


Abstract
Chronic exposure of mice and rats to cigarette smoke affects T-cell responsiveness that may account for the decreased T-cell proliferative and T-dependent antibody responses in humans and animals exposed to cigarette smoke. However, the mechanism by which cigarette smoke affects the T cell function is not clearly understood. Our laboratory has shown that chronic exposure of rats to nicotine inhibits the antibody-forming cell response, impairs the antigen-mediated signaling in T cells, and induces T cell anergy. To determine the mechanism of cigarette smoke-induced immunosuppression and to compare it with chronic nicotine exposure, rats were exposed to diluted, mainstream cigarette smoke for up to 30 months or to nicotine (1 mg/kg b.wt./24 h) via miniosmotic pumps for 4 weeks, and evaluated for immunological function in vivo and in vitro. This article presents evidence suggesting that T cells from long-term cigarette smoke-exposed rats exhibit decreased antigen-mediated proliferation and constitutive activation of protein tyrosine kinase and phospholipase C-γ1 activities. Moreover, spleen cells from smoke-exposed and nicotine-treated animals have depleted inositol-1,4,5-trisphosphate-sensitive Ca2+ stores and a decreased ability to raise intracellular Ca2+ levels in response to T cell antigen receptor ligation. These results suggest that chronic smoking causes T cell anergy by impairing the antigen receptor-mediated signal transduction pathways and depleting the inositol-1,4,5-trisphosphate-sensitive Ca2+ stores. Moreover, nicotine may account for or contribute to the immunosuppressive properties of cigarette smoke.
Cigarette smoking is a major health risk factor and significantly increases the incidence of heart disease, cancers of various organs, and acute and chronic respiratory tract infections (for review, see Johnson et al., 1990; Sopori and Kozak, 1998). It is postulated that this increased susceptibility reflects cigarette smoke-induced impairment of the immune system (Holt and Keast, 1977). Cigarette smoke (SM) affects a wide range of immunological functions in humans and experimental animals, including both the humoral and cell-mediated immune responses (Sopori et al., 1994; Sopori and Kozak, 1998). Although chronic SM affects T cell responses in rodents (Thomas et al., 1973; Holt et al., 1976; Chang et al., 1990), monkeys (Sopori et al., 1985), and humans (Silverman et al., 1975; Peterson et al., 1983), the molecular mechanism through which SM affects the lymphocyte function is largely unknown. Chronic exposure of rats to nicotine, one of the major components of SM, inhibits the antibody-forming cell (AFC) response, and this immunosuppression is causally related to impairment of antigen-mediated signaling in T cells (Geng et al., 1995, 1996; Sopori et al., 1998). In addition to nicotine, SM contains thousands of other bioactive substances, so it is not known whether the molecular mechanisms are similar for the immunosuppression by whole SM and nicotine. To examine the molecular mechanism for SM-induced immunosuppression, F344 rats were exposed chronically to mainstream SM for several hours per day, and their spleen cells investigated for the T cell antigen receptor (TCR)-mediated proliferation, the AFC response to the T-dependent antigen sheep red blood cells (SRBC), and the TCR-mediated signal transduction pathway. Results indicate that chronic exposure to mainstream SM causes aberrant antigen-mediated signaling in T cells. Furthermore, our studies show that changes in the signaling were associated with constitutive activation of protein tyrosine kinase (PTK) and phospholipase C-γ1 (PLC-γ1) activities. In addition, chronic exposure to SM or nicotine was observed to deplete inositol-1,4,5-trisphosphate (IP3)-sensitive intracellular Ca2+ stores, and this loss may account for the immunosuppression and the loss of T cell function in SM animals.
Materials and Methods
Animals.
Pathogen-free female F344/Crl rats 4 to 5 weeks old were obtained from Charles River Laboratories (Raleigh, NC). Animals were placed one per cage into whole-body inhalation chambers (H2000; Laboratory Products, Maywood, NJ) and supplied with filtered air (FA) during quarantine and conditioning. The chambers were maintained at 12 ± 2 air changes per hour, at a temperature of 24 ± 2°C and relative humidity of 40 to 70%. Room lights were on a 12-h on/off cycle. Food (Wayne Lab Blox; Allied Mills, Chicago, IL) and water were available ad libitum, except food was withheld from all rats during the daily 6-h exposures. Rats were randomly assigned by weight to experimental groups. Exposure to SM was begun when the animals were 6 to 7 weeks old.
Antibodies and Chemicals.
