





Abstract
Altitude illness remains a major cause of mortality. Reduced chemosensitivity, irregular breathing leading to central apnoeas/hypopnoeas, and exaggerated pulmonary vasoconstriction may compromise oxygenation. All factors could enhance susceptibility to acute mountain sickness (AMS).
We compared 12 AMS-susceptible individuals with recurrent and severe symptoms (AMS+) with 12 “AMS-nonsusceptible” subjects (AMS-), assessing sleep-breathing disorders in simulated altitude as well as chemoresponsive and pulmonary vasoconstrictive responses to hypoxia.
During exposure to simulated altitude, mean blood oxygen saturation during sleep was lower in AMS+ subjects (81.6±2.6 versus 86.0±2.4%, p<0.01), associated with a lower central apnoea/hypopnoea index (18.2±18.1 versus 33.4±24.8 events·h−1 in AMS+ and AMS- subjects, respectively; p=0.038). A lower hypoxic (isocapnic) chemoresponsiveness was observed in AMS+ subjects (0.40±0.49 versus 0.97±0.46 L·min−1·%; p<0.001). This represented the only significant and independent predictive factor for altitude intolerance, despite a higher increase in pulmonary artery systolic pressure in response to hypoxia, a lower lung diffusing capacity and a higher endothelin-1 level at baseline in AMS+ subjects (p<0.05). AMS+ subjects were more hypoxaemic whilst exhibiting fewer respiratory events during sleep owing to lower hypoxic (isocapnic) chemoresponsiveness.
In conclusion, the reduction in peripheral hypoxic chemosensitivity appears to be a major causative factor for altitude intolerance.
The term high-altitude illness encompasses acute mountain sickness (AMS), high-altitude cerebral oedema (HACE) and high altitude pulmonary oedema (HAPE). Although HAPE represents the major cause of mortality due to high altitude [1], AMS is the most frequently encountered syndrome, and is usually considered as an early stage of HAPE and HACE. When subjects rapidly ascend to moderate altitude (2,000–3,000 m), up to 25% will suffer from AMS [2]. When mountaineers ascend to high or very high altitude (>4,500 m), the AMS incidence may exceed 60% [3].
High altitude illness is partly preventable by gradual and stepwise ascent. However, certain individuals are at greater risk of developing high-altitude illness and are more likely to be reproducibly affected during ascents. The ability to predict AMS susceptibility would be highly beneficial, but no predictive test is currently agreed upon by specialists [4].
The mechanisms of high altitude illness are still not fully understood, which precludes any clinical or biological prediction of AMS susceptibility. Characteristics of established AMS and HACE, however, suggest that factors lowering blood oxygenation may play a major role in their development [5, 6]. Thus, a relative hypoventilation induced by low hypoxic chemosensitivity during wakefulness has been suggested as an important feature of AMS susceptibility [7]. Reduced lung diffusing capacity following altitude exposure may also compromise oxygenation [8, 9]. Early fluid retention [10] and increased hypoxic pulmonary vasoconstriction (HPV) in a less adaptable pulmonary circulation (e.g. a smaller pulmonary vascular bed) have also been shown as being important. Both endothelin-1 levels [11] and exhaled nitric oxide, as well as their changes in response to hypoxic conditions [12], may further affect HPV.
Neither periodic breathing during sleep [13] and central chemosensitivity (assessed by ventilatory response to carbon dioxide [14]) have been found to be important predictors of AMS susceptibility. Nocturnal oxygenation, however, may play a role in AMS susceptibility. Whether altitude-induced central sleep apnoea syndrome favours AMS by reducing oxygenation has not been studied. Finally, these factors have usually been investigated separately, making it difficult to determine their individual contribution to high-altitude illness and their ability to predict individual AMS susceptibility.
Our working hypothesis was that AMS susceptibility was associated with low hypoxic chemosensitivity, lower nocturnal oxygenation during sleep and higher pulmonary vasoreactivity when compared with altitude-tolerant subjects.
