





Abstract
Cholinergic tone contributes to airflow obstruction in chronic obstructive pulmonary disease. Accordingly, anticholinergics are effective bronchodilators by blocking the muscarinic M3 receptor on airway smooth muscle. Recent evidence indicates that acetylcholine also contributes to airway inflammation. However, which muscarinic receptor subtype(s) regulates this process is unknown.
In this study, the contribution of the M1, M2 and M3 receptor subtypes to cigarette smoke-induced airway inflammation was investigated by exposing muscarinic receptor subtype deficient mice to cigarette smoke for 4 days.
In wild-type mice, cigarette smoke induced an increase in macrophages, neutrophils and lymphocytes in bronchoalveolar lavage fluid. Neutrophilic inflammation was higher in M1-/- and M2-/- mice compared to wild-type mice, but lower in M3-/- mice. Accordingly, the release of keratinocyte-derived chemokine (KC), monocyte chemotactic protein-1 and interleukin-6 was higher in M1-/- and M2-/- mice, and reduced in M3-/- mice. Markers of remodelling were not increased after cigarette smoke exposure. However, M3-/- mice had reduced expression of transforming growth factor-β1 and matrix proteins. Cigarette smoke-induced inflammatory cell recruitment and KC release were also prevented by the M3-receptor selective antagonist 1-dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP) in wild-type mice.
Collectively, our data indicate a pro-inflammatory role for the M3 receptor in cigarette smoke-induced neutrophilia and cytokine release, yet an anti-inflammatory role for M1 and M2 receptors.
Abstract
Inhibition of the muscarinic M3 receptor prevents inflammation in response to cigarette smoke exposure in mice http://ow.ly/p7UdG
Introduction
Chronic obstructive pulmonary disease (COPD) is an inflammatory disease characterised by progressive airflow limitation that is not fully reversible [1]. The most common cause of COPD in the Western world is tobacco smoking. Inflammation plays a central role in the disease, and contributes to the airway fibrosis, mucus hypersecretion and emphysema that is observed in patients with COPD [1]. Macrophages and neutrophils are particularly increased in patients with COPD and this increase is related to an increased production of specific cytokines and chemokines, including interleukin (IL)-8, IL-6, IL-1β, IL-17, monocyte chemotactic protein (MCP)-1 and tumour necrosis factor (TNF)-α [2]. Moreover, COPD is associated with an increased production of growth factors, including transforming growth factor (TGF)-β and vascular endothelial growth factor (VEGF), which are thought to contribute to the remodelling of the airways [3].
Acetylcholine is the primary parasympathetic neurotransmitter that induces bronchoconstriction in the airways. Parasympathetic activity is increased in patients with COPD, and this appears to be the major reversible component of airway obstruction [4]. Therefore, treatment with anticholinergics, inhibiting muscarinic receptor activation, is an effective bronchodilator therapy in COPD.
Genes encoding five muscarinic receptor subtypes are present in the human genome (M1–M5). The M1, M2 and M3 receptors are abundantly expressed in the lungs and have been extensively studied in the context of vagal neurotransmission. Their primary roles are bronchoconstriction and mucus secretion, which are mainly regulated via M3 receptors on airway smooth muscle and glands, respectively. Furthermore, the M1 receptor facilitates neurotransmission in the parasympathetic ganglia and regulates electrolyte and water secretion by mucus-producing cells. The M2 receptor is an autoinhibitory pre-junctional receptor on vagal nerves inhibiting acetylcholine release and an abundant post-junctional receptor on airway smooth muscle [5–7].
It is now known that acetylcholine can exert many additional, non-neuronal effects in the airways. Muscarinic receptors are expressed by almost all cell types in the lungs, including epithelial and inflammatory cells [8, 9]. Strikingly, these cells express all the necessary components to synthesise and release acetylcholine by themselves, including choline acetyl transferase (ChAT), the synthesising enzyme of acetylcholine. This is referred to as non-neuronal acetylcholine and may contribute to airway inflammation [9–11]. Indeed, in vitro studies have revealed a variety of effects of acetylcholine on these cell types [11], including an induced release of the potent neutrophil chemoattractants IL-8 and leukotriene (LT)B4 from airway epithelial, smooth muscle and inflammatory cells [12–15]. Recent evidence from in vivo studies also demonstrated a pro-inflammatory role for acetylcholine under pathophysiological conditions. In a cigarette smoke-induced mouse model of COPD, tiotropium partly prevented the increase in total cells and neutrophils in the bronchoalveolar lavage fluid (BALF). Furthermore, the release of various cytokines, including IL-6, keratinocyte-derived chemokine (KC, the mouse orthologue of IL-8), LTB4 and MCP-1, was inhibited by tiotropium [16]. Our group recently demonstrated that lipopolysaccharide-induced neutrophilic inflammation could be completely prevented by tiotropium in a guinea pig model of COPD [17]. Similar findings indicating a pro-inflammatory role for acetylcholine have been observed in animal models of asthma, acute lung injury and fibrosis [6].
Together, these studies clearly indicate a role for acetylcholine in inflammation, which may have implications for anticholinergic therapy in patients with COPD. Therapy with anticholinergics is presently focused on the M3 receptor, since this receptor subtype mediates bronchoconstriction. Although in vitro studies suggest a role for the M3 receptor in cytokine release [12, 18], no information is available with respect to the muscarinic receptor subtypes involved in the pro-inflammatory effects of acetylcholine in vivo. Therefore, the aim of this study was to investigate the role of the M1, M2 and M3 receptor subtypes in cigarette smoke-induced airway inflammation using muscarinic receptor subtype-deficient mice. We hypothesised that the M3 receptor plays a predominant pro-inflammatory role. In order to study this, we assessed inflammatory cell counts and mediator release in the lavage fluid. Furthermore, we analysed the expression of genes associated with remodelling.
