





Abstract
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease and is currently the fourth leading cause of death worldwide. Chronic inflammation and repair processes in the small airways are characteristic of COPD. Despite extensive efforts from researchers and industry, there is still no cure for COPD, hence an urgent need for new therapeutic alternatives. MicroRNAs are such an option; they are small noncoding RNAs involved in gene regulation. Their importance has been shown with respect to maintaining the balance between health and disease. Although previous reviews have discussed the expression of microRNAs related to lung disease, a detailed discussion regarding the function of differential miRNA expression in the pathogenesis of COPD is lacking.
In this review we link the expression of microRNAs to different features of COPD and explain their importance in the pathogenesis of this disease. We further discuss their potential to contribute to the development of future therapeutic strategies.
Abstract
Complexity of miRNAs in COPD: an up-to-date overview of the role of miRNAs in the different features of COPD http://ow.ly/Pk4FP
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic heterogeneous disease of the lungs characterised by persistent and excessive inflammation, leading to tissue remodelling, alveolar lesions, airflow limitation and accelerated lung function decline [1]. Over 300 million people suffer from COPD. It is currently the fourth leading cause of death worldwide and predicted by the World Health Organization to become the third leading cause by 2030 [2]. Cigarette smoking is the predominant risk factor for COPD, while exposure to other noxious gases has also been identified as a risk factor. Although smoking cessation can delay disease progression, there is no cure for COPD and current medication cannot reverse the long-term decline in lung function.
Inhaled cigarette smoke first encounters the airway epithelium, which forms a continuous and highly regulated barrier. The exposure of airway epithelium to noxious particles like cigarette smoke causes the release of pro-inflammatory mediators, including damage-associated molecular patterns and cytokines like interleukin (IL)-1, IL-8 and tumour necrosis factor (TNF)-α [1, 3, 4]. Inflammatory cytokines cause lung infiltration by inflammatory cells that further amplify the inflammation and release proteases and reactive oxidative species that damage the parenchymal lung tissue, contributing to emphysema development in COPD patients [5]. Repeated injury leads to fibroblast activation, which causes excessive extracellular matrix deposition and remodelling of the small airways in COPD [6]. However, fibroblasts appear unable to provide adequate tissue repair after smoke-induced damage in the parenchyma of COPD lungs [5].
Besides the inflammatory and structural cells in the lung and pro-inflammatory mediators that contribute to the pathogenesis of COPD, microRNAs (miRNAs) have recently been implicated in COPD pathogenesis [7]. MiRNAs are small noncoding RNAs of ∼19–25 nucleotides. MiRNAs can cause post-transcriptional gene repression either by increasing mRNA degradation or by inhibiting protein translation of specific mRNA targets.
Currently, over 2400 mammalian miRNAs (listed in miRBase release 21 [8, 9]) are known to be expressed in a wide variety of cell types and have been predicted to target about 60% of mammalian mRNAs [6].
There is large overlap in gene regulation by miRNAs, as several miRNAs can regulate a single gene, and multiple genes can be affected by a single miRNA [7]. Potential miRNA targets can be predicted with bioinformatics tools that recognise complementary elements in the 3′ untranslated region (UTR) of genes and in the seed sequence of the miRNAs [10]. However, evidence suggests that this sequence pairing is not necessarily a reliable predictor of a miRNA function, since it only compares the sequences; it does not consider other variables and interactions that are present in vivo [10]. Therefore, experimental validation of miRNA targets is crucial to understand mRNA function. The most direct way of miRNA target validation is by luciferase assays, where the 3′UTR of the mRNA of interest is cloned into the 3′ region of a luciferase reporter plasmid for further evaluation of target gene expression in cells that are transfected with these constructs [10]. Another, indirect, way of validating miRNA targets is by assessing the effect of miRNA up- or downregulation on protein expression of predicted targets. In the present review, differences between validated and predicted targets are indicated.
MiRNA expression levels can be determined by different techniques such as small RNA sequencing, microarray hybridisation and quantitative reverse transcriptase PCR (qRT-PCR). The first two techniques are unbiased screening approaches that provide a large number of results at once, giving an estimate of the overall changes in miRNA expression profile between different samples. In general, the key findings of these unbiased approaches are validated using a miRNA-specific qRT-PCR approach, which is a more sensitive and quantitative method.