Antibodies to phosphotyrosine (PY), PLC-γ1, and horseradish peroxidase-conjugated anti-mouse IgG were purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Anti-rat αβ-TCR and anti-CD3 were obtained from either Upstate Biotechnology or PharMingen (San Diego, CA). Goat anti-mouse IgG was obtained from Sigma Chemical Co. (St. Louis, MO). Chemluminescence Western blotting detection reagents were purchased from Amersham Life Sciences (Arlington Heights, IL). Nicotine base [(−)-nicotine] was purchased from ICN Biochemicals (Aurora, OH). All other reagents, unless mentioned otherwise, were bought from Sigma Chemical Co.
Exposure to SM.
Diluted, mainstream SM was generated from 1R3 research cigarettes (Tobacco and Health Research Institute, Lexington, KY) by modified AMESA Type 1300 automated smoking machines (AMESA, Geneva, Switzerland) as previously described (Chen et al., 1992). Cigarettes were held at 24°C and 50 to 70% relative humidity before use, then puffed twice per minute at a 70-ml puff volume taken over 2 s. Each cigarette was puffed six or seven times; the fresh smoke was diluted with FA and delivered to the exposure chambers. The mass concentration of total particulate matter (TPM) in SM was determined gravimetrically, CO concentrations were measured periodically with an infrared analyzer (Beckman Industries, La Habra, CA), and smoke particulate size was measured by cascade impaction (Finch et al., 1994). Exposures were conducted daily, 6 h/day for 8 or 28 to 30 months. The mean levels of TPM were within 5% of the target concentration of 250 mg TPM/m3, and the smoke particulate size was 0.52 μm (±.05 S.D.). Control (CON) animals were held in a separate chamber in the same room but were exposed to FA.
Health Status of SM-Exposed Animals.
Animals in this study were exposed as a part of a larger study of the carcinogenicity of SM. Sera from rats before and after exposures were examined (Microbiological Associates, Bethesda, MD) and found to be free from antigens against common rodent pathogens.
Compared with CON, SM animals were clinically normal except that body weights were reduced and the pelt was tinted brown. Nonneoplastic changes in the lungs of SM rats included macrophage aggregations and focal inflammatory lesions, with neutrophils, lymphocytes, and epithelial hyperplasia. These lesions were generally found adjacent to the pleura and scattered throughout the parenchyma distal to the terminal bronchiole-alveolar duct junction (Finch et al., 1995). Lung tumors were found in a few rats exposed to SM (K. Nikula, J. Mauderly, E. Barr, and G.L.F., unpublished data). No significant abnormal lesions were observed grossly at necropsy in any of the rats used in this study.
Determination of Serum Cotinine Levels.
To examine whether animals exposed to SM had nicotine levels comparable with heavy human cigarette smokers, we determined the serum cotinine levels of SM animals. Because the half-life of nicotine is very short, particularly in rodents (3–9 min) (Mactutus, 1989), serum/urine cotinine levels are normally used to ascertain the level of SM/nicotine exposure. Therefore, to determine the level of nicotine exposure in SM animals, we assayed serum cotinine of these animals by a radioimmunoassay with the nicotine metabolite kit (Diagnostic Products Corp, Los Angles, CA) according to the manufacturer's directions and as described previously (Geng et al., 1995). Cotinine levels of six randomly selected SM sera were 963 ± 103 ng/ml compared with undetectable levels in sham-exposed animals. These serum cotinine levels compare to human smokers smoking approximately two to three packs of high-tar, high-nicotine cigarettes per day (Geng et al., 1995).
Nicotine Treatment.
(−)-Nicotine base was administered s.c. for 3 to 4 weeks via constant-release miniosmotic pumps (Alzet Corp., Palo Alto, CA) at the rate of ∼1 mg/kg b.wt./day (Geng et al., 1995). These nicotine exposures correspond to humans smoking approximately two packs of cigarettes per day (Geng et al., 1995).
Immunizations.
Animals were injected i.v. with 5 × 108 SRBC 4 days before sacrifice as described previously (Sopori et al., 1989).
Preparation of Lymphoid Cells and AFC Assay.
