


Abstract
We investigated the cytochrome P-450-dependent metabolism of 1,1-dichloroethylene (DCE) by human lung and liver microsomes and compared the results from analogous experiments in mice. Metabolites were identified by HPLC analysis of their glutathione conjugates and/or hydrolyzed products and were detected by using [14C]DCE. The role of human CYP2E1 in the metabolic reactions was examined by comparing p-nitrophenol hydroxylase activities with levels of metabolites formed and by using the CYP2E1-selective inhibitor diallyl sulfone. The major products formed in microsomal incubations containing NADPH were the DCE-epoxide-derived glutathione conjugates 2-(S-glutathionyl)acetyl glutathione and 2-S-glutathionyl acetate. Lower levels of the acetal of 2,2-dichloroacetaldehyde were also detected. In lung samples from eight patients, the amounts of epoxide-derived conjugates formed ranged from 15.6 ± 4.23 to 34.9 ± 12.75 pmol/mg protein/min. The levels in murine lung were higher at 40.0 ± 3.8 pmol/mg protein/min. In liver samples from five patients, conjugate levels ranged from 46.5 ± 8.3 to 240.0 ± 10.5 pmol/mg protein/min, whereas levels in murine liver were 83.0 ± 6.2 pmol/mg protein/min. Conjugate levels formed in human liver correlated with the relative levels of p-nitrophenol hydroxylase activity present, but this relationship was equivocal in human lung. Diallyl sulfone inhibited the formation of the glutathione conjugates (20–65%) in liver samples from all four patients, whereas only one of five human lung samples exhibited this inhibition (27%). These results demonstrated that the DCE-epoxide is a major metabolite formed by human microsomes and is mediated by CYP2E1 in liver and in some individuals in lung.
1,1-Dichloroethylene (DCE), a chemical used in the manufacture of plastics and a widespread water contaminant, causes pulmonary and hepatocellular injury in experimental animals (Coleman et al., 1976; Forkert and Reynolds, 1982;Forkert et al., 1986). Previous studies have determined that the mechanism of DCE-induced injury involves cytochrome P-450-catalyzed metabolism to reactive intermediates (Okine and Gram, 1986; Forkert et al., 1987). Exposure to DCE causes dose-dependent increases in covalent binding and concomitant decreases in cellular glutathione (GSH) in both lung and liver (Forkert and Moussa, 1991; Moussa and Forkert, 1992). The severity of tissue injury correlates with the extent of binding and parallels the decline in GSH, suggesting that binding of reactive intermediates to critical macromolecules mediates the cytotoxicity. Moreover, the hepatotoxic effects of DCE were exacerbated by procedures that lower GSH levels (Jaeger et al., 1973, 1974; McKenna et al., 1978;Anderson et al., 1980). Hence, conjugation of DCE metabolites with GSH represents a detoxication reaction.
Previous studies in rat liver have identified the primary metabolites formed in microsomal incubations from DCE as 2,2-dichloroacetaldehyde, DCE-epoxide, and 2-chloroacetyl chloride (Lieber and Guengerich, 1983;Costa and Ivanetich, 1984; Liebler et al., 1985, 1988). In our recent studies in murine lung and liver, the major metabolic products formed in microsomal incubations supplemented with GSH were the conjugates 2-(S-glutathionyl) acetyl glutathione [B] and 2-S-glutathionyl acetate [C] (Dowsley et al., 1995, 1996). These products are believed to arise from conjugation of GSH with the DCE-epoxide (Dowsley et al., 1995) (Fig.1). The acetal of 2,2-dichloroacetaldehyde was detectable in lung microsomal incubations. However, S-(2,2-dichloro-1-hydroxy)ethyl glutathione ([A]), the product of GSH conjugation with 2,2-dichloroacetaldehyde, was not detected in our experiments, suggesting that this reaction is unlikely to contribute to GSH depletion or to be responsible for DCE-induced cytotoxicity (Dowsley et al., 1995). This assumption is consistent with results from studies that showed that isolated hepatocytes incubated with 2,2-dichloroacetaldehyde sustained less toxic effects than incubation with the parent compound (Kainz et al., 1993). S-(2-chloroacetyl)-glutathione ([D]) and chloroacetic acid, the GSH-conjugated and hydrolysis products of 2-chloroacetyl chloride, respectively, were formed at minimal levels in both liver and lung microsomal incubations (Dowsley et al., 1995,1996). Hence, the DCE-epoxide is the major metabolite formed from DCE in vitro and is an efficient scavenger of GSH, suggesting that it is the most plausible candidate for mediating the toxic effects of DCE. This is supported by results from several studies that showed a strong correlation between susceptibility to lung or liver injury in rodents and the rate of production of the DCE-epoxide (Dowsley et al., 1995;Forkert et al., 1996a, b). For example, mice are more susceptible than rats to DCE-induced injury. The LD50 value for an oral dose of DCE is 7-fold lower in mice than in rats (Jones and Hathway, 1978). Our studies have also shown that the DCE-epoxide is produced in microsomal incubations at a level that is 6-fold higher in mice than in rats (Dowsley et al., 1995). These data supported the proposal that the DCE-epoxide may be responsible for DCE-induced toxicity by depleting GSH and binding to cellular macromolecules.