Methods
Animals
Homozygous, inbred, specific-pathogen-free breeding colonies of M1-/-, M2-/- and M3-/- mice and C57Bl/6NTac wild-type mice with the same genetic background were obtained from Taconic (Ry, Denmark). The M1-/-, M2-/- and M3-/- mice used were generated on a 129 Sv/J background and backcrossed for ≥10 generations onto the C57Bl/6NTac background [19–21]. Knock-out animals did not differ from wild-type controls in overall health, fertility and longevity [19–21], although the weight of M3-/- mice was less than that of wild-type mice (online supplementary table E1). Exposure to cigarette smoke did not affect the weight of the mice (online supplementary table E1). Animals were housed conventionally under a 12-h light–dark cycle and received food and water ad libitum. All experiments were performed in accordance with the national guidelines and approved by the University of Groningen Committee for Animal Experimentation (Groningen, the Netherlands).
Animal model
Male mice (n=8–9 per group, 10–12 weeks old) were exposed to cigarette smoke from Kentucky 3R4F research cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, KY, USA) on four consecutive days by whole-body exposure. Each cigarette was smoked without a filter in 5 min at a rate of 5 L·h−1 in a ratio with 60 L·h−1 air using a peristaltic pump (45 rpm) (323 E/D; Watson Marlow, Rotterdam, The Netherlands). Cigarette smoke was directly distributed into a 6-L perspex box. On day 1, mice were exposed to the mainstream smoke of one cigarette in the morning and three cigarettes in the afternoon. On days 2–4, mice were exposed to five cigarettes in the morning and five in the afternoon (fig 1). Control animals were handled in the same way but exposed to fresh air only. Because of the capacity of the experimental set-up, not all animals used for this study could be included in a single experiment. Therefore, experiments were performed on three occasions (n=19–24 animals per experiment). Air- and cigarette smoke-exposed wild-type mice were included in every experiment to minimise variability. 16 h after the last cigarette smoke exposure, animals were euthanised by intraperitoneal pentobarbital injection (400 mg·kg−1; University Medical Center Groningen, Groningen, The Netherlands), after which the lungs were immediately lavaged, resected and snap frozen in liquid nitrogen.

Experimental procedure. Male C57Bl/6NTac mice were exposed to cigarette smoke twice daily on four consecutive days by whole body exposure. 16 h after the last smoke exposure a bronchoalveolar lavage was performed and lungs were harvested for lung tissue homogenates and cryosection. Cig: cigarette(s); TGF: transforming growth factor; ChAT: choline acetyl transferase.
Muscarinic antagonist administration
In a substudy, the M3 receptor selective antagonist 4-DAMP (1-dimethyl-4-diphenylacetoxypiperidinium iodide) (1 mg·kg−1) was administered to wild-type mice (n=7) by i.p. injection, 30 min prior to each cigarette smoke exposure. The same experimental protocol was used as described above.
Analysis of BALF
After euthanising the mice, the lungs were gently lavaged through a tracheal cannula with 1 mL PBS containing 5% bovine serum albumin and protease inhibitors (inhibiting chymotrypsin, thermolysin, papain, pronase, pancreatic extract and trypsin; F. Hoffman-La Roche, Basel, Switzerland) and another four times with 1 mL PBS. Cells were pelleted and the supernatants of the first fraction were stored at -20°C for measurement of cytokines and growth factors by ELISA. For each animal individually, bronchoalveolar lavage cells of the different fractions were combined, resuspended in 500 μL PBS, and total cell numbers were determined. For cytological examination, cytospin preparations were stained with May–Grünwald and Giemsa (both Sigma, St Louis, MO, USA) and a differential cell count was performed by counting ≥400 cells in duplicate in a blinded fashion. KC, MCP-1, IL-6, IL-1β, IL-17, TNF-α and VEGF release was determined in BALF supernatants by a MILLIPLEX assay (Millipore, Billerica, MA, USA). TGF-β in BALF was determined by an ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
ChAT staining
To identify ChAT expression, 5-μm-thick cryosections (n=4) were stained with a specific rabbit anti-ChAT antibody provided by W. Kummer (Institute for Anatomy and Cell Biology, Justus-Liebig-University, Giessen, Germany). The antibody was visualised using a horseradish peroxidase-linked secondary antibody and diaminobenzidine (1 mg·mL−1). Airways within each section were photographed digitally.
Analysis of gene expression in lung tissue
Total RNA was extracted from lung tissue (right superior lobe) or bronchalveolar lavage cells using the RNeasy Mini Kit (Qiagen, Venlo, the Netherlands) according to the manufacturer’s instructions. Lung homogenates were prepared by pulverising the tissue under liquid nitrogen. Equal amounts of total mRNA were then reverse transcribed and cDNA was subjected to real-time quantitative PCR (Westburg, Leusden, the Netherlands). Real-time PCR was performed with denaturation at 94°C for 30 s, annealing at 59°C for 30 s and extension at 72°C for 30 s for 40 cycles followed by 10 min at 72°C. Real-time PCR data were analysed using the comparative cycle threshold method. The amount of the target gene was normalised to the endogenous reference gene 18S ribosomal RNA. Several other housekeeping genes, including β2-microglobulin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), were tested for the influence of the experimental procedure on the expression. The expression of all housekeeping genes, including 18S, was stable in the tested conditions. The specific forward and reverse primers used are listed in online supplementary table E2.
Statistical analysis
Data are presented as mean±sem. Statistical differences between means were calculated using one- or two-way ANOVA, followed by Newman–Keuls multiple comparison tests. Differences were considered significant at p<0.05.
Results
Characterisation of the mice
The genotypes of the knockout mice were confirmed by PCR analysis of mouse ear DNA (fig. 2a and online supplementary table E3). We also examined whether the deletion of one muscarinic receptor gene affected the expression levels of the two other muscarinic receptors expressed in the lung. As shown in figure 2b, no such compensatory changes were observed. Muscarinic receptors were highly expressed in lung tissue homogenates, with M2>M3>M1 (fig. 2b).