MATERIAL AND METHODS
Study population
A total of 24 mountaineers participated in the study. All gave informed consent and the study was approved by the Grenoble Ethics Committee (CPP Sud-Est V, Grenoble, France). Two distinct groups of subjects were recruited for the study: 1) 12 AMS-susceptible subjects (AMS+) with recurrent and severe AMS (Lake Louise score ≥6) despite appropriate acclimatisation; and 2) 12 carefully matched subjects who had always felt well during exposure to equivalent altitudes (AMS-). Subjects were recruited after responding to requests on specialised websites: the International Federation of Mountain Guides Association, www.grenoble-montagne.com, the French Alpine Club and the French Federation of Climbing and Mountaineering. None of the subjects had previously participated in altitude research. 80 subjects initially volunteered to participate in the study and a structured telephone interview was given to all subjects by two of the investigators (H. Nespoulet and B. Wuyam), wherein all described their altitude experience. The interviews took place between 2 and 6 months after the last ascent. Tolerance to altitude was assessed by asking specific questions pertaining to clinical manifestations occurring at high altitude. Answers to specific questions and quotations were recorded in respect to the five common symptoms of AMS (headache, fatigue, sleep, vomiting and dizziness). Circumstances of occurrence were checked to exclude obvious acclimatisation mistakes. All AMS-sensitive subjects had experienced recurrent and severe AMS (i.e. Lake Louise score ≥6), predominantly at a threshold altitude of between 2,500 and 4,000 m. Lake Louise scores were additionally and retrospectively completed during the inclusion visit (Grenoble: 210 m altitude). Diagnosis of HAPE was based on retrospective evaluation of the clinical symptoms (dry cough, haemoptoic sputum and highly disproportionate breathlessness), as well as pulmonary alveolar images on chest radiograph immediately after descent (n=4). AMS+ and AMS- subjects were matched with respect to sex, age, body mass index, physical fitness and experience of high altitude. All subjects were natives of low altitude regions and none had gone to altitudes >2,000 m during the 2 weeks before the study. All were free of significant cardiac, pulmonary or neurological pathology at the time of the study. All females completed the tests during the first week following menses. Measurements were made at Grenoble University Hospital, Grenoble.
Measurements
All subjects were asked to avoid caffeine and other respiratory stimulants during all experimental sessions. Subjects were allowed to have regular light meals before all sessions.
Sleep
One full-night polysomnography was performed in normoxia followed by a second one in hypoxia (inspiratory oxygen fraction (FI,O2) 14.5%, corresponding to 3,000 m), either on two consecutive nights (n=22) or within a few days (<7 days, n=2). Recordings were performed in a hypoxic tent, as previously described by Gilmartin et al. [15]. Subjects were blinded to the condition studied (normoxia or hypoxia). Details of sleep monitoring and criteria used for scoring apnoeas and hypopnoeas are reported in the online supplementary material.
Ventilatory responses during wakefulness
Subjects were studied while supine in a quiet room. Conditions of resting eupnoeic ventilation were carefully controlled as follows: motor rest without anticipation of exercise, absence of noise, light of moderate intensity, absence of specific mental tasks and relaxed wakefulness. Subjects breathed through a mouthpiece and wore a nose clip. The dead space of the apparatus was minimised and was identical in all conditions [16]. None of the subjects reported any respiratory or general discomfort at any time. All tests were performed on a Friday, between 15.00 h and 19.00 h.
Hypoxic ventilatory response (HVR) was assessed according to Lake Louise recommendations [17]. In a quiet environment, after habituation to the respiratory valve, subjects breathed spontaneously during hypoxic conditions for 20 min. Minute ventilation (V′E), end-tidal carbon dioxide tension (PET,CO2), arterial oxygen saturation measured by pulse oximetry (Sp,O2) were recorded. The hypoxic gas was delivered by the Altitrainer® system (SMTEC, Bern, Switzerland) using variable nitrogen fractions, whilst carbon dioxide (CO2) was adjusted within the inspired air (iso- or poikilocapnic) using a specific device (Isocap®; SMTEC, Bern, Switzerland). FI,O2 was set to achieve 80% Sp,O2. Acute HVR (isocapnic HRV at 5 min; i/pHVR5 min) and decline (isocapnic hypoxic ventilatory decline at 20 min; i/pHVD20 min) were derived under isocapnic and poikilocapnic conditions (i.e. with and without control of PET,CO2, respectively).