Scheme of the proposed pathway of DCE metabolism.
We identified CYP2E1 as a major cytochrome P-450 isozyme involved in the metabolism of DCE in murine lung and liver (Dowsley et al., 1995,1996; Forkert et al., 1996a,b). Our previous studies in mice indicated that the degree of DCE-induced Clara cell cytotoxicity is linked to magnitudes of CYP2E1-dependent bioactivation of DCE to the epoxide (Forkert et al., 1996a). Moreover, treatment of mice with diallyl sulfone (DASO2) inhibited CYP2E1 and protected against the Clara cell damage induced by DCE (Forkert et al., 1996b). The expression of CYP2E1 in human lung and liver (Wrighton et al., 1986; Ekström et al., 1989; Wheeler et al., 1992; Lucas et al., 1993) has raised the possibility that DCE exposure results in generation of DCE-epoxide in these tissues. It is of importance to determine the capacity of human liver and lung CYP2E1 to metabolize DCE to reactive intermediates, including the DCE-epoxide, so as to be able to better assess the potential risk to humans exposed to DCE and other low-molecular-weight chemicals that are substrates for CYP2E1. The objectives of the present study were to investigate the P-450-mediated metabolism of DCE by human lung and liver microsomes. We also compared the metabolic profiles with those in murine lung and liver to evaluate relevance of the murine model for future studies with DCE. The role that human CYP2E1 plays in DCE bioactivation is examined through preincubation of human microsomes with DASO2, a CYP2E1-specific inhibitor.
Materials and Methods
Chemicals and Reagents.
1,1-Dichloroethylene (>99% purity), phosphoric acid (85% v/v), and GSH were purchased from Aldrich Chemical Co. (Montreal, Quebec, Canada). Glucose-6-phosphate and glucose-6-phosphate dehydrogenase were purchased from Sigma Chemical Co. (St. Louis, MO). NADP+ was obtained from BDH Chemical Co. (Toronto, Ontario, Canada). DASO2 was obtained from Parish Chemical Co. (Orem, UT). [14C]DCE (99% pure by GLC, specific activity 11.3 nCi/nmol) was obtained from Amersham Corp. (Arlington Heights, IL) and was diluted to 300 μCi/ml for our experiments. The DCE-epoxide-derived GSH conjugates [B] and [C] were synthesized as described previously (Dowsley et al., 1995, 1996) and were used as standards for metabolite identification. Other chemicals and reagents were purchased from standard suppliers.
Animal Treatment.
Female CD-1 mice, weighing 20 to 25 g, were obtained from Charles River Canada (St. Constant, Quebec, Canada). The mice were kept in an animal facility maintained on a 12-h light/dark cycle and were provided with food (Purina Rodent Chow, Ralston Purina International, Strathroy, Ontario, Canada) and water on an ad libitum basis. The mice were housed for 7 days after arrival to acclimatize to laboratory conditions and then were sacrificed by cervical dislocation.
Preparation of Microsomes.
Human lung tissue (10–50 g) was obtained from Kingston General Hospital (Kingston, Ontario, Canada) from consenting patients undergoing surgical lobectomies. The protocol for the studies in human lung was approved by Queen’s University Human Ethics Committee. Tissues distant from the primary lesions were surgically excised, placed on ice, and immediately transferred to a biohazard facility. Human liver tissue was obtained from the Department of Surgery (University of Arizona, Tucson, AZ). The liver samples were rapidly frozen in liquid nitrogen, shipped on dry ice, and stored at −80°C. The liver tissue was thawed in a laminar flow hood before homogenizing.