Characterisation of the mice. a) SYBR Safe-stained agarose gel showing the PCR products for genotyping, including the wild-type, muscarinic M1, M2 and M3 receptor bands for M1-/-, M2-/- and M3-/- mice, respectively. b) Gene expression of muscarinic receptors in lung tissue homogenates depicted as amplification cycle number (Ct) values corrected for 18S (n=5 mice per group). Note that data are expressed as ΔCt values, thus lower values mean higher expression levels and every unit lower on the y-axis represents a two-fold increase in expression. NA: not applicable.
The non-neuronal cholinergic system
It has been proposed that the non-neuronal cholinergic system (NNCS) might contribute to airway inflammation [11]. To investigate the effect of cigarette smoke exposure, gene expression of different components of the cholinergic system was analysed in lung tissue homogenates of wild-type mice, including the choline transporters high-affinity choline transporter-1 and choline transporter-like protein-1, the acetylcholine synthesising enzyme ChAT, the acetylcholine degrading enzyme acetylcholinesterase and the different muscarinic receptor subtypes. As depicted in figure 3a, these components were expressed in the murine lungs and cigarette smoke exposure did not affect their expression level. Similarly, the muscarinic receptor expression in the bronchoalveolar lavage cells from wild-type mice was not altered after cigarette smoke exposure (fig. 3b). In lung tissue, M2 receptors were expressed at the highest levels (M2>M3>M1), whereas in the cells from the BALF, M3 receptors were expressed at the highest levels (M3R>M1R>M2R) (online supplementary fig. E1). ChAT expression, detected by immunohistochemical staining, was localised to the epithelium and smooth muscle layer of the airway wall in wild-type mice (fig. 3c). Exposure to cigarette smoke did not alter the expression or localisation of ChAT (fig. 3c).
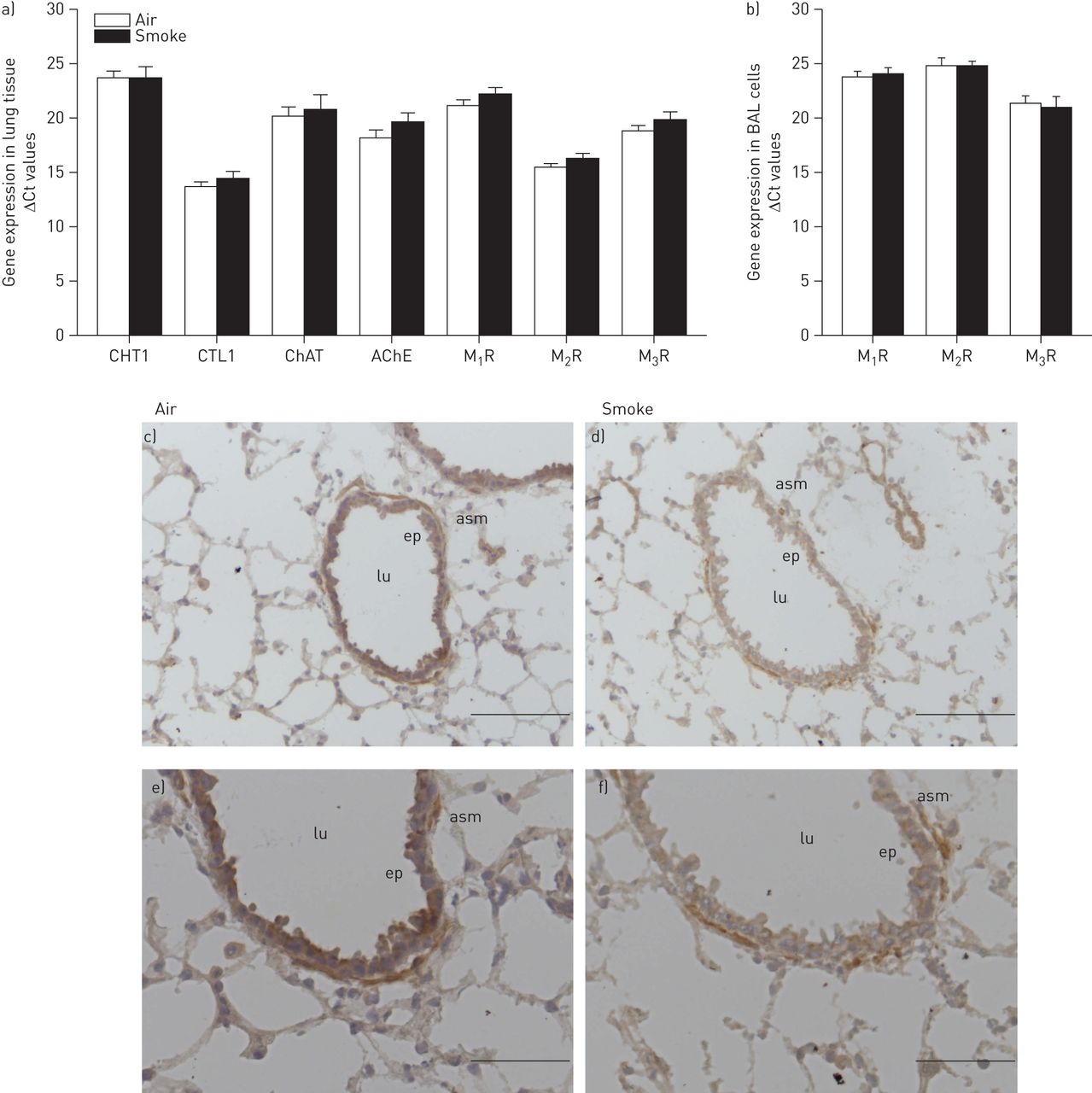
Effect of cigarette smoke exposure on the expression of the non-neuronal cholinergic system. Mice were treated as described in figure 1. 16 h after the last smoke exposure lung tissue and bronchoalveolar lavage fluid (BALF) were harvested. Gene expression in a) lung tissue homogenates and b) bronchoalveolar lavage cells was analysed (n=3–5 mice per group). Amplification cycle number (Ct) values corrected for 18S are depicted, expressed as mean±sem. Note that data are expressed as ΔCt values, thus lower values mean higher expression levels and every unit lower on the y-axis represents a two-fold increase in expression. High-affinity choline transporter (CHT)1, choline transporter like protein (CTL)1, choline acetyl transferase (ChAT), acetylcholinesterase (AChE), muscarinic M1 receptor (M1R), M2 receptor (M2R) and M3 receptor (M3R). c–f) Cryosections of wild-type mice exposed to air and smoke stained for ChAT. A representative picture of n=4 animals is shown. lu: airway lumen; ep: epithelium; asm: airway smooth muscle. c and d) Scale bars=100 μm; e and f) Scale bars=50 μm.