Preparation of spleen cell suspensions and purification of T cells were carried out as described elsewhere (Sopori et al., 1989). Briefly, spleens were passed through stainless steel mesh, and red blood cells were lysed by treatment with NH4Cl solution. T cells were purified by passing spleen cells over a nylon wool column. The nylon-wool nonadherent cell fraction (T cells) was >90% CD3+ by flow cytometry. Enriched B cells were obtained by negative selection; cells were treated with mouse anti-rat CD3 monoclonal antibody (mAb), and CD3+ cells were removed by incubating in dishes coated with anti-mouse IgG (Sopori et al., 1985). The nonadherent fraction (B cells) was >88% by flow cytometry analysis. The primary direct AFC response was determined essentially by the method of Cunningham and Szenberg (1968) as described elsewhere (Sopori et al., 1989). Various concentrations of spleen cells were mixed with 2% SRBC and 25 μl of guinea pig complement in a final volume of 150 μl of complete medium (RPMI-1640 containing 10% fetal calf serum, 2 mM glutamine, 50 mM 2-mercaptoethanol, and 10 μg/ml gentamicin). The cell suspension was injected into Cunningham slides, sealed with petroleum wax, and incubated for 45 min at 37°C. Results were expressed as AFC/106 spleen cells.
Assay for Anti-CD3-Induced Proliferation.
Anti-CD3-induced T cell proliferation was assayed as described previously (Geng et al., 1995). Briefly, 2 × 105 cells were cultured in 0.2 ml of complete medium in microtiter wells in the presence or absence of 5 μg/ml anti-CD3. The cultures were incubated at 37°C in the presence of 5% CO2 and harvested after 3 days. Proliferation was assayed by overnight pulsing of the culture wells with 0.5 μCi of [3H]thymidine before harvesting.
Assay for Intracellular Ionized Ca2+ and IP3-Sensitive Ca2+ Stores.
The intracellular concentration of ionized Ca2+ was determined as previously described (Razani-Boroujerdi et al., 1994). Briefly, cells were loaded with acetoxymethyl ester of indo-1 (Molecular Probes, Eugene, OR). Before the assay, cells were suspended in 2 ml of PBS containing 1 mM Ca2+ and 1 mM Mg2+, and the cytoplasmic Ca2+ concentration [Ca2+]i was determined by spectrofluorometry in a PTI Deltascan fluorometer (Photo Technology International, South Brunswick, NJ). The baseline Ca2+ concentration of the cells was recorded before the addition of 2.5 μg/ml mouse anti-rat αβ-TCR and 2.5 μg/ml of the second antibody (goat anti-mouse IgG). To determine the concentration of Ca2+ within IP3-sensitive intracellular Ca2+ stores, indo-1-labeled cells were suspended in Ca2+, Mg2+-free PBS containing 100 μM EGTA, and treated with 1 mM thapsigargin. The excitation wavelength for indo-1 is 355 nm, and emission was measured at 410 and 485 nm. After subtracting the background, [Ca2+]iwas calculated as described elsewhere (Razani-Boroujerdi et al., 1994).
PTK Assay.
Tyrosine phosphorylation of proteins was determined by immunoblotting with anti-PY mAb by previously published methods (Geng et al., 1996). Briefly, 1 × 107 spleen cells were suspended in 0.5 ml of complete medium and, where indicated, incubated at 37°C with 2 μg/ml anti-αβ-TCR for 2 min. After centrifugation, the cell pellet was lysed with lysis buffer (10 mM Tris-HCl, pH 7.8, containing 1% Nonidet P-40, 150 mM NaCl, 0.1 mM sodium vanadate, 50 mM NaF, 30 mM sodium pyrophosphate, 2 mM iodoacetate, 5 μM ZnCl2, 1.5 mM phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 10 μg/ml antipain, 5 μg/ml aprotinin, and 3 μg/ml pepstatin A). After debris was removed by centrifugation, lysates were boiled in Laemmli sample buffer (60 mM Tris-HCl, pH 6.8, containing 2% SDS, 10% glycerol, 10 mM EDTA, and 0.001% bromophenol blue) for 5 min. Lysate protein (10–20 μg) was electrophoresed on 7.5% SDS-polyacrylamide gel electrophoresis and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Blots were blocked by incubating at room temperature with 5% skim milk protein (Upstate Biotechnology) in 10 mM Tris-HCl, pH 7.4, containing 150 mM NaCl for 1.5 h. Blots were washed and probed with anti-PY or anti-PLC-γ1. Blots were developed with horseradish peroxidase-conjugated anti-mouse IgG and visualized by chemluminescence.
Results
Chronic Whole-Body Exposure to SM Suppresses AFC Response without Affecting Lymphocyte Subpopulations.