For preparation of murine microsomes, lungs from 25 mice or livers from 10 mice were pooled. The human tissues were not pooled but were retained as individual samples. The tissues were homogenized in 4 volumes of cold phosphate-buffered KCl (100 mM K2HPO4, 1.15% KCl, 1.15 mM EDTA, pH 7.4), and microsomes were prepared as described previously (Forkert et al., 1987; Matsubara et al., 1987). Microsomal pellets were resuspended in phosphate-buffered KCl in a volume of 0.2 ml/g tissue weight (mouse) or 0.1 ml/g tissue weight (human). Aliquots of the microsomal suspension were dispensed into Eppendorf tubes, frozen in liquid nitrogen, and stored at −70°C. Protein concentrations of the microsomal samples were determined by the method of Lowry et al. (1951)using BSA as the standard.
Microsomal Incubations.
Microsomal incubations were performed as described in our previous studies (Dowsley et al., 1995,1996). Reactions were performed at 25°C for 30 min in a total volume of 0.5 ml of phosphate buffer containing 1.5 mM EDTA. Incubation mixtures contained 5.0 mg/ml microsomal protein, [14C]DCE (2 mM, specific activity 7.5 nCi/nmol), 10.0 mM MgCl2, 15 mM GSH, and an NADPH-generating system (15.0 mM glucose-6-phosphate, 2 units/ml glucose-6-phosphate dehydrogenase, and 0.8 mM NADP+). The reactions were initiated with DCE and were terminated by chilling the samples in an ice bath. The microsomal proteins were precipitated with perchloric acid (70%, 50 μl/ml) and centrifugation. Both the protein concentrations and the incubation time were within the linear range for formation of metabolites.
Preincubation of Microsomes with DASO2
The involvement of CYP2E1 in the formation of DCE metabolites in human lung and liver incubations was examined through preincubation of the microsomes with the CYP2E1-selective inhibitor DASO2 (Brady et al., 1991; Forkert et al., 1996). Previous studies have shown that DASO2 inhibits the CYP2E1 enzyme in both lung (Forkert et al., 1996) and liver (Brady et al., 1991). Microsomes were preincubated with 20 mM DASO2 in the presence of an NADPH-generating system for 30 min at 37°C. The samples were then washed and centrifuged at 105,000g for 30 min to recover the microsomes. The microsomal pellet was homogenized and resuspended in 100 mM phosphate-buffered KCl, pH 7.4. The conditions for DCE incubation were the same as those described for the microsomal incubations with DCE. Control incubations consisted of incubations performed without DASO2.
p-Nitrophenol Hydroxylase Activity.
Microsomes from human and murine liver and lung were resuspended in 100 mM K2HPO4 buffer, pH 6.8, using 3.0 mg/ml of microsomal protein. p-Nitrophenol (PNP) hydroxylase activity was used as a selective enzyme marker for CYP2E1 and was determined according to the method of Koop (1986). Incubations were performed in a total volume of 2 ml and contained 3.0 mg/ml protein and an NADPH-generating system (7.5 mm glucose-6-phosphate, 5.0 mM MgCl2, 2 U of glucose-6-phosphate dehydrogenase, and 0.4 mM NADP+). The reaction mixtures were preincubated for 3 min at 37°C; subsequently, PNP in dimethyl sulfoxide (4 μl, 200 μM) was added and allowed to react for an additional 10 min. The reaction was terminated by cooling the samples in an ice bath. The microsomal proteins were precipitated by the addition of perchloric acid (70%, 50 μl/ml) and centrifugation. The supernatant was obtained, NaOH (100 μl, 0.9 M) was added to a 1.0-ml aliquot, and the formation of 4-nitrocatechol was determined spectrophotometrically at 546 nm. Levels of 4-nitrocatechol formed were determined by relating absorbance to a standard calibration curve of known amounts of 4-nitrocatechol. The assay was carried out under linear conditions of time and protein concentrations.
Metabolite Identification.
Synthesized standards of DCE metabolites were characterized by reversed-phase HPLC analysis using a C18 column (5 μm, 4.6 × 250 mm, Microsorb-MV; Rainin Instruments Co., Inc., Woburn, MA). The mobile phase consisted of 0.2% H3PO4, pH 2.0, and was run isocratically at a flow rate of 1.0 ml/min. The column effluent was monitored at 200 nm. For analysis of microsomal incubations, 100-μl aliquots of the supernatant from each microsomal incubation were injected onto the HPLC. Fractions (0.25 ml) of the column effluent were collected, and levels of radioactivity were determined by liquid scintillation spectroscopy (Beckman model LS 7000 liquid scintillation counter). Identification of the metabolites was achieved through radiochemical detection of the fractions eluting from the column with retention times corresponding to synthesized standards. Concentrations of the metabolites were estimated by summing the radioactivity associated with each peak and converting the data to picomolar amounts using the specific activity of the [14C]DCE.