Cigarette smoke-induced inflammatory cell recruitment
To study the contribution of muscarinic receptor subtypes to cigarette smoke-induced airway inflammation, cell counts were determined in the BALF of wild-type, M1-/-, M2-/- and M3-/- mice. All mice exposed to cigarette smoke had a two-fold increase in the number of inflammatory cells compared to air-exposed control animals (fig. 4a). The predominant cell type after cigarette smoke exposure was the macrophage, which almost doubled in number in all strains (fig. 4b). Only small increases in lymphocytic infiltration were observed after cigarette smoke exposure, which were comparable in all strains (fig. 4c). However, neutrophilic infiltration showed significant differences between the strains (fig. 4d). A four-fold increase in the amount of neutrophils was observed in wild-type mice compared to air-exposed animals. However, in M1-/- and M2-/- mice, the increase in neutrophil number upon cigarette smoke exposure was much higher, up to 24-fold in M1-/- mice. In striking contrast, no significant increase in neutrophil numbers was observed in M3-/- mice after cigarette smoke exposure compared to air-exposed control animals (figure 4d).
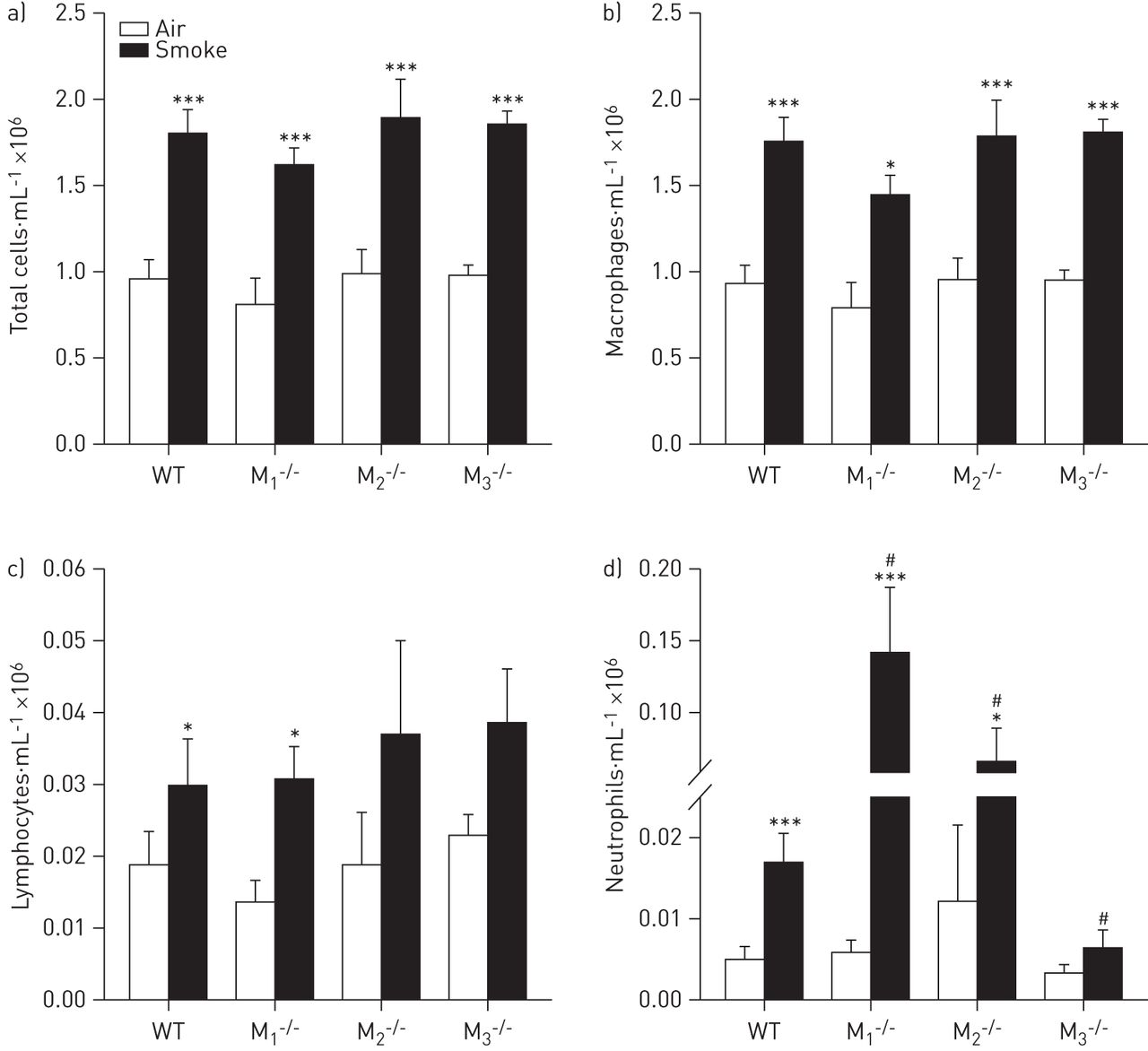
Inflammatory cell counts after cigarette smoke exposure. Mice were treated as described in figure 1. 16 h after the last smoke exposure bronchoalveolar lavage was performed and a) total cells, b) macrophages, c) lymphocytes and d) neutrophils were determined in the bronchoalveolar lavage fluid. Data are presented as mean±sem. n=8–9 mice per group. Data were analysed using two-way ANOVA: a) total cells F[1,56]=89.25, p<0.001 for sham versus smoke treatment; F[3,56]=1.233, p=0.307 for the muscarinic receptor subtypes; F[3,56]=0.0493, p=0.985 for the interaction; b) macrophages F[1,56]=78.60, p<0.001 for sham versus smoke treatment; F[3,56]=1.93, p=0.135 for the muscarinic receptor subtypes; F[3,56]=0.27, p=0.849 for the interaction; c) lymphocytes F[1,56]=12.70, p<0.001 for sham versus smoke treatment; F[3,56]=0.587, p=0.587 for the muscarinic receptor subtypes; F[3,56]=0.05, p=0.984 for the interaction; d) neutrophils F[1,56]=17.65, p<0.001 for sham versus smoke treatment; F[3,56]=6.37, p<0.001 for the muscarinic receptor subtypes; F[3,56]=5.90, p=0.001 for the interaction. WT: wild type. Individual comparisons were made using a Student–Newman–Keuls multiple comparisons post hoc test. *: p<0.05; ***: p<0.001 compared to air-exposed control mice; #: p<0.05 compared to wild-type cigarette smoke-exposed mice.