The hypercapnic ventilatory response (HCVR) test protocol was based on the method of Katayama et al. [18]. Ventilation and CO2 fraction were measured with an max 229 analyser (SensorMedics, Yorba Linda, CA, USA). HCVR sensitivity was determined as the slope of V′E plotted against PET,CO2 and the PET,CO2 threshold of the ventilatory response (intercept) was also determined.
Resting baseline V′E and PET,CO2 were analysed during a 2-min period of relaxed wakefulness at rest, after adaptation to the mouthpiece with steady-state ventilation ensured.
Echocardiography
Transthoracic echocardiography (TTE) was performed during normoxia at rest, and after 1 h of hypoxic exposure with FI,O2 adjusted in order to achieve 80% Sp,O2. Targeting a set value of Sp,O2 minimised the effect of inter-individual differences in arterial oxygenation. The pulmonary artery systolic pressure (Ppa,sys) was derived from the maximum velocity of tricuspid regurgitation by Doppler TTE, using the modified Bernoulli equation and pulmonary artery diameter estimation (see online supplementary material) [19]. Pulmonary vascular resistance (PVR) was calculated using Abbas' equation [20], and cardiac output was calculated using Huntsman’s equation [19]. Four subjects were excluded from the analysis due to insufficient Doppler profiles during hypoxic exposure, leaving the sample size for this parameter at n=20.
Blood analyses
Blood samples were taken in the supine position, upon awakening, after both study nights. Endothelin-1 and Big-endothelin were assessed by ELISA kits. Renin, aldosterone and vasopressin analyses were performed by radio-immunoassays kits (table S5).
Pulmonary measurements
Measurements for lung function and diffusing capacity of the lung for carbon monoxide (DL,CO) and nitric oxide (DL, NO) were performed in a BodyBox 5500 (Medi-Soft, Dinant, Belgium). Standardisation of DL,CO and DL,NO procedures were those reported by the European Respiratory Society/American Thoracic Society [21] and Aguilaniu et al. [22], respectively. DL,CO per unit of alveolar volume (transfer coefficient; KCO), membrane diffusion capacity (DM) and pulmonary capillary blood volume (Vc) were derived [23].
Protocol
The entire study was conducted in the laboratory. During the initial visit, subjects underwent medical examination (visit 1). On the second visit, isocapnic and poikilocapnic HVR were measured and a maximal incremental exercise test in normoxia (maximal oxygen uptake) was performed. Measurements of pulmonary vascular pressures (using TTE during normoxia followed by 1-h hypoxic exposure), lung function tests, measurement of hypercapnic ventilatory response to CO2 and lung diffusing capacity measurements were performed at visit 3. Visits 4 and 5 consisted of the sleep studies during normoxia and hypoxia respectively, followed by blood sample collections. This design avoided any influence of acclimatisation to hypoxia.
Statistical analysis
Statistics were carried out with NCSS software (NCSS, Kaysville, UT, USA). Results are expressed as the mean±sd. For continuous variables, comparisons between the AMS+ and AMS- groups were performed using paired t-test or Wilcoxon paired test, depending on the normality of distribution. A value of p<0.05 was considered to be statistically significant. Univariate conditional logistic regressions were made to determine the odds ratio for the occurrence of AMS. Values of each variable were recoded in relation to the median value. Predictive parameters were considered as significant for a probability <0.05 and if the odds ratio inferior or superior limits did not include 1.0. To test whether HAPE and HACE and/or AMS were different in ventilatory and circulatory responses, a specific analysis was performed, excluding HAPE subjects and their matched AMS subjects.
RESULTS
Physical characteristics and fitness
12 subjects with typical and recurrent altitude illness, i.e. severe AMS, with an average Lake Louise score of 9.7 and HAPE (n=4) or HACE (n=1) and matched AMS nonsusceptible subjects are presented in table 1. None of the subjects were taking medication affecting the control of breathing. One subject was on diuretic therapy for hypertension during altitude exposure and normoxic/hypoxic test sessions.