Instrumentation.
HPLC experiments were conducted on a Beckman System Gold Programmable Solvent Module 126 HPLC with a Beckman System Gold Module 168 UV detector. UV spectra for all other assays were determined with a Hewlett Packard model 8452 diode array UV spectrophotometer.
Statistical Analysis.
Data are expressed as mean ± S. D. Statistical analysis was performed with one-way ANOVA followed by the Tukey test to identify significant differences between experimental groups (p < .05).
Results
Metabolism of DCE by Human Lung and Liver Microsomes.
Human lung tissue was obtained from patients undergoing surgical lobectomies. Of the eight patients examined, six were women and two were men. The age of these patients ranged from 56 to 73 years. All were smokers or former smokers. Human liver tissue was procured from organ transplant donors. Of the nine patients studied, four were women and five were men. The age of these patients ranged from 1 to 69 years. These patients did not have a history of alcohol consumption. The smoking histories of these patients were not known.
In the human lung microsomal incubations, three major peaks were observed in the radiochromatograms (Fig.2A). These were identified by coelution with authentic standards as the acetal of 2,2-dichloroacetaldehyde and conjugates [B] and [C] in order of increasing elution time. These peaks were not observed when NADP+ was omitted from the incubation mixtures. In the absence of GSH, conjugates [B] and [C] were also not present, whereas two peaks eluting at 3.0 and 4.0 min appeared (data not shown). These peaks corresponded to the retention times of formaldehyde and glycolic acid, respectively, and are the hydrolysis products of the DCE-epoxide (Liebler et al., 1985). However, these were low in comparison with the levels of conjugates [B] and [C] formed. Microsomes from eight human lung subjects (HL-1 to HL-8) were used to assess the metabolism of DCE in this tissue. The rate of formation of the DCE-epoxide-derived GSH conjugates [B] and [C] ranged from 15.6 ± 4.2 (HL-8) to 34.9 ± 12.7 (HL-5) pmol/mg protein/min (Table 1). Levels of the acetal of 2,2-dichloroacetaldehyde were lower and were less variable between microsomes from the different patients, with a mean rate of formation of 5.2 ± 0.9 pmol/mg protein/min. HPLC analysis of human liver microsomal incubations from five patients (HLv-1 to HLv-5) yielded similar metabolites to those detected in the human lung microsomal incubations but at higher levels (Fig. 2B). There also was considerable variability in the extents to which the DCE-epoxide-derived GSH conjugates were formed in the liver microsomes from the five patients (Table 2). The total amount of [B] and [C] ranged from 46.5 ± 8.3 (HLv-1) to 240.0 ± 10.5 (HLv-2) pmol/mg protein/min; the highest level of conjugates produced was about 5-fold of that at the lowest level. The mean rate of formation of the acetal in the human liver samples (8.0 ± 1.5 pmol/mg protein/min) was only slightly higher than those detected in the human lung samples and was less variable between the patients. Compared with the formation of the GSH conjugates, the amounts of acetal produced in the liver were relatively low (Table 2; Fig. 2B).

HPLC analysis of human lung (A) and liver (B) microsomal incubations. Reactions were conducted in a total volume of 0.5 ml of 50 mM phosphate buffer, pH 7.4, at 25°C for 30 min. The reaction mixtures contained 5.0 mg/ml microsomal protein, 2 mM [14C]DCE (specific activity 7.5 nCi/nmol), 15.0 mM GSH, and an NADPH-generating system. Microsomal proteins were precipitated, and the supernatant was subjected to HPLC analysis. Radioactivity was determined in fractions (0.25 ml) eluting from the column. The peaks were identified as the GSH conjugates [B] and [C] and the acetal of 2,2-dichloroacetaldehyde.
Formation of DCE metabolites in human and murine lung microsomal incubations
Formation of DCE metabolites in human and murine liver microsomal incubations
Comparison of DCE Metabolism and CYP2E1 Activity in Mice and Humans.