Cigarette smoke-induced cytokine release
Subsequently, inflammatory cytokine release in BALF was determined. Levels of KC in BALF of wild-type mice exposed to cigarette smoke were 15-fold higher compared to air-exposed animals (fig. 5a). No significant differences in MCP-1 and IL-6 release were observed in cigarette smoke-exposed wild-type mice (fig. 5b and c). In M1-/- mice, cigarette smoke-exposed mice had higher levels of KC, MCP-1 and IL-6 compared to air-exposed mice. Interestingly, the concentration of all these cytokines was significantly higher when compared to wild-type cigarette smoke-exposed mice. In cigarette smoke-exposed M2-/- mice, KC release was also significantly higher, both compared to air-exposed mice and to wild-type cigarette smoke-exposed mice, whereas MCP-1 and IL-6 release were not significantly different. In M3-/- mice, no increase in the release of any of these cytokines was observed after cigarette smoke exposure, and KC release was significantly lower compared to wild-type cigarette smoke-exposed mice (fig. 5). Levels of IL-17, IL-1β and TNF-α were below detection limit in all strains (not shown).

Inflammatory cytokine release after cigarette smoke exposure. Mice were treated as described in figure 1. 16 h after the last smoke exposure a bronchoalveolar lavage was performed. Release of a) keratinocyte-derived chemokine (KC); b) monocyte chemotactic protein (MCP)-1; and c) interleukin (IL)-6 in bronchoalveolar lavage fluid was determined. Data are presented as mean±sem. n=8–9 mice per group. Data were analysed using two-way ANOVA: a) KC F[1,56]=32.85, p<0.001 for sham versus smoke treatment; F[3,56]=4.40, p=0.008 for the muscarinic receptor subtypes; F[3,56]=3.61, p=0.019 for the interaction; b) MCP-1 F[1,56]=13.91, p<0.001 for sham versus smoke treatment; F[3,56]=7.49, p<0.001 for the muscarinic receptor subtypes; F[3,56]=5.89, p=0.001 for the interaction; c) IL-6 F[1,56]=9.32, p=0.003 for sham versus smoke treatment; F[3,56]=8.69, p<0.001 for the muscarinic receptor subtypes; F[3,56]=5.63, p=0.002 for the interaction. WT: wild type; ND: not detectable. Individual comparisons were made using a Student–Newman–Keuls multiple comparisons post hoc test. *: p<0.05; ***: p<0.001 compared to air-exposed control mice; #: p<0.05; ###: p<0.001 compared to wild-type cigarette smoke-exposed mice.
Cigarette smoke-induced growth factor and extracellular matrix expression
We next determined the release of the growth factors TGF-β1 and VEGF in the BALF. Small increases in TGF-β1 protein release were observed in all strains after cigarette smoke exposure (1.4–2.0-fold), which was significant in wild-type mice (fig. 6a). Remarkably, there was significantly less TGF-β1 in the BALF of M3-/- mice compared to wild-type mice, irrespective of air or cigarette smoke exposure (56% and 46% lower, respectively). VEGF release was not altered (fig. 6b). Expression of TGF-β1 at the mRNA level in lung tissue of M3-/- mice was reduced to a similar extent compared to protein levels (fig. 6b). At the transcriptional level, TGF-β1 expression was significantly increased 1.4-fold in M1-/- mice and 1.7-fold in M2-/- mice after cigarette smoke exposure (fig. 6c).