Regulation by miRNAs is a form of epigenetic regulation that involves inherited alterations in gene expression, which cannot be attributed to modifications in the DNA sequence. Aside from the regulation by noncoding RNAs of which miRNAs are part, epigenetic mechanisms include histone modification and DNA methylation [11]. These mechanisms do not only individually affect gene expression, but can also interact to affect disease pathogenesis. For example, the methylation of CpG islands in the promoter regions of miRNAs can inhibit miRNA transcription, while miRNAs can target and inhibit the activity of DNA methyltransferases [12, 13].
The presence and stable expression of miRNAs in serum, plasma, sputum, urine and several organ tissues and the role of their dysregulation in disease indicate their importance, allowing further exploration of their use as biomarkers and therapeutic targets [14]. The first successful trials using miRNAs as therapeutic targets in liver disease have been reported [15].
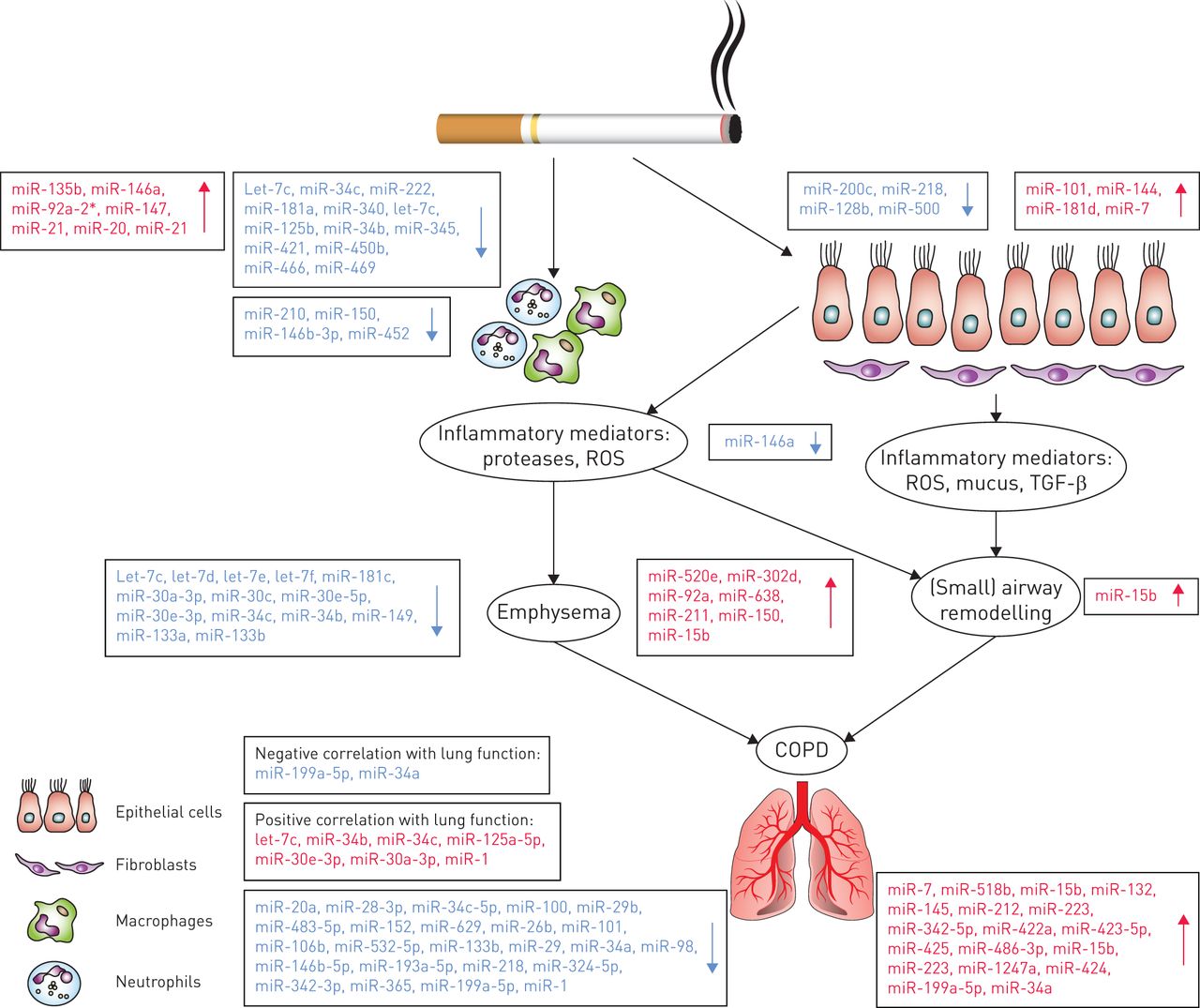
Although some recent reviews have discussed the role of miRNAs in lung diseases, a detailed review focusing on the specific role of miRNAs in COPD pathogenesis is lacking. Hence, in this review we will provide an overview of the studies published on differential miRNA expression in COPD and healthy individuals, and we will discuss the role of specific miRNAs involved in different aspects of COPD pathogenesis. We will cover the following topics: 1) the effects of cigarette smoke exposure on miRNA expression; 2) the potential roles of miRNAs in the excessive inflammatory response, the presence of emphysema and the tissue repair response in COPD; 3) the association of miRNAs with lung function; and 4) the potential of miRNAs as therapeutic targets in COPD. Overviews of the discussed miRNAs can be found in tables 1 and 2 and a schematic overview is shown in figure 1.