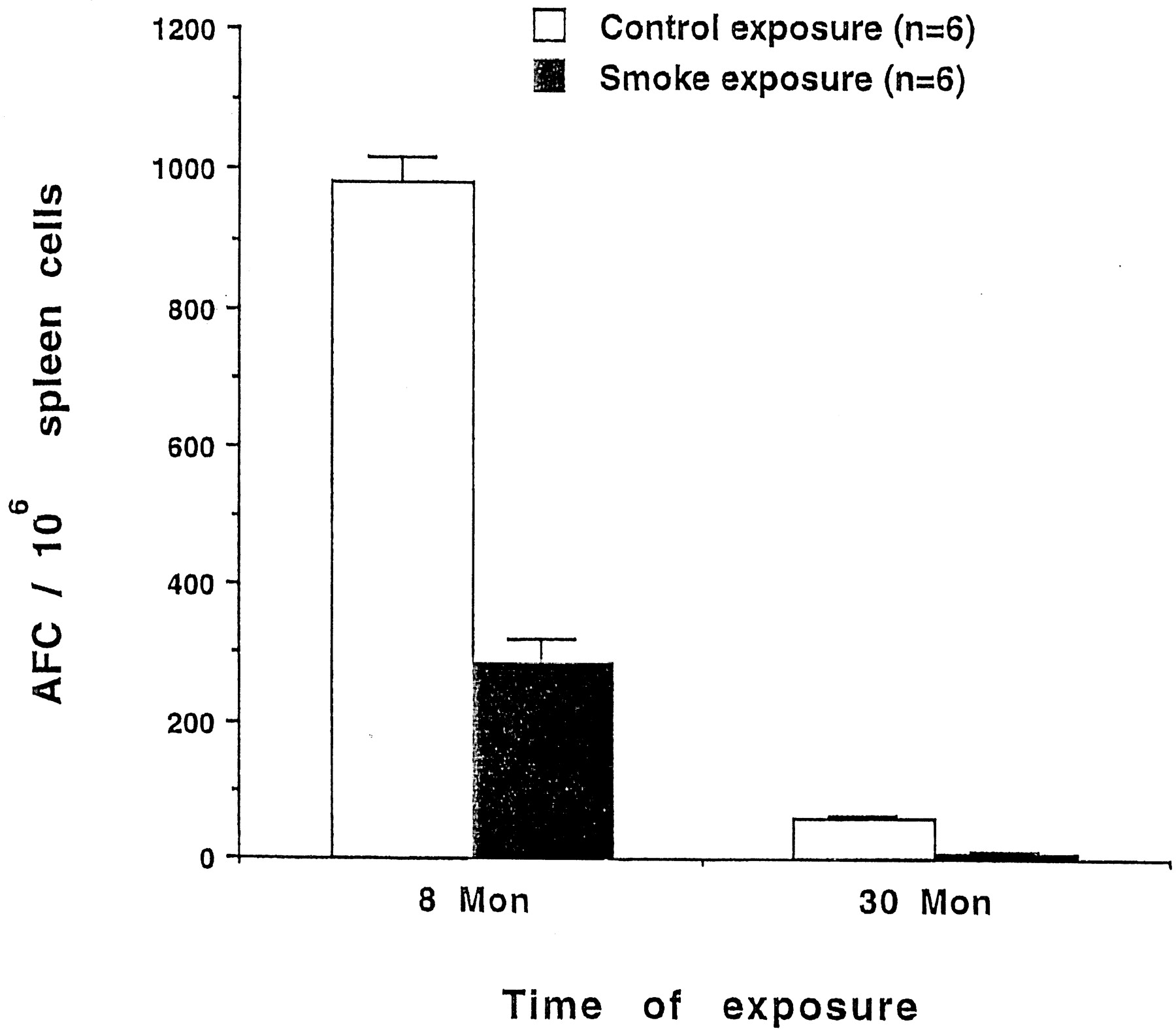
Nose-only exposure of rats twice per day to mainstream SM for >6 months results in a decreased AFC response (Sopori et al., 1989; Savage et al., 1991). To examine whether the whole-body exposure to diluted mainstream SM was effective in modulating the immune system, rats in the present study were exposed to SM or FA (CON) for 8 or 30 months. As reported previously (Savage et al., 1991), the percentages of T and B cells in the spleens of 8-month CON and SM animals were not significantly different (data not shown). The anti-SRBC AFC response of the spleen cells after 8- and 30-month exposures to SM was determined 4 days after SRBC immunization. As shown in Fig. 1, the anti-SRBC AFC response of SM rats exposed for 8 months was significantly lower than the CON animals in this group. CON animals in the 30-month group have very few antibody-producing cells as a result of aging, whereas SM animals in this group have an even lower AFC response. A significant reduction in the AFC response, however, was not observed in animals exposed to SM for <3 months (data not shown). Thus, long-term, whole-body exposure to SM suppresses the AFC response without significantly altering the total number or subpopulations of lymphocytes.