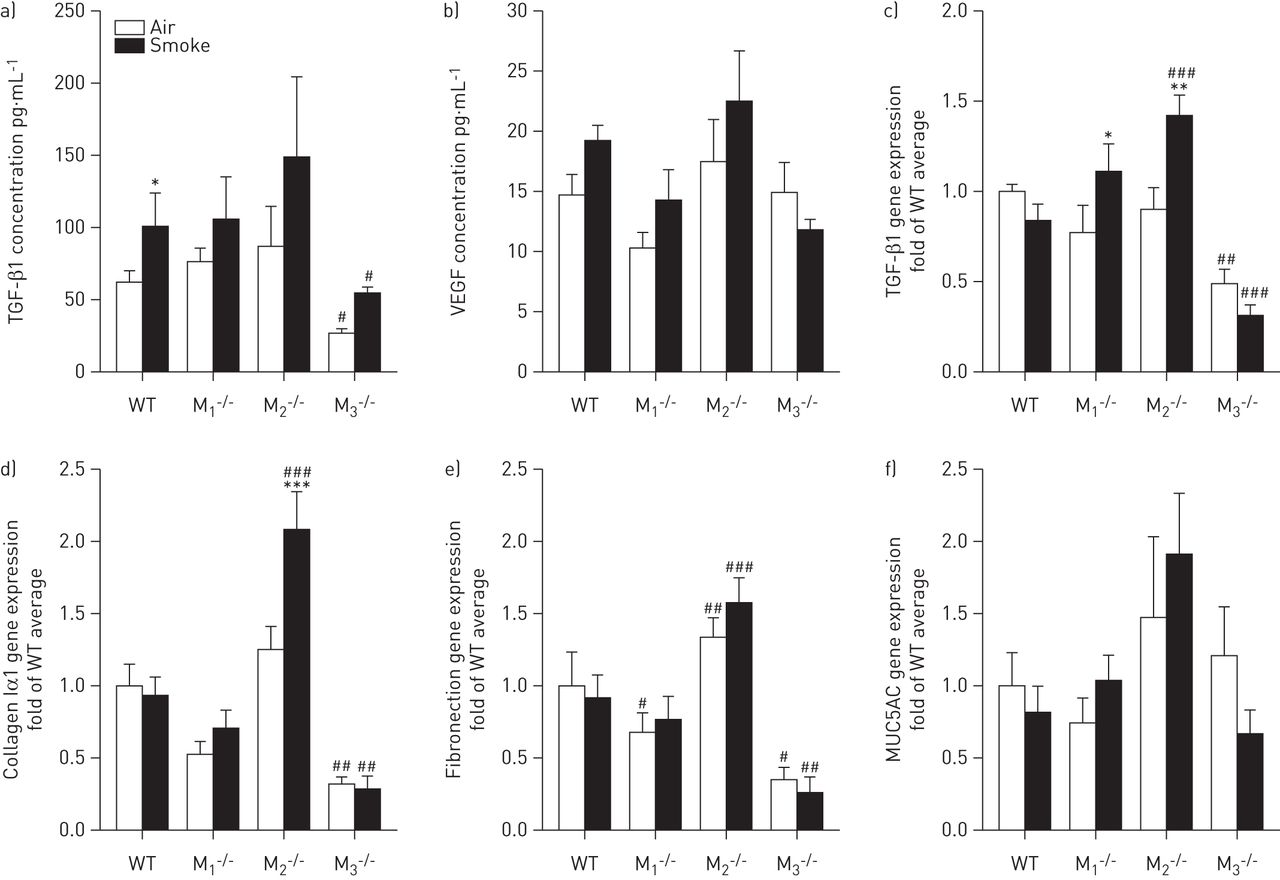
Parameters of remodelling after cigarette smoke exposure. Mice were treated as described in figure 1. 16 h after the last smoke exposure a bronchoalveolar lavage was performed and lungs were collected. Release of a) transforming growth factor (TGF)-β1 and b) vascular endothelial growth factor (VEGF) in bronchoalveolar lavage fluid was determined. In lung tissue homogenates, gene expression of c) TGF-β1, d) collagen Iα1, e) fibronectin and f) MUC5AC was determined. Data are presented as mean±sem. n=8–9 mice per group. Statistics were performed on log-transformed data. Data were analysed using two-way ANOVA: a) TGF-β1 protein F[1,56]=4.72, p=0.034 for sham versus smoke treatment; F[3,56]=3.10, p=0.034 for the muscarinic receptor subtypes; F[3,56]=0.18, p=0.908 for the interaction; b) VEGF protein F[1,56]=2.15, p=0.148 for sham versus smoke treatment; F[3,56]=3.84, p=0.014 for the muscarinic receptor subtypes; F[3,56]=1.13, p=0.347 for the interaction; c) TGF-β1 mRNA F[1,56]=3.16, p=0.081 for sham versus smoke treatment; F[3,56]=18.38, p<0.001 for the muscarinic receptor subtypes; F[3,56]=5.66, p=0.002 for the interaction; d) collagen Iα1 F[1,56]=6.91, p=0.011 for sham versus smoke treatment; F[3,56]=43.02, p<0.001 for the muscarinic receptor subtypes; F[3,56]=5.97, p=0.001 for the interaction; e) fibronectin F[1,56]=0.03, p=0.862 for sham versus smoke treatment; F[3,56]=36.10, p<0.001 for the muscarinic receptor subtypes; F[3,56]=1.37, p=0.262 for the interaction; f) MUC5AC F[1,56]=4.95×10−4, p=0.982 for sham versus smoke treatment; F[3,56]=3.19, p=0.030 for the muscarinic receptor subtypes; F[3,56]=1.04, p=0.381 for the interaction. Individual comparisons were made using a Student–Newman–Keuls multiple comparisons post hoc test. WT: wild-type. *: p<0.05; ***: p<0.001 compared to air-exposed control mice; #: p<0.05; ###: p<0.001 compared to WT control mice.
In addition, we analysed the gene expression of the matrix proteins collagen Iα1 and fibronectin in lung tissue. No increased expression after cigarette smoke exposure was observed (fig. 6d and e), with the exception of collagen Iα1 in M2-/- mice, which was increased after cigarette smoke exposure. Furthermore, expression levels of collagen Iα1 and fibronectin were higher in cigarette smoke-exposed M2-/- mice compared to cigarette smoke-exposed wild-type mice. In line with the findings for TGF-β1, both matrix proteins were expressed at a significantly lower level in M3-/- mice compared to wild-type mice, irrespective of air or cigarette smoke exposure (68% and 65% lower, respectively). This was confirmed at the protein level for fibronectin, which was 38.6±8.9% lower in M3-/- mice than in wild-type mice (p<0.05). Finally, cigarette smoke exposure had no significant effect on MUC5AC gene expression in any of the analysed strains (fig. 6f).