{kind=link}
Differential regulation of microRNAs in chronic obstructive pulmonary disease (COPD). A schematic representation of the pathophysiology of COPD with the various differentially regulated microRNAs highlighted. ROS: reactive oxidative species; TGF: transforming growth factor.
MicroRNAs involved in different features of chronic obstructive pulmonary disease (COPD)
MicroRNAs involved in more than one feature of chronic obstructive pulmonary disease (COPD)
The effects of cigarette smoke exposure on miRNA expression
As cigarette smoking is the major risk factor for COPD, we first describe the effects of cigarette smoke exposure on miRNA expression profiles. Several studies using different models have shown that direct exposure to cigarette smoke or exposure to cigarette smoke extract (CSE) affects the miRNA expression in the lungs.
An in vitro study by Hassan et al. [16] evaluated the effect of CSE on the human bronchial epithelial cell line 16HBE and showed that 24-h CSE exposure induces upregulation of miR-101 and miR-144 expression. These two miRNAs are predicted to target the cystic fibrosis transmembrane conductance regulator [16], which has previously been shown to have a reduced function upon cigarette smoke exposure [36]. Interestingly, they also found a higher miR-101 expression in slides of lung tissue samples from COPD patients compared with controls, suggesting that this miRNA may also play a role in COPD pathogenesis [16]. Another in vitro study showed that CSE exposure of human bronchial epithelial cells leads to a decrease in miR-200c expression [17]. This miRNA was suggested to maintain the epithelial phenotype as it was shown to target the expression of the E-cadherin transcriptional repressors ZEB1 and ZEB2 [37, 38]. Therefore, a decrease in miR-200c expression may contribute to an increase in epithelial to mesenchymal transition (EMT). EMT represents the loss of epithelial properties and the acquisition of mesenchymal cell characteristics, associated with pathological processes and tissue remodelling. EMT has previously been linked to COPD and cigarette smoke exposure [39, 40]. The expression of miR-7 was also found to be altered by CSE. MiR-7 expression was significantly increased in CSE-exposed human airway smooth muscle cells, while this was not observed for 16HBE cells and MRC5 fibroblasts [18]. Overexpression of miR-7 in human airway smooth muscle cells led to decreased protein levels of exchange protein directly activated by cAMP (Epac)1, a predicted target for miR-7 that has previously been shown to be decreased upon CSE exposure and to have a low expression in COPD lung tissue samples [41].
In addition to in vitro studies, several animal models have evaluated the effect of cigarette smoke exposure on the miRNA profile. A microarray analysis of a rat model of 4 weeks’ cigarette smoke exposure showed downregulation of 24 miRNAs and upregulation of one miRNA (miR-294) in lung homogenates [19]. From the downregulated miRNAs, the differential expression of three (let-7c, miR-34c and miR-222) was validated by qRT-PCR. These miRNAs are predicted to regulate genes involved in cell proliferation and p53 regulation. It is noteworthy that these miRNAs have human homologues and thus may be involved in the response to cigarette smoke exposure in humans as well. In a mouse model of 4 days’ cigarette smoke exposure, miR-135b was shown by qRT-PCR to be increased in lung tissue samples [20]. The authors suggest that the upregulation of miR-135b may act as a counter-regulatory mechanism for cigarette smoke-induced inflammation, by regulating the IL-1 receptor (IL-1R1) expression. Xie et al. [21] studied the effects of 4 and 15 weeks’ cigarette smoke exposure in a rat model and demonstrated differential expression of 30 and 37 miRNAs, respectively, by miRNA array in lung homogenates. Upon validation by qRT-PCR, miR-146a, miR-92a-2*, miR-147, miR-21 and miR-20 were shown to significantly increase at 4 and 15 weeks, while miR-181a significantly decreased upon cigarette smoke exposure. Of interest, both miR-146a and miR-181a have been linked to “inflammaging”, an ageing-related state characterised by systemic chronic inflammation [42]. Since the authors also found significantly increased serum levels of miR-21 and decreased miR-181a expression in heavy smokers and COPD patients compared with healthy controls [21], they suggested that a high ratio of serum miR-21 to miR-181a expression may be a risk factor for the development of COPD and can be associated with the risk of developing COPD in heavy smokers.