Chronic SM inhibits the AFC response to SRBC. Animals were exposed for 8 or 30 months to mainstream SM or FA and, 4 days before sacrifice, immunized with SRBC. Spleen cells were assayed for the AFC response. Assays were run in duplicates and the error bars represent standard deviation. Note the decreased AFC response in 30-month compared to 8-month CON animals.
Smoking Inhibits TCR-Mediated Proliferation.
Chronic smoking has been associated with decreased proliferative response to the T cell mitogens, Con A, and/or phytohemagglutinin (for review, see Sopori et al., 1994). Although both these mitogens are known to activate T cells through the TCR, in this experiment we used anti-CD3 to directly ligate the TCR. In rat spleen cells, this leads to the proliferation of T cells even in the absence of CD28 activation (Geng et al., 1995). As seen in Fig. 2, proliferation in response to anti-CD3 was significantly reduced in spleen cells from 8-month SM-exposed animals. Thus, chronic smoking affects the antigen-mediated activation of T cells.

Chronic smoking inhibits anti-CD3-induced spleen cell proliferation. The figure shows counts per minute ± standard deviation of [3H]thymidine incorporation of spleen cells in response to anti-CD3 from four 8-month SM-exposed and three CON animals, as described in Materials and Methods. Cultures were run in six-well replicates and, for background response, wells contained 5 μg/ml normal mouse IgG. Differences in the SM and CON groups were significant (P < .01).
Chronic Smoking Is Associated with Inhibition of TCR-Mediated Ca2+ Response.
A number of observations indicate that SM affects T cell function in humans and experimental animals (for review, see Sopori et al., 1994). To examine the possibility that chronic whole-body exposure to SM affects antigen-mediated signaling in T cells, spleen cells from 30-month CON and SM groups were treated with anti-TCR mAb followed by goat anti-mouse IgG (second antibody), and the [Ca2+]i was measured by spectrofluorometry. Results from a representative experiment (Fig.3) indicate that the ability of spleen cells to increase intracellular Ca2+ level after TCR ligation was significantly reduced after exposure to SM. Thus, chronic smoking may alter the antigen-mediated signaling in T cells.

SM inhibits Ca2+ mobilization in response to the TCR ligation. Spleen cells from CON or SM-treated animals were loaded with indo-1 and, at the indicated times (arrows), stimulated with mouse anti-rat αβ-TCR (α-TCR) followed by goat anti-mouse IgG as the second antibody. [Ca2+]i was computed as described in Materials and Methods.
SM Causes Tyrosine Phosphorylation of PLC-γ1.
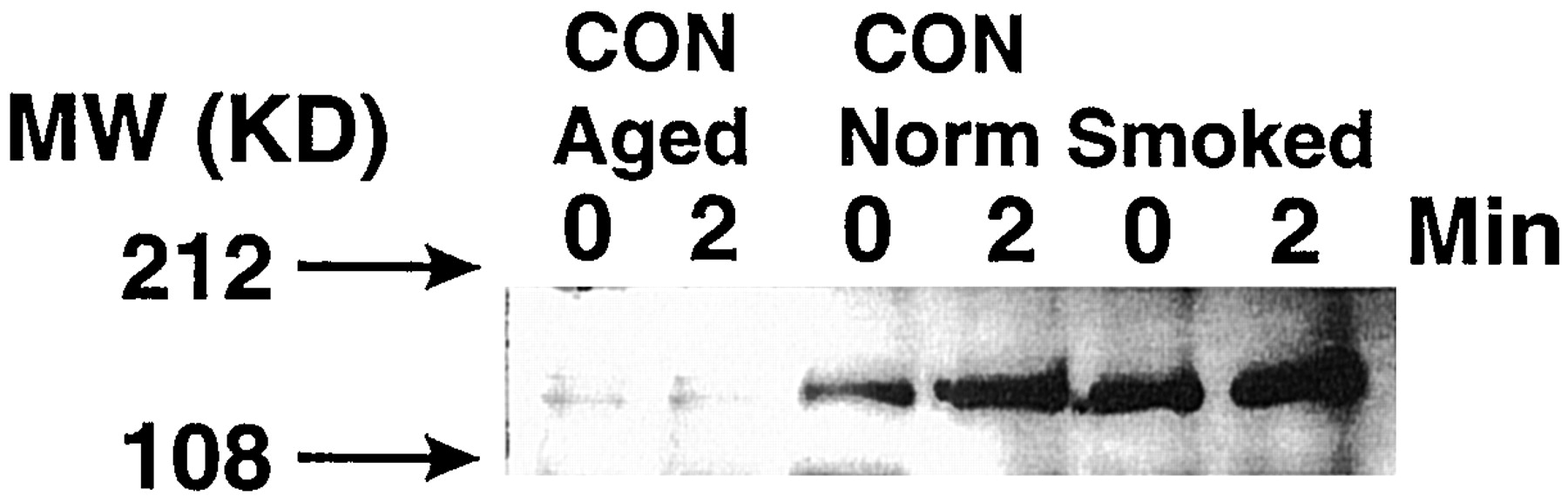