Inhibition of cigarette smoke-induced inflammation by an M3 antagonist
To investigate whether the pro-inflammatory role of the M3 receptor can also be observed using a pharmacological intervention, wild-type mice were pre-treated with the M3 receptor selective antagonist 4-DAMP 30 min prior to every cigarette smoke exposure. This resulted in an inhibition of inflammatory cell number in the BALF by 30% compared to untreated animals (fig. 7a). Similar inhibitory effects of pre-treatment with 4-DAMP were observed on macrophage accumulation (fig. 7b), whereas cigarette smoke-induced lymphocytic and neutrophilic inflammation were completely prevented (fig. 7c and d). This was accompanied by the absence of cigarette smoke-induced KC release after pre-treatment with 4-DAMP (fig. 7e).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cigarette smoke-induced inflammation after pretreatment with 4-DAMP (1-dimethyl-4-diphenylacetoxypiperidinium iodide). The same experimental model was used as described in figure 1; however, prior to each smoke exposure mice were treated with the selective muscarinic M3 antagonist 4-DAMP. 16 h after the last smoke exposure a bronchoalveolar lavage was performed and a) total cells, b) macrophages, c) lymphocytes, d) neutrophils and e) keratinocyte-derived chemokine (KC) release were determined in the bronchoalveolar lavage fluid. Results are expressed as mean±sem. n=7–13 mice per group. Data were analysed using one-way ANOVA: a) total cells F[2],[30]=12.98, p<0.001 between groups; b) macrophages F[2],[30]=11.92, p<0.001 between groups; c) lymphocytes F[2],[30]=5.87, p=0.007 between groups; d) neutrophils F[2],[30]=8.23, p=0.001 between groups; e) KC F[2],[30]=9.53, p<0.001 between groups. Individual comparisons were made using a Student–Newman–Keuls multiple comparisons post hoc test. *: p<0.05; ***: p<0.001 compared to air-exposed control mice; #: p<0.05 compared to cigarette smoke-exposed mice.
Discussion
In this study we demonstrate that the M3 receptor plays a profound pro-inflammatory role in cigarette smoke-induced inflammation and that this is the primary muscarinic receptor subtype involved in the pro-inflammatory effects of acetylcholine. Inhibition of the M3 receptor, by total knockout of the receptor or by a pharmacological approach, prevented neutrophilic inflammation and cytokine release in the lavage fluid of cigarette smoke-exposed mice. In striking contrast, knockout of the M1 and M2 receptors resulted in increased neutrophils and cytokine release in the BALF, indicating an anti-inflammatory role of these receptor subtypes in cigarette smoke-induced inflammation. This study is the first to demonstrate the differential regulation of inflammation by muscarinic receptors in vivo and implies an anti-inflammatory role for M3 selective anticholinergics.
Neutrophils are considered to be one of the major cell types involved in COPD [2].Various studies suggest an important role for acetylcholine in regulating neutrophilic inflammation. Activation of muscarinic receptors can contribute to neutrophil influx by inducing neutrophil chemotactic activity from macrophages [22, 23] and LTB4 release from sputum cells of COPD patients [15]. Furthermore, IL-8 is released from epithelial and airway smooth muscle cells in response to muscarinic receptor stimulation [12, 14]. These findings are supported by in vivo studies in which cigarette smoke- and lipopolysaccharide-induced neutrophilia was inhibited by the muscarinic receptor antagonist tiotropium [16, 17].
The regulatory effects of muscarinic receptors appeared to be specific for neutrophils in the muscarinic receptor deficient mice, whereas macrophages and lymphocytes were not altered. Our results on neutrophilic inflammation are in line with various in vitro studies demonstrating that the pro-inflammatory effects of muscarinic receptor activation are mainly dependent on the M3 receptor subtype. Thus, it has been shown that 4-DAMP and DAU5884, M3 receptor selective antagonists, inhibited methacholine and cigarette smoke-induced IL-8 release from airway smooth muscle cells [12]. In addition, alveolar macrophage-mediated migration of neutrophils from COPD patients was inhibited by 4-DAMP [18]. Moreover, the M3 receptor was the primary receptor subtype expressed by inflammatory cells within the BALF as shown in our study. It is also known that inflammatory cells express M3 receptors and that this receptor mediates pro-inflammatory effects [8]. We therefore believe that the pro-inflammatory effect of the M3 receptor, as found in our study, is dependent on regulation of cytokine release by structural cells, in combination with direct activation of M3 receptors on inflammatory cells.
In contrast to the findings in M3-/- mice, neutrophilic inflammation was increased in M2-/- mice compared to wild-type mice. The M2 receptor is located pre-junctionally on pre- and post-ganglionic nerves and acts as an inhibitory autoreceptor limiting acetylcholine release [5]. Furthermore, the M2 receptor is expressed post-junctionally by smooth muscle cells and fibroblasts [6, 24]. With acetylcholine acting as a pro-inflammatory mediator inducing chemokine release from structural and inflammatory cells via the M3 receptor, increased levels of acetylcholine in the M2-/- mice, due to loss of its autoinhibitory role, may therefore explain the observed aggravated neutrophilia. In support of such a role, M2 receptor expression appeared to be low on inflammatory cells in the BALF, but high in lung tissue, suggesting that the effects of M2 receptor deficiency are not due to direct effects on inflammatory cells. The literature supports this notion, indicating that the pro-inflammatory effects of acetylcholine in macrophages, epithelial cells and airway smooth muscle cells are not mediated by M2 receptors [12, 23, 25].
A role for M2 receptors as pre-junctional autoreceptors driving exaggerated acetylcholine release and inflammation has significant implications. Acetylcholine has long been known as a classical neurotransmitter. More recent findings suggest that acetylcholine can also be released from non-neuronal sources, including epithelial, airway smooth muscle and inflammatory cells [9, 10]. It is not yet known to which extent this non-neuronal acetylcholine affects airway inflammation [11]. The release of non-neuronal acetylcholine is not known to be affected by M2 receptors in an autoinhibitory way. Our data therefore imply an important role for neuronal acetylcholine in cigarette smoke-induced inflammation. Furthermore, we did not find any upregulation of expression of components of the NNCS in the lungs of cigarette smoke-exposed mice, in contrast to the previously reported increase in cultured human airway epithelial cells after exposure to cigarette smoke extract [13]. Future studies investigating the contribution of neuronal and non-neuronal acetylcholine to inflammation are clearly warranted.