Studies that have analysed samples from human smokers have also contributed to understanding the effect of cigarette smoke exposure on the miRNA expression profile. Van Pottelberge et al. [22] analysed the miRNA expression profile in induced sputum of nonsmoking and smoking controls and smoking COPD patients; their analysis was based on comparing the miRNA expression profile of these groups in a screening cohort versus a validation cohort. When comparing smoking and nonsmoking controls in the screening cohort, they found 34 differentially expressed miRNAs, most being decreased in smokers. In their validation cohort, only one of these miRNAs, miR-340, could be validated. When comparing smoking COPD patients versus nonsmoking controls, they initially found eight differentially expressed miRNAs of which two, let-7c and miR-125b, were validated. Another human ex vivo study performed miRNA array analysis on human alveolar macrophages from bronchoalveolar lavage (BAL) of smoking and nonsmoking individuals. Their analysis consisted of three subject cohorts, two of which were used for the screening and validation of the array results. This study showed an overall decrease in miRNA expression in smokers [23], with a decrease of 43 miRNAs and an increase of 11 miRNAs at a two-fold cut-off, in the first cohort. Three of the downregulated miRNAs (miR-210, miR-150 and miR-146b-3p) were validated by qRT-PCR in two of the cohorts. The most downregulated miRNA, miR-452, was followed up in vitro, where its expression was shown to be inversely related to the expression of its predicted target, matrix metalloproteinase (MMP)-12. This is of interest, since MMP-12 has been found to be increased in sputum of COPD patients [43] and contributes to the development of emphysema [44]. In addition to these studies, miRNA profiling on bronchial airway epithelial cells of current and never-smokers revealed differential expression of 28 miRNAs, most of which were downregulated in the current smokers [24]. Of these, miR-218, miR-128b, miR-500 and miR-181d were validated using qRT-PCR, and all were downregulated with smoking except miR-181d, which was upregulated. MiR-218 was found to be most strongly correlated with genes that were strongly upregulated by cigarette smoke exposure. Within the negatively correlated genes, there was an overrepresentation of miR-218 predicted target genes involved in cell structure, cell–cell adhesion, and cell signalling and ion transport pathways [24].
Considering all the evidence from these studies, we propose that the cumulative effects of cigarette smoke on lung tissue damage and inflammation are reflected by an effect on the miRNA profile, and that smokers with and without COPD develop differential miRNA profiles. Interestingly, Izzotti et al. [25] demonstrated that cigarette smoke-induced miRNA changes in mouse lungs were dose dependent and only partly reversible, as the expression levels (by miRNA microarray) of several miRNAs, including miR-34b, miR-345, miR-421, miR-450b, miR-466 and miR-469, did not revert to basal levels after 1 week of smoking cessation. These findings suggest that the stability of alterations in some miRNAs after smoking cessation might be dependent on the cigarette smoke dose. In particular, irreversible changes could contribute to the development of COPD, although it is important to take into consideration that, due to the high variability of the genetic background that exists between humans, a high-dose long-term cigarette smoke exposure will not always lead to COPD. Again, it is also important to note that effects of smoking cessation in an animal model might not be comparable to those observed in humans. In line with this study, in collaboration with Bossé et al. [45], we have previously shown that smoking deregulates the levels of many genes in human lung tissue and that for most of the genes, expression levels are reverted to control levels upon smoking cessation. However, some genes, including serpin peptidase inhibitor clade D, member 1 (SERPIND1), showed no or a very slow reversal to basal levels.
Although most of the results from these studies do not point at the same miRNAs, some miRNAs have been associated with multiple features of COPD (table 2), and may thus be relevant for the pathogenesis of COPD.
Differential miRNA expression in COPD
To date, seven studies have reported differential miRNA expression in COPD patients in comparison with (smoking) control subjects. Overlap between these studies is rather limited and this is mainly due to different types of samples (i.e. serum, plasma, BAL, whole blood or lung tissue), small sample sizes, differences in (statistical) methods and inclusion criteria used.