We compared the rates of formation of the DCE-epoxide-derived conjugates and the acetal of 2,2-dichloroacetaldehyde in incubations with murine lung and liver microsomes with the amounts formed in those with human lung and liver microsomes. The specific activity of the [14C]-DCE used was the same in all of the microsomal incubations. The mean rate of formation of the GSH conjugates in murine lung microsomal incubations was 1.7-fold higher than the level in the human lung samples from the eight patients investigated (Table 1). However, lung samples from patients HL-1, HL-5, and HL-6 formed the conjugates at rates that were 74%, 70%, and 87% of the levels found in murine lung, respectively. The mean level of GSH conjugates formed by liver microsomes from the five patients was 1.8-fold higher than those formed by murine liver microsomes (Table 2). Levels in murine liver were higher than in samples from livers of patients HLv-1 and HLv-4 but lower than those from patients HLv-2, HLv-3, and HLv-5; therefore, in the microsomal samples examined here, murine liver microsomes formed levels of GSH conjugates that were in the midrange of the rates detected in the human liver incubations. The acetal was formed in both murine lung and liver microsomal incubations, but the levels were low compared with the amounts of GSH conjugates detected (Tables 1 and 2). However, acetal levels were slightly higher in microsomes from mice than in those from humans (Tables 1 and 2).
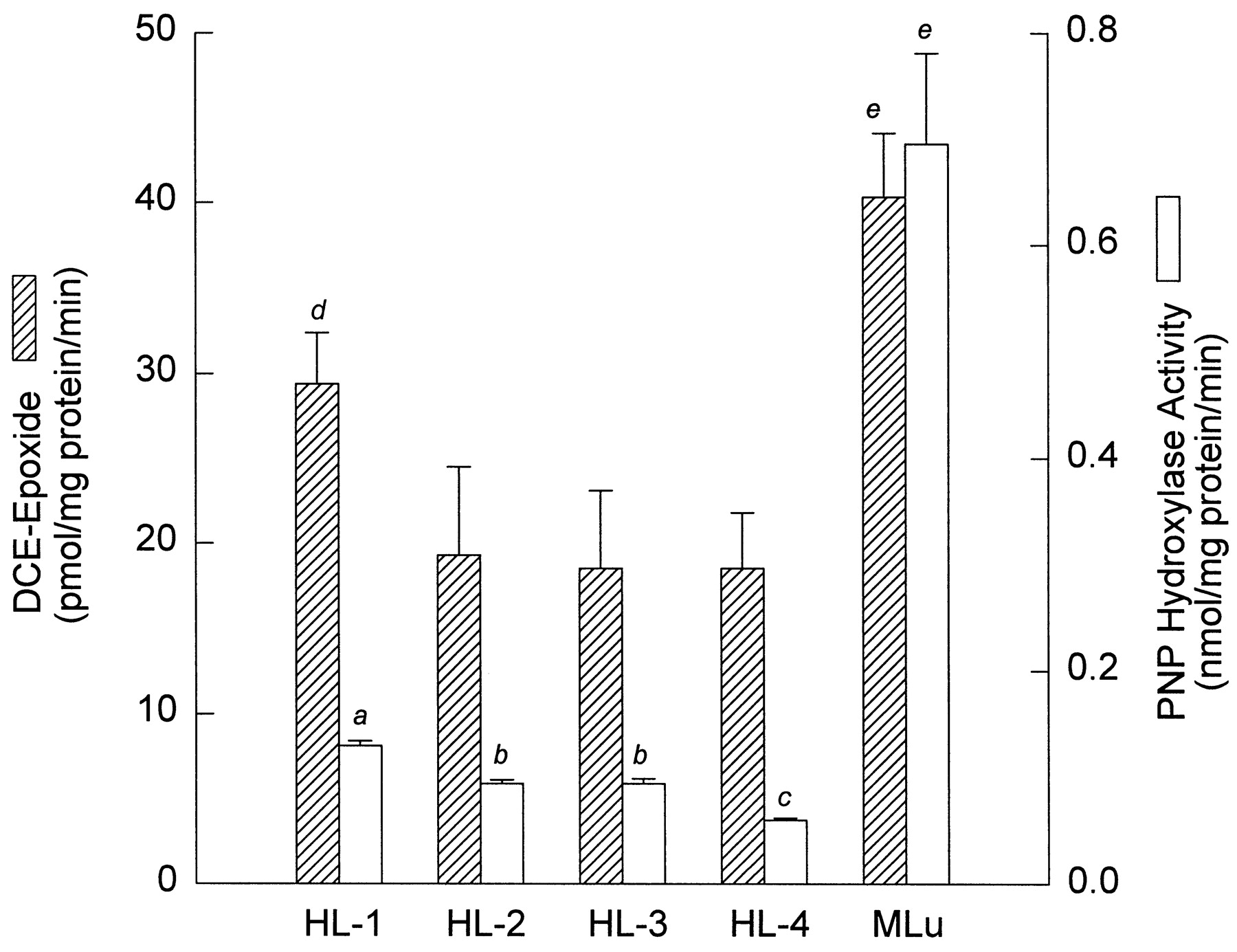
Our previous studies established an important role for CYP2E1 in the metabolism of DCE to the epoxide in murine liver and lung microsomes (Dowsley et al., 1995, 1996). In the present investigation, we measured PNP hydroxylase activities of microsomal samples from the lungs of four patients (HL-1 to HL-4) and the livers of four patients (HLv-1 to HLv-3 and HLv-9) and compared the levels with those obtained in lungs and livers of mice. The results are summarized in Figs.3 and 4. Activities of lung PNP hydroxylase from the four patients had a mean rate of 0.10 ± 0.03 nmol/mg protein/min. The mean rate in murine lung microsomes (0.70 ± 0.08 nmol/mg/min) was about 7-fold higher than those of human lung (Fig. 3). The four human liver samples showed a large range in the amounts of hydroxylase activity. Patient HLv-9 had the lowest level (1.26 ± 0.05 nmol/mg protein/min), whereas the quantities in HLv-2 (2.59 ± 0.07 nmol/mg protein/min) and HLv-3 (2.87 ± 0.07 nmol/mg protein/min) were similar to each other and were both higher. In mice, the hydroxylase activity in liver microsomes was significantly higher than that in lung microsomes (Figs. 3 and 4) but significantly lower than that detected in liver microsomes from patients HLv-2 and HLv-3 (Fig. 4).