Surprisingly, neutrophilic inflammation was also enhanced in M1-/- mice. It is well known that M1 receptors facilitate neurotransmission in the parasympathetic ganglia [5]. However, based on this, one would expect that M1 receptor deficiency causes reduced acetylcholine release leading to inhibition of inflammation. Reinheimer et al. [26] reported that the M1 receptor selective antagonist pirenzepine can antagonise the inhibitory effect of acetylcholine on histamine release from human mast cells. Lack of this inhibitory M1 receptor might explain the increased neutrophil chemotaxis, since mast cell numbers are increased upon smoking and higher in patients with COPD [27, 28]. Alternatively, as the M1 receptor also controls electrolyte and water secretion by airway epithelial cells [29], lack of M1 receptor expression may result in a reduced ability of M1-/- mice to clear their lungs of smoke particles after cigarette smoke exposure, leading to aggravated inflammatory and injury responses. The sharp induction of the damage response-associated cytokines IL-6 and MCP-1 in M1-/- mice, which is absent in wild-type mice, supports this hypothesis.
The results of this study on transgenic mice suggest a primary role for the M3 receptor in regulating cigarette smoke-induced inflammation, implying that M3 subtype selectivity of anticholinergics would be beneficial. Indeed, we show that pharmacological inhibition of the M3 receptor using 4-DAMP partly prevented accumulation of inflammatory cells in BALF, accompanied by a strong inhibition of cigarette smoke-induced KC release. 4-DAMP is selective for M3 receptors over M2 (∼16-fold) [7] and, to a lesser extent, M1 receptors (approximately three-fold) [30]. Interestingly, the anti-inflammatory effects of pharmacological inhibition of the M3 receptor are more pronounced than effects of knockout of this receptor. Similar discrepant data are reported on muscarinic receptor mediated contraction ex vivo, which can be fully inhibited with a M3 receptor selective antagonist in wild-type mice, whereas only partial inhibition of contraction is observed in M3-/- mice [31, 32]. Although the mechanism behind this discrepancy is unclear it appears that compensating mechanisms are operative in the knock-out mice that limit the impact of the M3 receptor deficiency.
Although tiotropium is known to be kinetically selective for the M3 receptor (dissociation half-life 27 h), it has a dissociation half-life from the M1 receptor of 10.5 h [33]. Steady-state binding affinity of tiotropium for the M1, M2 and M3 receptors is not different [34]. The half-life ratio of ipratropium and of aclidinium and glycopyrrolate, two anticholinergics under development, for the M3 versus the M1 receptor is comparable to tiotropium [33]. Our study suggests that an even more selective compound solely inhibiting M3 receptors is desirable and may lead to improved effects on cigarette smoke-induced inflammation.
Interestingly, in our study basal expression of TGF-β1, collagen Iα1 and fibronectin was significantly lower in M3-/- mice compared to wild-type mice. In M2-/- mice, expression of these components was increased after cigarette smoke exposure. This suggests that in addition to inflammation, acetylcholine regulates important aspects of lung structure via the M3 receptor. Indeed, M3 receptors are expressed by structural cells in the airways, including epithelial cells and airway smooth muscle cells [24]. Moreover, in vitro studies have demonstrated a role for the M3 receptor in the regulation of airway smooth muscle proliferation [35] and in vivo studies have shown a protective effect of anticholinergics on matrix protein deposition [17, 36]. The exact roles of the individual muscarinic receptor subtypes in this process are not yet clear and remain to be elucidated [6].
Evidence for the pro-inflammatory role of acetylcholine from in vitro and in vivo studies is increasing [6]. However, the translational utility of these observations is not yet clear, since in patients with COPD, effects on inflammation or on the rate of decline in lung function after anticholinergic therapy have not been demonstrated. It is known from the UPLIFT (Understanding Potential Long-term Impacts on Function with Tiotropium) study that the use of tiotropium is associated with a reduction in the number of exacerbations, generally seen as inflammatory events [37]. Patients who have more exacerbations demonstrate increased levels of inflammatory markers at stable state [38, 39]. However, in two recent studies, no direct evidence for an anti-inflammatory effect was found, since IL-6 and IL-8 levels in the sputum of patients with COPD were not decreased after anticholinergic therapy [40, 41]. However, both studies have substantial limitations as discussed by the authors. In the study by Perng et al. [41], the treatment group was small and patients were only treated with tiotropium for 12 weeks. In the study by Powrie et al. [40], the amount of sputum was reduced after tiotropium treatment, which might have resulted in increased cytokine concentrations. Future studies using different methods to assess inflammation could therefore resolve the question of whether anticholinergics indeed have anti-inflammatory properties in patients with COPD. At present, there is no evidence for such a role.
In conclusion, the results of our study demonstrate that inhibition of the M3 receptor prevents inflammation in response to cigarette smoke exposure in mice. This confirms the previously established pro-inflammatory role of acetylcholine in the pathophysiology of airway diseases, and demonstrates that this is solely mediated via M3 receptors, since knockout of the M1 and M2 receptor aggravated inflammation compared to wild-type mice. This study therefore opens new perspectives on M3 receptor selective anticholinergics to specifically target airway inflammation.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support statement: We would like to thank the Netherlands Asthma Foundation for financial support (grant 3.2.08.014).
Conflict of interest: Disclosures can be found alongside the online version of this article at www.erj.ersjournals.com
- Received July 19, 2012.
- Accepted January 21, 2013.
- ©ERS 2013
References