Sleep, breathing and blood oxygenation
Except for one subject presenting with moderate obstructive sleep apnoea with an apnoea/hypopnoea index (AHI) of 27 events·h−1, nocturnal ventilation was normal in both groups during normoxia. The mean values in AMS+ and AMS- subjects, respectively, were as follows: AHI 7±7 versus 9±9 events·h−1, p=0.3; Sp,O2 94±1 versus 95±1%, p=0.2. There was no difference in sleep architecture or sleep efficiency between groups. During sleep in hypoxic conditions (table 2), respiratory events (apnoeas and hypopnoeas) increased with predominantly central events (64% and 74% of total events in AMS+ and AMS- subjects, respectively). AMS+ subjects had a significantly lower AHI than AMS- subjects (18.2±18.0 versus 33.4±24.8 events·h−1, p=0.038), whilst exhibiting lower mean Sp,O2 and minimum Sp,O2 levels than AMS- subjects (81.6±2.6 versus 86.0±2.4% (p<0.01) and 73.6±3.0 versus 78.0±2.6% (p<0.01), respectively). The time spent at low levels of Sp,O2 was higher in AMS+ subjects than in AMS- subjects (fig. 1). Despite lower AHI, sleep efficiency was also reduced in AMS susceptible subjects compared with AMS- subjects (83.5±9.2% versus 90.0±6.0%, p=0.02) due to increased wakefulness after sleep onset (44.9±31.1 versus 70.4±45.1 min, p=0.03). Stage I (10.8% versus 14.8%, p<0.01) and micro-arousals associated with respiratory events (12.9±15.6 versus 23.8±19.2 events·h−1) were lower in AMS+ subjects, which was consistent with the AHI difference between groups. Also rapid eye movement sleep was slightly longer in AMS- than in AMS+ subjects (26.6 versus 21.1% of total sleep time, p<0.01), as shown in table 2. Details of sleep data are available in table S1.

Nocturnal blood oxygenation difference between acute mountain sickness-susceptible (AMS+) and AMS-nonsusceptible (AMS-) subjects in hypoxia. Nocturnal arterial oxygen saturation measured by pulse oximetry (Sp,O2) in a) a representative AMS+ and b) AMS- (tolerant) subject. Note the lower oscillations and mean Sp,O2 in the AMS+ subject. Mean apnoea/hypopnoea index (AHI) in c) AMS+ and d) AMS- subjects. e) Mean distribution of nocturnal Sp,O2 in AMS+ and AMS- subjects. TST: total sleep time.
AMS+ subjects exhibited a lower central CO2 chemosensitivity than AMS- subjects (fig. 2), with a right shift of the response due to a lower slope (2.6±1.5 versus 4.0±1.8 L·min−1·mmHg−1, p=0.02) and an increased PET,CO2 threshold (48.9±2.8 versus 45.8±4.0 mmHg, p=0.04). AMS+ subjects also had higher eupnoeic PET,CO2 (40.2±2.5 mmHg versus 35.7±2.8 mmHg, p<0.001) than AMS- subjects.

Schematic representation of differences in slope and threshold of the ventilatory response to carbon dioxide. V′E: minute ventilation; PET,CO2: end-tidal carbon dioxide pressure. *: p<0.05.