During the antigen-mediated activation of T cells, the increase in [Ca2+]i is dependent on the generation of IP3 (Weiss and Littman, 1994; Clapham, 1995). In the antigen-activated T cells, the formation of IP3 from phosphatidylinositol 4,5-bisphosphate is catalyzed by activated (tyrosine-phosphorylated) PLC-γ1 (Weiss and Littman, 1994). To ascertain whether the decreased Ca2+ response in 30-month SM T cells reflected the lack of activated PLC-γ1, lysates from spleen cells, before and after a 2-min treatment with anti-TCR antibody, were immunoprecipitated with anti-PLC-γ1 antibody. The immunoprecipitates were run on Western blots and probed with anti-PY or anti-PLC-γ1 mAb. Results from one of the five representative experiments (Fig. 4, lane 1) show that, in 30-month CON animals, the background level of PY-PLC-γ1 was substantially reduced, and anti-TCR treatment did not significantly increase the tyrosine phosphorylation (Fig. 4, lane 2). However, anti-TCR treatment of spleen cells from younger (8-month-old) CON animals results in strong tyrosine phosphorylation of PLC-γ1 (Fig. 4, lanes 3 and 4). These data suggest that aging affects both the basal and the anti-TCR-induced levels of PY-PLC-γ1 in CON animals. Interestingly, however, 8-month SM spleen cells showed strong tyrosine phosphorylation of PLC-γ1 even before the addition of anti-TCR (Fig.4, lane 5), and the anti-TCR treatment did not significantly alter the extent of this tyrosine phosphorylation (Fig. 4, lane 6). However, Western blots of anti-PLC-γ1 immunoprecipitates, probed with anti-PLC-γ1, did not show significant variations between the groups, indicating that SM affects only the tyrosine phosphorylation but not the total content of PLC-γ1. Thus, chronic exposure to mainstream SM constitutively activated tyrosine phosphorylating activity, and this enzyme activity was less susceptible to age-related changes.

SM causes tyrosine phosphorylation of PLC-γ1. Lysates were prepared from spleen cells before (time zero) or 2 min after the addition of anti-TCR antibody. Lysates were precipitated by anti-PLC-γ1 antibody, run on gels, and the Western blots probed with anti-PY antibody as described in Materials and Methods. Samples are from 30-month CON (lanes 1 and 2), 8-month CON (lanes 3 and 4), and 30-month SM animals (lanes 5 and 6).
SM Causes Chronic Activation of PTK Activity.
TCR engagement activates Src-like PTK activity leading to tyrosine phosphorylation of PLC-γ1 (Weiss and Littman, 1994). To determine whether the activation of PLC-γ1 in SM cells correlated with increased PTK activity, lysates from spleen cells from 30-month CON animals and SM animals were treated with anti-TCR for 0 or 2 min and run on Western blots. The blots were probed with anti-PY antibody. Data from one of the four experiments is shown in Fig. 5, and suggest that cells from SM-exposed rats expressed strong PTK activity even before anti-TCR treatment (time zero) and, unlike 30-month CON cells, this activity was not further enhanced by incubation with anti-TCR. These results indicate that chronic exposure to SM appears to cause a constitutive activation of the PTK activity, which was observed even in aged rats. It is therefore likely that tyrosine phosphorylation of PLC-γ1 seen in SM animals (Fig. 4) results from the constitutive activation of PTK activity in these animals.

Long-term exposure to SM constitutively activates PTKs. Western blot analysis of 30-month CON and SM lysates with and without the 2-min stimulation with anti-TCR antibody.
Chronic Exposure to SM or Nicotine Depletes IP3-Sensitive [Ca2+]i Stores.
