combining the data from both groups (Fig. 5); dexamethasone did not suppress LPS induced phospho-p38 or phospho-HSP27 at any time point (p > 0.05 for all comparisons).

The effects of dexamethasone (DEX) on phospho-p38 MAPK and phospho-HSP27 in LPS-stimulated alveolar macrophages. Data shown are median ± range of total p38 (A), phospho-p38 (B), and phospho-HSP27 (C) protein band density normalized to β-actin. LPS stimulation alone (LPS) or preincubated with dexamethasone (1000 nM) for 2 h (LPS + DEX) at time points up to 60 min are shown (n = 6 patients with COPD and smokers combined). Representative Western blots for COPD and smokers (S) show total p38 (A), phospho-p38 (B), and phospho-HSP 27 (C) protein bands below respective conditions on the charts.
Discussion
Combination treatment with BIRB-796 and dexamethasone provided synergistic suppression of a range of inflammatory mediators produced by LPS-stimulated alveolar macrophages from patients with COPD and both smoking and nonsmoking controls. The maximal effect of dexamethasone was increased by the BIRB-796. In addition, there was a synergistic dose-sparing effect when BIRB-796 and dexamethasone were combined, because equivalent responses were achieved at lower doses of these drugs combined than expected from the monotherapy responses.
Macrophages and PBMCs have previously been used to study combination effects with a p38 MAPK inhibitor and corticosteroid (Kent et al., 2009; Bhavsar et al., 2010). The current study performed a more detailed analysis of the interaction between p38 MAPK inhibitors and corticosteroids by using an experimental design generating full dose-response curves for both drugs and adopting a synergy analysis. Long-term treatment with high-dose inhaled corticosteroids can lead to side effects such as osteoporosis, hypertension, and cataracts (Roland et al., 2004). Strategies using lower corticosteroid doses would limit these side effects. Combination therapy with a corticosteroid and a p38 MAPK inhibitor could enable lower doses of corticosteroids to be used in clinical practice, but without loss of efficacy. Our data suggest that such a combination approach will lead to increased efficacy using lower doses.
p38 MAPK inhibitors and corticosteroids both reduce inflammatory gene transcription (Kent et al., 2009), albeit by different mechanisms, because corticosteroids transrepress transcription factor activity at the promoter regions of inflammatory genes (Adcock et al., 2004), whereas p38 MAPK inhibitors suppress the enzymatic phosphorylation of transcription factors (Rolli et al., 1999; Zhu and Lobie, 2000; Wiggin et al., 2002). p38 MAPK inhibitors further differentiate from corticosteroids by acting post-transcriptionally and on protein translation (Winzen et al., 1999; Neininger et al., 2002). We observed more than an additive effect of these different molecular mechanisms when combination treatment was used. We speculate that these synergistic effects could be caused by the inhibition of p38 MAPK-mediated GR phosphorylation allowing effective binding of dexamethasone to GR (Irusen et al., 2002) and inhibition of p38 MAPK phosphorylation of histones, thus altering the chromatin structure to facilitate GR action as seen for NF-κB (Saccani et al., 2002).
The activation of p38 MAPK in PBMCs from patients with severe asthma is resistant to corticosteroids in vitro (Hew et al., 2006; Bhavsar et al., 2010). Likewise, the administration of prednisolone to patients with COPD did not suppress the ex vivo activation of p38 MAPK in whole blood (Singh et al., 2010). We provide similar observations using alveolar macrophages, from both patients with COPD and controls. The lack of effect of dexamethasone on p38 MAPK or HSP27 activation is a mechanism that is likely to play a role in the incomplete suppression of inflammatory mediators such as IL-8 by corticosteroids, which we observed in the current and previous studies (Armstrong et al., 2009; Kane et al., 2009). Cytokines such as TNFα that are more sensitive to corticosteroids in comparison to IL-8 may be more dependent on NF-κB signaling and less dependent on p38 MAPK signaling.
A gene expression study in healthy mouse alveolar macrophages showed that approximately half of the LPS-regulated genes were insensitive to corticosteroids (Ogawa et al., 2005). This indicates that corticosteroids have a limited effect on some aspects of the macrophage innate immune response, even in healthy cells. This important observation suggests that these aspects of the innate immune response will also be corticosteroid-insensitive in COPD cells. We observed that IL-8 and RANTES production were incompletely suppressed (<65%) in both patients with COPD and controls, whereas even lower suppression was observed for IL-1α, IL-1β, and MDC (<50%) in all groups.