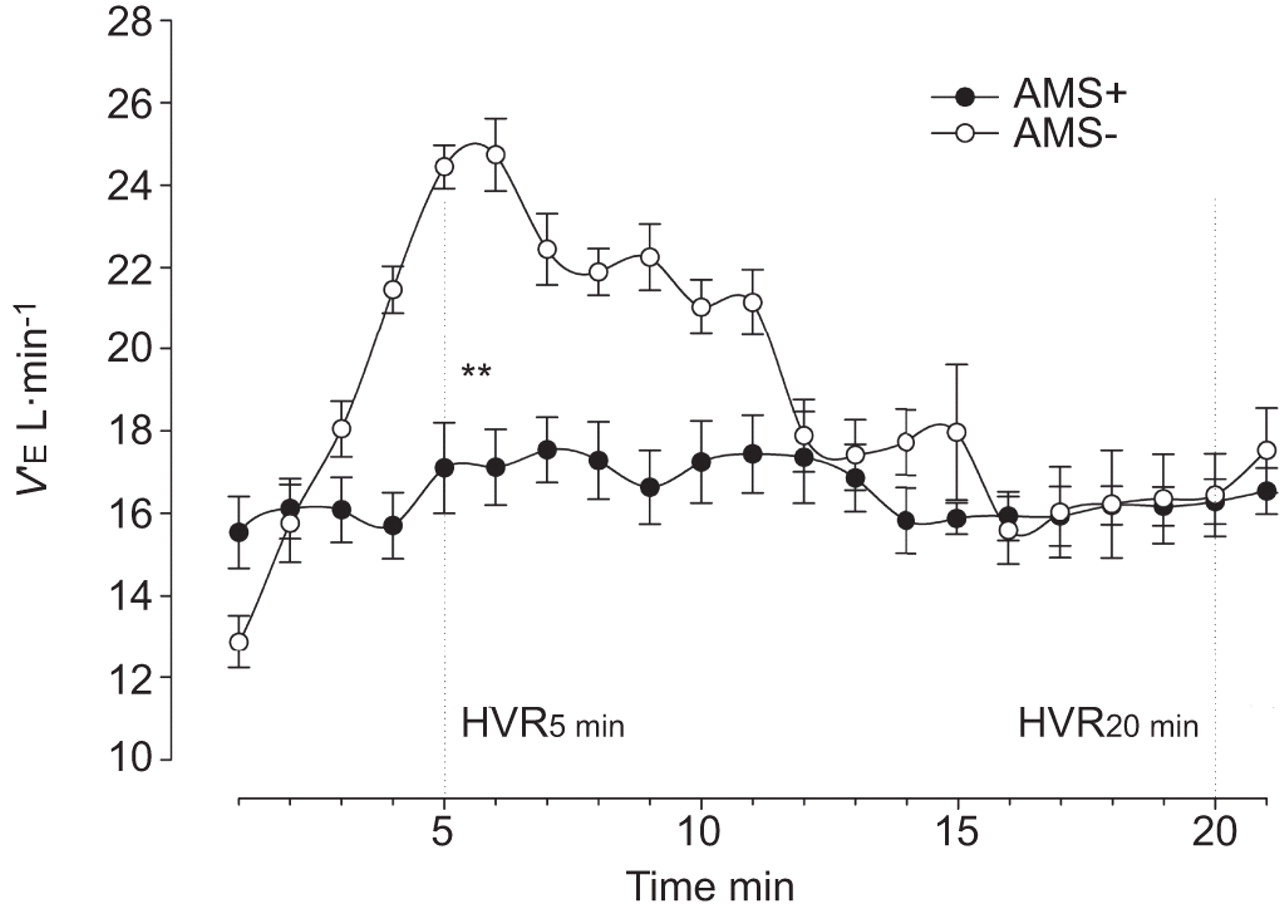
Peripheral oxygen chemosensitivity assessed by the isocapnic HVR at 5-min (iHVR5 min) was reduced in AMS+ compared with AMS- subjects (0.40±0.49 versus 0.97±0.46 L·min−1·%, p<0.001) (fig. 3). Hypoxic ventilatory decline in isocapnic hypoxia was not apparent in AMS+ (0.40±0.49 at 5 min versus 0.13±0.24 L·min−1·% at 20 min, p=0.2) compared with AMS- subjects (0.97±0.46 at 5 min versus 0.40±0.43 L·min−1·% at 20 min, p<0.001). Poikilocapnic HVR at 5-min (pHVR5 min: 0.34±0.43 versus 0.21±0.16 mmHg·%, p=0.2), poikilocapnic ventilatory decline (pHVR20 min: 0.40±0.43 versus 0.13±0.24, p=0.2) and iHVR20 min (0.58±0.45 versus 0.34±0.52, p=0.1) were not significantly different.

Mean “isocapnic” hypoxic ventilatory response (HVR) in acute mountain sickness susceptible (AMS+) and AMS nonsusceptible (AMS-) subjects. V′E: minute ventilation. **: p<0.01.