Formation of DCE-epoxide and PNP hydroxylase activity in microsomes of human (HL) and murine (MLu) lung. Levels of the epoxide were estimated from the total levels of [B] and [C]. Data are expressed as the mean ± S.D. of triplicate determinations for each human lung sample, whereas values for murine lung are derived from triplicate determinations from each of three separate microsomal preparations. Details of the microsomal incubations with DCE and the measurements for PNP hydroxylase activity are described inMaterials and Methods. aSignificantly different from HL-2, HL-3, HL-4, and MLu (p < .05). bSignificantly different from HL-1, HL-4, and MLu (p < .05). cSignificantly different from HL-1, HL-2, HL-3, and MLu (p < .05).dSignificantly different from HL-3 and HL-4 (p < .05). eSignificantly different from all human lung samples (p < .05).

Formation of DCE-epoxide and PNP hydroxylase activity in microsomes from human (HLv) and murine (MLv) liver. Levels of the epoxide were estimated from the total levels of [B] and [C]. Data are expressed as the mean ± S.D. of triplicate determinations for each human lung sample, whereas values for murine lung are derived from triplicate determinations from each of three separate microsomal preparations. Details of the microsomal incubations with DCE and the measurements for PNP hydroxylase activity are described inMaterials and Methods. *Significantly different from HLv-1, HLv-9, and MLv (p < .05). †Significantly different from HLv-1, HLv-9, and MLv.
Effects of DASO2 on Formation of DCE-EpoxideDerived GSH Conjugates in Human Lung and Liver Microsomes.
We observed a relationship between epoxide-derived GSH conjugate formation and levels of PNP hydroxylase activity in microsomal incubations in murine lung and liver (Figs. 3 and 4). These findings prompted us to confirm the role of CYP2E1 in catalyzing the formation of the [B] and [C] conjugates in human liver and lung incubations. We measured the effect of DASO2, a potent CYP2E1-specific inhibitor, on the production of [B] and [C] in human lung and liver microsomes. Five human lung samples and four human liver samples were examined. In the human lung experiments, samples from only one of five patients (HL-1) exhibited inhibition (27%) of formation of the epoxide-derived GSH conjugates in the microsomal incubations (Table3). In contrast to the lung, DASO2 caused significant inhibition of conjugate formation in all of the human liver microsomal samples (Table 4).
Effects of DASO2 on DCE-epoxide formation in human lung and liver microsomes
Discussion
Considerable data have accumulated to implicate the DCE-epoxide as the ultimate toxic species involved in the hepatotoxic and pneumotoxic effects of DCE in mice (Dowsley et al., 1995, 1996; Forkert et al., 1996a,b). The DCE-epoxide-derived GSH conjugates [B] and [C] are the major products formed by murine liver and lung microsomes (Dowsley et al., 1995, 1996). Previous studies have identified species-, sex-, and age-dependent differences in formation of the epoxide, as assessed by levels of [B] and [C] formed (Jones and Hathway, 1978; Dowsley et al., 1995; Forkert et al., 1996a,b). The findings showed a strong relationship between the rate of production of the DCE-epoxide and susceptibility to DCE-induced injury. Here, we extended the findings from this previous work in experimental animals to studies in the human, and investigated the capacity of human lung and liver microsomes to bioactivate DCE to reactive intermediates and to determine whether the DCE-epoxide is a major product formed.