We previously observed no difference between patients with COPD and controls for the effects of dexamethasone on LPS-stimulated alveolar macrophage cytokine production after 4 h of cell culture (Armstrong et al., 2009). The current study provides similar data across a range of cytokines and chemokines after 24-h cell culture. So what is the role of alveolar macrophages in the limited clinical effect of corticosteroids in patients with COPD? There are more alveolar macrophages in patients with COPD compared with controls (Di Stefano et al., 1998; Hogg et al., 2004), which can contribute to raised levels of inflammatory cytokines and chemokines. Corticosteroids provide only partial repression of these inflammatory mediators; for example, IL-8 is particularly corticosteroid-insensitive (Armstrong et al., 2009; Kent et al., 2009). The inflammatory drive in COPD may be primarily through these steroid-resistant mediators. Our data show that combination treatment with a corticosteroid and p38 MAPK inhibitor can enhance suppression of such inflammatory mediators.
Cosio et al. (2004) showed that LPS-stimulated alveolar macrophages from patients with COPD were less sensitive to the effects of dexamethasone compared with cells from smokers, which has been linked to a reduction in histone deacetylase 2 activity in COPD macrophages. It is probable that a variety of molecular mechanisms are responsible for the limited effects of corticosteroids seen in clinical practice in patients with COPD. Our data point toward the following mechanism: corticosteroids have a limited effect on macrophage cytokine production both in healthy and COPD cells, with corticosteroid-insensitive p38 MAPK signaling involved in cytokine production in these cells.
The clinical development of an oral program for BIRB-796 was stopped presumably because of an unacceptable therapeutic index with significant systemic side effects (Genovese, 2009). Novel p38 MAPK inhibitors that are in clinical development for the treatment of COPD hopefully will have fewer side effects. This may be achieved by inhaled delivery, thus limiting systemic exposure (Chopra et al., 2008), or else, as suggested by our data, through the dose-sparing effect of a combination with a corticosteroid, to increase the safety margin.
The anti-inflammatory effects of BIRB-796 on macrophage function were lower than dexamethasone. The pharmacological characteristics of these individual drugs has a major influence on such data, because different results may be obtained using other drugs in these classes with different potencies and maximal effects. Nevertheless, there are other data showing that corticosteroids have a greater effect on cytokine production than p38 MAPK inhibitors, both in vitro using cultured macrophages (Kent et al., 2009; Bhavsar et al., 2010) and in a clinical trial using stimulated whole blood from patients with COPD (Singh et al., 2010). Because the anti-inflammatory effect of p38 MAPK inhibitors seems to be lower than corticosteroids in these cell models, it is possible that the optimum use of p38 MAPK inhibitors in the treatment of COPD is in combination with corticosteroids. Our observations are based on Toll-like receptor 4 activation, and it remains to be determined whether this observation is also replicated in other cell models.
Cytokine production from LPS-stimulated macrophages seemed lower in patients with COPD compared with controls, but it did not reach significance. Previous studies have shown that smoking reduces cytokine production from macrophages (Birrell et al., 2008; Kent et al., 2008; Armstrong et al., 2009; Kane et al., 2009). Our study was probably not large enough to address this question.
In summary, we have shown that the p38 MAPK inhibitor BIRB-796 and the corticosteroid dexamethasone have synergistic anti-inflammatory effects on COPD alveolar macrophages. For p38 MAPK inhibitors in clinical development, these data provide a strong rationale for combination clinical trials with inhaled corticosteroids.
Authorship Contributions
Participated in research design: Armstrong, Booth, Wreggett, and Singh.
Conducted experiments: Armstrong, Booth, and Cadden.
Contributed new reagents or analytic tools: Harbron and Wreggett.
Performed data analysis: Armstrong, Harbron, and Lea.
Wrote or contributed to the writing of the manuscript: Armstrong, Harbron, Lea, Booth, Cadden, Wreggett, and Singh.
Footnotes
This research was funded by AstraZeneca.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.111.180737.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- COPD
- chronic obstructive pulmonary disease
- ANOVA
- analysis of variance
- BIRB-796
- 1-(5-tert-butyl-2-p-tolyl-2H-pyrazol-3-yl)-3(4-(2-morpholin-4-yl-ethoxy)naphthalen-1-yl)urea
- BAL
- bronchoalveolar lavage
- FEV1
- forced expiratory volume in 1 s
- GR
- glucocorticoid receptor
- LPS
- lipopolysaccharide
- MAPK
- mitogen-activated protein kinase
- NF-κB
- nuclear factor-κB
- HNS
- healthy nonsmokers
- IL
- interleukin
- TNF
- tumor necrosis factor
- ELISA
- enzyme-linked immunosorbent assay
- RANTES
- regulated on activation normal T cell expressed and secreted
- MCP-3
- monocyte chemoattractant protein 3
- MDC
- macrophage-derived chemokine
- GM-CSF
- granulocyte macrophage–colony-stimulating factor
- PBMC
- peripheral blood mononuclear cell
- HSP27
- heat shock protein 27.

- Received March 1, 2011.
- Accepted May 18, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}