Pulmonary vasoreactivity
Ppa,sys, cardiac output and PVR are presented in figure 4. As expected, cardiac output and PVR increased after 1 h of hypoxic exposure. The mean increase in PVR in response to hypoxia (ΔPVR) was not different between groups (+0.31±0.23 versus +0.33±0.19 Wood units in AMS+ and in AMS- subjects, respectively; p>0.05), although AMS+ subjects showed an increase in Ppa,sys (ΔPpa,sys) in hypoxia (versus normoxia), which was not observed in AMS- subjects (+6.42±1.85 versus +0.66±0.19 mmHg, p=0.04) (fig. 4). This was essentially attributable to normoxic Ppa,sys values in the latter group (AMS- subjects). No significant difference in pulmonary vasoconstrictive response to hypoxia could be shown between groups in this experimental condition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pulmonary circulation characteristics a) pulmonary arterial systolic pressure (Ppa,sys), b) cardiac output and c) pulmonary vascular resistance (PVR) in normoxia (Nx) and difference regarding hypoxia (Hx) in acute mountain sickness-suseptible (AMS+) and AMS nonsusceptible (AMS-) subjects. *: p<0.05.
Pulmonary diffusion
None of the subjects displayed abnormal lung function, and both groups had similar ventilatory volumes (forced expiratory volume in 1 s and forced vital capacity). Significant differences in carbon monoxide and nitric oxide diffusion characteristics were observed at rest, in normoxia. Comparative to AMS- subjects, AMS+ subjects showed lower lung Vc (86.2±13.8% versus 95.0±22.8% pred, p=0.02), a lower DL,CO (89.8±15.2% versus 101.8±16.2% predicted, p=0.037) and a lower KCO (79.4±9.9% versus 87.5±8.9% pred, p=0.02). AMS+ also had a higher DL,CO/DL,NO (5.3±0.2 versus 5.0±0.2, p=0.03), which suggests the possibility of a vascular restriction (table 3).
Specific analysis of AMS/HACE subjects, excluding HAPE subjects and their matched nonsusceptible subjects, did not show any significant difference in change in Ppa,sys (ΔPpa,sys) between hypoxic and normoxic exposure.
Pulmonary vasoactive biomarkers and fluid retention
The plasma endothelin level was markedly increased in AMS+ versus AMS- subjects, both after the night in normoxia (6.4±1.8 versus 3.6±1.0 fmol·mL−1, p=0.02) and after the night in hypoxia (7.1±1.9 versus 3.7±1.1 fmol·mL−1, p=0.02). AMS+ subjects had a significantly higher increase in Big-endothelin after the normoxic night than AMS- subjects (1.03±0.29 versus 0.89±0.14 fmol·mL−1, p=0.04). There was no difference in the morning plasma vasopressin level between groups in normoxia (3.8±1.4 versus 4.5±1.8 pg·mL−1, p=0.12) and a lower level in AMS+ compared with AMS- subjects in hypoxia (3.0±2.4 versus 4.3±2.5 pg·mL−1, p=0.04). AMS+ subjects had significantly higher plasma bicarbonate ion levels than AMS- subjects (27.1±1.7 versus 25.4±2.6 mmol·L−1, p=0.02) owing to reduced ventilation in hypoxia, which was not found after the normoxic night (25.6±2.5 versus 25.3±1.7 mmol·L−1, p=0.2). No difference was found between groups in renin and aldosterone levels after both normoxic and hypoxic nights.
Among factors independently associated with high altitude susceptibility (hypoxic ventilatory response, capillary volume, change in pulmonary arterial pressure with hypoxia, left ventricular ejection fraction and central chemosensitivity), iHVR5 min was the only significant parameter in univariate conditional logistic regression analysis involved in AMS development. iHVR5 min was a protective factor (lower limit 0.016, upper limit 0.999; p<0.05). Above the median value of 0.58 L·min−1·%, an odds ratio of 0.125 for the occurrence of AMS was observed (p<0.05). All details of the statistical analysis are available in table S4.