Our results demonstrated that human liver microsomes possess a strong capacity to metabolize DCE to the epoxide-derived conjugates [B] and [C] (Fig. 2; Table 2). The major products formed were [B] and [C], whereas the acetal of 2,2-dichloroacetaldehyde was detected at low levels (Table 2). There was considerable variation, spanning a 5-fold range, in the levels of the GSH conjugates generated in liver microsomes from the different patients. Liver microsomes from three patients (HLv-2, HLv-3, and HLv-5) metabolized DCE to the epoxide-derived conjugates at levels that were 2.5- to 3-fold higher than that in murine liver microsomes. These data suggested that humans exposed to DCE may be at risk and that some individuals may sustain hepatotoxic effects greater than those seen in the mouse. Importantly, this risk factor in humans will also depend on individual differences in cellular GSH levels for detoxication. GlutathioneS-transferase activity in humans probably will not play an important role because the DCE-epoxide is highly reactive toward GSH nonenzymatically (Liebler et al., 1985). We previously determined that production of the DCE-epoxide-derived GSH conjugates in liver microsomes was 6-fold higher in mice than in rats (Dowsley et al., 1995). Because conjugate formation in the human liver experiments described herein were within the range of the levels in murine liver, it appears that the mouse is a better model than the rat for assessing human risk to DCE-induced hepatotoxicity.
We also detected formation of DCE metabolites in microsomes from the eight human lung samples. As was found in the human liver experiments, [B] and [C] were the major products detected, indicating that the DCE-epoxide is also the major metabolite formed in human lung microsomes (Fig. 2, Table 1). The acetal of 2,2-dichloroacetaldehyde was also detected and amounted to only about 25% of the total amount of [B] and [C] formed. The relative level of acetal was lower in human liver microsomes and was formed at about 7% of the level of the epoxide-derived GSH conjugates (Table 2). The relatively higher levels of the acetal in human lung versus human liver are consistent with findings obtained in murine lung in previous studies (Dowsley et al., 1996). Nevertheless, the quantities of acetal produced were low in both human lung and liver, suggesting that DCE epoxidation is the preferential route of metabolism. The toxicological significance of this finding is unclear, although most of the available evidence indicated that 2,2-dichloroacetaldehyde is not an important metabolite in the DCE bioactivation pathway (Kainz et al., 1993; Dowsley et al., 1995). The eight human lung microsomal samples metabolized DCE to the epoxide-derived GSH conjugates at levels that spanned more than a 2-fold range (Table 1). The mean level was about 50% of the amount formed in lung microsomes from mice. This would suggest that susceptibility to DCE-induced lung injury is likely to be less severe in humans than in mice. However, the levels of epoxide formed in human lung microsomes were variable, suggesting that individuals capable of generating higher epoxide levels may be more vulnerable to DCE-induced pneumotoxicity.
Our previous studies have established a role for CYP2E1 in bioactivation and the ensuing toxicity of DCE in murine lung and liver (Lee and Forkert, 1994, 1995; Dowsley et al., 1995, 1996; Forkert et al., 1996a,b). Here, we evaluated the role of human CYP2E1 in DCE metabolism by comparing microsomal levels of PNP hydroxylation with the total amount of [B] and [C] formed. The large variations in hydroxylation in human liver were reflected in corresponding levels of [B] and [C] formed (Fig. 4), suggesting that quantities of the DCE-epoxide-derived GSH conjugates generated could be predicted from hydroxylase activity levels. In addition, preincubation with DASO2, which has been shown previously to selectively inhibit CYP2E1 (Forkert et al., 1996b), caused a reduction of 20 to 65% in the formation of the GSH conjugates in human liver microsomes (Table 3). These findings strongly suggested that human liver CYP2E1 catalyzes the formation of the DCE-epoxide. Hence, the mechanism of DCE metabolism in human liver resembles that delineated for murine liver, and this suggested that the mouse is an appropriate model for human studies of the chloroethylene. Thus, variations in CYP2E1 levels are likely to be manifested in the rate of formation of the epoxide. Interestingly, levels of human liver CYP2E1 can vary at least 50-fold among individuals (Wrighton et al., 1986; Lucas et al., 1993). This variability may reflect the existence of genetic polymorphisms (Hayashi et al., 1991) and/or exposure to inducing agents such as acetone and ethanol (Johansson et al., 1988, 1990; Badger et al., 1993; Lucas et al., 1993; Takahashi et al., 1993). Alcoholics or individuals exposed to CYP2E1-inducing agents may be at greater risk to the hepatotoxic effects of DCE due to greater production of the epoxide. Conversely, exposure to CYP2E1 inhibitors, such as the garlic derivative DASO2, may lower DCE activation as well as the associated risk.