If SM constitutively stimulates the PLC-γ1 activity, this should increase [Ca2+]i through IP3 by releasing Ca2+ from the endoplasmic IP3-sensitive Ca2+ stores and the stimulation of Ca2+ influx (Clapham, 1995). However, in spite of activated PLC-γ1, T cells from SM-exposed animals did not mobilize Ca2+ in response to TCR ligation (Fig. 3). It is possible that in SM cells, the constant presence of higher levels of IP3 depleted the IP3-sensitive Ca2+ stores in these cells. To evaluate the status of IP3-sensitive Ca2+ stores in SM cells, indo-1-labeled cells from 30-month SM and CON groups were suspended in Ca2+-free medium (to stop Ca2+ influx) and treated with thapsigargin to irreversibly empty IP3-sensitive Ca2+ stores (Thastrup et al., 1990; Razani-Boroujerdi et al., 1994). Results presented in Fig. 6A show that thapsigargin did not raise the [Ca2+]i in SM cells to levels seen with CON spleen cells. Similar results were obtained with cells from animals chronically (4 weeks) treated with nicotine (Fig.6B). These data suggest that chronic exposure to SM/nicotine depleted intracellular IP3-sensitive Ca2+ stores, resulting in the failure to mobilize Ca2+normally in these cells.

Effect of 30-month SM treatment on the levels of Ca2+ in the IP3-sensitive intracellular Ca2+stores. Indo-1-labeled 30-month CON and SM cells were incubated with thapsigargin (TPG) in Ca2+-free medium to release Ca2+ from the IP3-sensitive Ca2+ stores. A, 30-month SM-exposed and CON animals. B, 4-week nicotine-treated and CON animals.
Discussion
Previous results have shown that chronic exposure to mainstream SM affects T cell responses (for review, see Johnson et al., 1990; Sopori et al., 1994, 1998). We have previously reported that chronic exposure to nicotine causes T cell unresponsiveness (anergy) and appears to be related to impaired antigen-mediated signaling and suppression of intracellular Ca2+ response (Geng et al., 1996). Surprisingly, however, spleen cells from rats treated chronically with nicotine have significantly higher background intracellular levels of IP3 and are unable to move normally into the S phase of the cell cycle (Geng et al., 1995; 1996).
Protein phosphorylation is a primary post-translational mechanism for the regulation of essentially all cellular processes (Cohen, 1989). Stimulation of T cells through antigen-specific receptor by an antigen or antireceptor antibodies initiates a series of biochemical events that can result in profound effects, including T cell activation, tolerance, and/or differentiation (Sloan-Lancaster and Allen, 1996). TCR engagement activates a complex cascade of discrete signaling pathways. One of the earliest events after stimulation with an Ag is the activation of a number of PTKs, which leads to the activation of PLC-γ1 via a tyrosine phosphorylation-dependent mechanism (Weiss et al., 1991; Weiss and Littman, 1994). The activation of PLC-γ1 results in the hydrolysis of phosphatidylinositol 4,5-bisphosphate, yielding the second messenger IP3. It is well recognized that IP3 is responsible for the TCR-induced rapid and sustained increase in the intracellular Ca2+ ions (Berridge, 1993). The main finding of the present study is that the immunosuppression caused by chronic exposure to mainstream SM is associated with the impairment of antigen-mediated signaling in T cells.
The inability of SM spleen cells to mobilize Ca2+normally in response to TCR ligation suggests that smoking affects the antigen-induced signaling cascade proximal to the rise in [Ca2+]i. Our results show that in SM cells tyrosine phosphorylation of PLC-γ1 is not increased by TCR ligation. However, in T cells from both 8- and 30-month SM-exposed animals, the background (time zero) level of tyrosine phosphorylation of PLC-γ1 is much higher than in CON cells. In fact, there is no significant difference in the level of tyrosine phosphorylation of PLC-γ1 between the activated (anti-TCR-treated) CON and time zero SM cells. This difference in the basal level of PLC-γ1 cannot be attributed to differences in T cell numbers between CON and SM-exposed animals because the proportion of T and B cells in the two groups of animals was comparable. Moreover, the total PLC-γ1 was not significantly different in various experimental groups, suggesting that chronic SM exposure primarily stimulates tyrosine phosphorylating enzyme activity(ies). These results support the previous finding of increased resting IP3 levels in chronically nicotine-treated T cells (Geng et al., 1996), suggesting a constitutive activation of PLC-γ1 in T cells from nicotine- and SM-exposed animals. Interestingly, T cell clones, which are made anergic in vitro through “incomplete” stimulation, have significantly higher intracellular IP3 levels than control T cells (Gajewski et al., 1994,1995).