DISCUSSION
The main result of the present study is that high peripheral hypoxic chemosensitivity appears to be a major protective factor for AMS. The presence of more central events with a higher AHI in the altitude-tolerant subjects is most likely to be explained by this higher chemosensitivity. Somewhat unexpectedly, this increase in central respiratory events during sleep was associated with an overall higher nocturnal oxygen level, i.e. higher Sp,O2 levels.
Lastly, hypoxic pulmonary vasoreactivity per se was not involved in AMS physiopathology in the present study. However, there was a reduced capillary bed and subtle differences in diffusion capacity of vascular origin.
Chemical control of breathing
In the present study, iHVR5 min, the index of peripheral hypoxic chemoresponsiveness without the influence of hypocapnia, represented a highly predictive factor for high altitude tolerance. Although hypoxic chemosensitivity has always been considered an important favouring factor, this remains controversial. Methodological issues have been raised [24]. As suggested by Roach et al. [6], studies where “arterial oxygen saturation (Sp,O2) after hypoxic exposure” was the criteria used to predict chemosensitivity and altitude tolerance led to coherent results (i.e. the lower the Sa,O2, the higher the severity of altitude intolerance). However, this was less clear when directly assessing the HVR (i.e. not only the Sa,O2 achieved, but also the level of ventilation). This may be related to hypocapnia [25], but also the time course of the response combining ventilatory stimulation and hypoxic ventilatory decline [7]. The HVR test used in the present study followed the Lake Louise recommendations [17] and, thus, we were able to separate the two parts of the response. iHVR5 min (i.e. measured after a 5-min hypoxic exposure) corresponds to the stimulating ventilatory phase of the response. Differences in iHVR5 min between groups cannot be attributed to baseline conditions of PET,CO2 [26], and the targeted isocapnia level, set at 1–2 mmHg above the individual’s resting normoxic PET,CO2 led to a similar level of PET,CO2 between groups during the test. Thus, our study emphasises the relevance of this index of pure hypoxic chemoresponsivness in predicting altitude tolerance (and AMS) when compared with all the other independent physiological factors tested (ventilatory decline, ΔPpa,sys between hypoxia and normoxia, baseline PVR in hypoxia, endothelin levels and central CO2 chemosensitivity). In our study, the risk of developing high-altitude illness was decreased by 80% (corresponding to OR 0.125) when iHVR5 min was higher than the median value of 0.58 L·min−1·%.
Sleep apnoea and altitude susceptibility
Periodic breathing and central events during sleep increase with altitude [27], but whether this higher incidence of central events alters altitude tolerance is still debated. Unlike the hypothesis suggesting that periodic breathing either precipitates [13] or is unrelated to [28] AMS, our results suggest that periodic breathing is associated with improved altitude tolerance. We suggest that this is related to an increase in the average nocturnal oxygen saturation.
The association between periodic breathing and improved altitude tolerance is related to the presence of a higher gain of the chemosensitive ventilatory response. It has been shown that enhanced ventilatory responses to carbon dioxide, to hypoxia or to both are critical factors for promoting periodic breathing during heart failure or in hypoxic conditions [29–31]. Thus, it is not surprising that the AMS- group, having higher chemoresponsiveness, exhibited more periodic breathing than the AMS+ group.
More unexpected was the effect of periodic breathing on nocturnal Sa,O2. The occurrence of hyperventilation after each reduction in ventilation, i.e. apnoeas and hypopnoeas, which characterises periodic breathing, resulted in an increased overall mean Sa,O2. A similar relationship between periodic breathing and nocturnal Sa,O2 was previously reported by Hackett et al. [32]. In this study, almitrine, an hypoxic ventilatory stimulant, led to a parallel increase in nocturnal Sp,O2 and periodic breathing during actual altitude exposure [32]. To the best of our knowledge, our study is the first not only to support this hypothesis but also, more importantly, to correlate with altitude tolerance. Other field studies performed at very high altitude tended to confirm that the higher the AHI, the higher the oxygen saturation achieved [27, 33]. This relationship may be related to hyperventilation-induced improvement in Sa,O2 associated with enhanced hypoxic chemosensitivity allowing hyperventilation after central events and, thus, to reach a higher oxygen saturation.