Although we observed substantial cytochrome P-450-mediated formation of the DCE-epoxide in our human lung microsomal incubations, it did not appear that CYP2E1 was as important in this tissue as in human liver. The formation of the DCE-epoxide-derived GSH conjugates [B] and [C] detected in human lung microsomes from individual patients did not correspond to the levels of PNP hydroxylase activity found in the same samples. Although the production of [B] and [C] in human lung was relatively substantial and amounted to about 50% of the level in murine lung, the levels of PNP hydroxylase activity were about 7-fold lower (Fig. 3). In addition, preincubation of microsomes with high concentrations of DASO2 was effective in inhibiting the formation of [B] and [C] in only one of five human lung microsomal samples (Table 3). Interestingly, the lung sample from patient HL-1 exhibited 27% inhibition in the levels of [B] and [C] formed after preincubation with DASO2, but it also had the highest levels of CYP2E1 and GSH conjugates formed of the four samples examined (Fig. 3). Nevertheless, the magnitude of CYP2E1 inhibition by DASO2 was relatively low, suggesting that this P-450 does not have an important role in the bioactivation of DCE in human lung. These data indicated that the metabolism of DCE by human lung CYP2E1 does not parallel that by murine lung CYP2E1, a P-450 that has a substantial role in DCE activation. This finding suggested that the mouse may not be a relevant model for investigations of DCE in human lung. It is noteworthy that our previous studies showed that 50% of the levels of [B] and [C] produced by murine lung microsomes are attributable to CYP2E1, as assessed by immunoinhibition studies with a CYP2E1-inhibitory antibody (Dowsley et al., 1996). We postulated that the remaining 50% are catalyzed by other P-450 isozymes as yet unidentified. The involvement of P-450 isozymes other than CYP2E1 in the formation of the DCE-epoxide in human and murine lung is under investigation in this laboratory. Taken together, our results indicated that the P-450-mediated metabolism of DCE differs in human lung and liver and that although CYP2E1 has a major role in DCE activation, the involvement of this P-450 in human lung appears to be relatively minor. It should be emphasized that the number of lung samples in our inhibitory studies is limited and that experiments with additional samples with CYP2E1 activity at least as high as HL-1 are needed to determine the full capacity of CYP2E1 to metabolize DCE in human lung tissue. Of relevance in this context is the identification of several genetic polymorphisms of human CYP2E1 (Hayashi et al., 1991). These polymorphic variations occur in the noncoding regions of CYP2E1; polymorphisms in the 5′ flanking region of human CYP2E1 have been shown to cause a 10-fold variation in the transcriptional regulation of the gene (Hayashi et al., 1991). This finding suggests that certain individuals may have higher expression of lung CYP2E1 than that found in our samples, and in these instances the contribution of CYP2E1 to the formation of the DCE-epoxide may be more significant than that in the present study.
In summary, our data showed that human lung and liver microsomes bioactivated DCE to the DCE-epoxide by a cytochrome P-450-dependent mechanism, as assessed by the NADPH-dependent formation of the GSH conjugates [B] and [C]. The epoxide-derived GSH conjugates are formed efficiently by human lung and liver microsomes and are catalyzed by CYP2E1 in human liver and in human lung from some individuals. These results suggested that DCE poses a potential risk to chemically induced lung and liver toxicities in humans.
Footnotes
-
Send reprint requests to: Dr. Poh-Gek Forkert, Department of Anatomy and Cell Biology, Kingston, Ontario, Canada K7L 3N6. E-mailforkertp{at}post.queensu.ca
-
↵1 This research was supported by Grant MT-11706 from the Medical Research Council of Canada (to P.G.F.), Grant RO1-CA73220-01 from the U.S. National Cancer Institute (to P.G.F.), the Arizona Disease Control Research Commission (to J.B.U.), and National Institute of Environmental Health Sciences Center Grant P30-ES-06694 (to J.B.U).
- Abbreviations:
- [A]
- S-(2,2-dichloro-1-hydroxy)ethyl glutathione
- [B]
- 2-(S-glutathionyl) acetyl glutathione
- [C]
- 2-S-glutathionyl acetate
- [D]
- S-(2-chloroacetyl)-glutathione
- DASO2
- diallyl sulfone
- DCE
- 1,1-dichloroethylene
- GSH
- glutathione
- PNP
- p-nitrophenol
- Received September 23, 1998.
- Accepted December 3, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}