Because PLC-γ1 requires PTK activity to be tyrosine phosphorylated, it is possible that SM activates PTKs in T cells. Although we have not identified the PTK enzyme(s) that specifically tyrosine phosphorylates PLC-γ1, the Western blot analysis of the lysates from SM cells shows significantly higher PTK activity than CON cells, and correlates with increased tyrosine phosphorylation of PLC-γ1. Similar activation of PTK activity has been shown in T cells chronically treated with nicotine (Geng et al., 1996). In fact, in several experimental models of T cell anergy, anergized T cells have higher basal levels of IP3, activated (tyrosine-phosphorylated) PLC-γ1, and increased or aberrant PTK activity (Boussiotis et al., 1997; Faith et al., 1997; Trebak et al., 1998). Thus, constitutive activation of PTKs and PLC-γ1 may indicate T cell anergy and account for the unresponsiveness of T cells from SM animals.
In spite of higher levels of tyrosine-phosphorylated PLC-γ1 in SM cells compared with CON cells, the TCR activation of 30-month SM cells produces a significantly lower increase in [Ca2+]i. However, interestingly, in 30-month CON cells, a significant Ca2+ response is observed despite reduced activation of PLC-γ1. It is not clear whether the age-related reduced levels of activated PLC-γ1 are sufficient to trigger a near-normal Ca2+ response or whether the age-related damage in cell membranes permits exaggerated Ca2+ influx with lower levels of PLC-γ1. Presence of lipotropic compounds, including nicotine in SM, may stabilize the plasma membrane.
In SM cells, activation of PLC-γ1 does not translate into significantly higher background levels of [Ca2+]i, a condition also observed in animals chronically treated with nicotine (Geng et al., 1996). Therefore, in T cells exposed to SM, the IP3 receptors on the [Ca2+]i stores are either insensitive to the elevated levels of IP3 or the stores do not contain sufficient releasable Ca2+ to significantly raise [Ca2+]i. To ascertain whether IP3-sensitive Ca2+ stores contain IP3-releasable Ca2+, SM spleen cells were treated with thapsigargin in Ca2+-free medium. Thapsigargin, a sesquiterpene lactone, inhibits the endoplasmic reticulum Ca2+-ATPases (Thastrup et al., 1990;Inesi and Sagara, 1992). This results in an uncompensated Ca2+ leak, depleting primarily the IP3-sensitive Ca2+ stores independently of IP3 formation (Jackson et al., 1988). Unlike CON cells, the amount of Ca2+ released from SM- and nicotine-treated cells by thapsigargin is significantly reduced, indicating that SM and nicotine deplete the IP3-sensitive Ca2+ stores in spleen cells. It is likely that as a result of activation of PLC-γ1, chronic presence of higher IP3 levels cause depletion of these Ca2+ stores. Recent reports suggest that these stores are critical for the communication between the cytoplasm and the nucleus (Greber and Gerace, 1995; Perez-Terzic et al., 1997) and affect various physiological and pathophysiological phenomena (Pesty et al., 1998; Takei et al., 1998; Wilcox et al., 1998), including the migration of G0/G1 cells into the S phase of the cell cycle (Takuwa et al., 1995). Although our results do not rule out the possibility that SM affects cell types other than T cells in the immune system, they do suggest that nicotine is a major immunosuppressive component in SM and may cause T cell anergy through constitutive activation of PTKs and the depletion of IP3-sensitive Ca2+ stores in T cells.
Acknowledgments
We thank Dolores Esparza, Vicki White, and the Institute's Exposure Operations and Necropsy units for help with animal exposures and necropsy, and Paula Bradley for assistance with preparation of the manuscript. This work was performed through Cooperative Agreement DE-FC04-96AC76406 with the U.S. Department of Energy in facilities fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International.
Footnotes
-
Send reprint requests to: Mohan Sopori, Ph.D., Pathophysiology Division, Box 5890, Lovelace Respiratory Research Institute, Albuquerque, NM 87185. E-mail: msopori{at}lrri.org
-
↵1 This work was supported in part by Grant DA04208 from the National Institute of Drug Abuse.
- Abbreviations:
- SM
- cigarette smoke
- AFC
- antibody-forming cell
- TRC
- T cell antigen receptor
- SRBC
- sheep red blood cells
- PTK
- protein tyrosine kinase
- PLC
- phospholipase C
- IP3
- inositol-1,4,5-trisphosphate
- PY
- phosphotyrosine
- TPM
- total particulate matter
- CON
- control
- FA
- filtered air
- mAb
- monoclonal antibody
- Received May 18, 1999.
- Accepted December 6, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}