Pulmonary circulation, pulmonary diffusion and altitude susceptibility
The present study does not show any difference in hypoxic pulmonary vasoconstriction between AMS+ and AMS- subjects. A similar increase in PVR was induced by hypoxia in both groups. A greater ΔPpa,sys in response to hypoxia was only observed in AMS+, which was not associated with greater changes in resistance. A higher cardiac output (nonsignificant, trend only) and a high Ppa,sys in normoxia found in AMS- subjects are the probable explanations for these findings. Pulmonary artery pressure variations in response to hypoxia remained higher in AMS+ subjects even when subjects with AMS and known pulmonary oedema (HAPE n=4) were excluded from the analysis. Although the pulmonary artery pressure response tended to be lower than previously reported in other studies [34], it is important to note that this is the first study examining altitude intolerance in which differences in response of arterial pulmonary pressure, cardiac output and PVR measured by Doppler TTE are investigated at a targeted Sp,O2 level, instead of iso-altitude or iso-FI,O2, as in other studies [34, 35]. This provides a way to measure hypoxic pulmonary vasoreactivity per se, i.e. independently from the oxygen desaturation achieved for a given FI,O2. Indeed, since desaturation for a given FI,O2 tends to be greater in AMS+ subjects (especially in HAPE-susceptible subjects [6]), this may lead to overestimate alterations in pulmonary vascular response during hypoxic exposure. In our study, the FI,O2 needed to reach the “target” Sp,O2 of 80% was higher in AMS+ than in AMS- subjects (12.9±0.7% versus 11.8±0.3%, p<0.05). Overall, our study does not confirm exaggerated hypoxic pulmonary vasoconstriction as a key factor for AMS susceptibility. Nevertheless, subtle differences in the pulmonary circulation at baseline were observed with reduced DL,CO and lower KCO. Previous functional studies have shown that reduced DL,CO was common in AMS+ subjects during altitude exposure [8]. In our study, diffusion defects of limited amplitude were observed in AMS+ subjects in normoxia at rest. Although clinically unimportant in normoxia, this could lead to lower blood oxygen saturation at altitude. Reduced Vc blood volume, measured using carbon dioxide/nitric oxide diffusion, was observed in AMS+ subjects. Combined carbon dioxide/nitric oxide diffusion suggested vascular involvement. DM was similar between groups. The interpretation of these changes and the precise significance in AMS pathophysiology deserve further investigations.
Lastly, after the hypoxic night, plasma endothelin-1 levels were higher in AMS+ than in AMS- subjects. This difference may be linked to an altered vasoconstrictive tone in AMS+ subjects. Unexpectedly, vasopressin level was lower in AMS+ than in AMS- subjects [10], which would tend to compensate for increased diuresis and renal excretion of bicarbonates.
In conclusion, the present study suggests that hyperventilation associated with high HVR is a major factor for preventing AMS occurrence. This is also the case during sleep, since periodic breathing and central sleep apnoeas result in a better preserved oxygenation, probably due to a more pronounced hyperventilation following the apnoeic events.
iHVR5 min below a “threshold” of 0·58 L·min−1·% is a highly predictive factor for AMS occurrence in the present study, but should be further tested in a prospective study including nonselected subjects.
Acknowledgments
The authors would like to thank B. Tollenaere, D. Jean and A. Favre Juvin for helpful discussions on altitude medicine, to N. Arnol for help in statistical analysis, to M. Mendelson and J. Loeppky for English editing, and to all subjects for their participation in this study.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
Funding was provided by the PHRC Inter-Régional 2007, Conseil Scientifique AGIRadom, Région Rhône-Alpes (Projet CIBLE 2010). The authors are grateful to the DRCI CHU de Grenoble (PHRC Régional 2007 “ALT” awarded to B. Wuyam), Conseil scientifique AGIRadom (awarded to B. Wuyam and H. Nespoulet), and the ANR “Tech-scan” (awarded to J-L. Pépin) for financial support.
Statement of Interest
None declared.
- Received April 29, 2011.
- Accepted January 20, 2012.
- ©ERS 2012
REFERENCES