Using a qRT-PCR miRNA array approach, Akbas et al. [14] showed that four miRNAs (miR-20a, miR-28-3p, miR-34c-5p and miR-100) were downregulated and one miRNA (miR-7) was upregulated in serum of COPD patients compared with smokers without COPD. Another study, evaluating miRNA expression levels in the plasma of healthy smokers and current smoking COPD patients, found nine differentially expressed miRNAs in COPD (miR-29b, miR-483-5p, miR-152, miR-629, miR-26b, miR-101, miR-106b, miR-532-5p and miR-133b) [26]. Using qRT-PCR, this study also showed that in ex-smoking COPD patients, miR-106b expression negatively correlates with the duration of COPD since diagnosis, whereas in current smoking COPD patients, miR-106b expression negatively correlates with years of smoking. Therefore, the authors suggest that plasma levels of miR-106b may reflect persistent and systemic changes even after smoking cessation in COPD.
A study by Leidinger et al. [27] focused on comparing miRNA profiles from whole blood of COPD patients, lung cancer patients and healthy volunteers. Here, 140 miRNAs were found to be differentially expressed between COPD and control, of which miR-518b was most significantly upregulated; this result, however, was not confirmed by qRT-PCR. Another study compared miRNA profiles by miRNA array in the cell fraction of BAL fluid of 23 COPD patients and 15 smokers and one ex-smoker without COPD. qRT-PCR validation was performed for a group of random miRNAs among those that were differentially expressed. The study found 10 miRNAs to be upregulated (miR-15b, miR-132, miR-145, miR-212, miR-223, miR-342-5p, miR-422a, miR-423-5p, miR-425 and miR-486-3p) and nine to be downregulated (miR-29, miR-34a, miR-98, miR-146b-5p, miR-193a-5p, miR-218, miR-324-5p, miR-342-3p and miR-365) in the COPD group when compared with the control group without COPD [28].
A miRNA profile analysis of lung tissue samples from COPD patients and smokers without airflow limitation showed differential expression of 70 miRNAs; 13 were downregulated and 57 were upregulated in COPD [29]. Four miRNAs (miR-15b, miR-223, miR-1274a and miR-424) were validated by qRT-PCR. Of those, miR-15b was highly expressed in lung tissue samples from COPD patients, with the highest expression in stage IV COPD patients compared with other stages of COPD and with control smokers. SMAD7, a protein of the transforming growth factor (TGF)-β pathway, which inhibits the production of collagen and other matrix proteins, was found to be one of the predicted targets for miR-15b that decreased upon miR-15b overexpression in the human bronchial epithelial cell line BEAS-2B. These results correlate with previous findings from our group, showing that SMAD7 protein expression is significantly lower in lung epithelium from stage II and IV COPD patients compared with control subjects, while other SMAD proteins did not significantly differ [46].
Another miRNA profiling study that analysed regulatory T-cells (Tregs) showed that miR-199a-5p is downregulated in Tregs of COPD patients compared with control smokers and healthy nonsmokers; this result was confirmed with qRT-PCR [30]. Several of the predicted targets of this miRNA are strongly related to the TGF-β pathway. Additional in vitro experiments with a miR-199-5p inhibitor showed that this miRNA has potential targets belonging to bone morphogenetic protein signalling, which promotes TGF-β-induced Treg differentiation [47]. Thus, the altered miR-199-5p expression might contribute to the dysregulation of pro-inflammatory T-cell-mediated inflammation in COPD.
The ultimate effects of the changes in the miRNome that differentiate COPD patients from smokers with normal lung function are possibly accumulating over time and may be related to the onset of disease and disease severity. With respect to the relationship of changes in miRNome to lung function, the expression of let-7c, miR-34b, miR-34c, miR-125a-5p, miR-30e-3p and miR-30a-3p has been positively correlated with forced expiratory volume in 1 s (FEV1) [22]. Furthermore, Lewis et al. [31] evaluated the role of miR-1 in the skeletal muscle dysfunction that accompanies COPD. MiR-1 expression, evaluated by qRT-PCR, was decreased in skeletal muscle of COPD patients compared with controls and positively correlated to FEV1. Mizuno et al. [32] evaluated the expression of miR-199-5p and miR-34a in COPD, since these miRNAs were thought to be playing a role in regulation of hypoxia-inducible factor-1α, previously shown to be altered in COPD. This study found by qRT-PCR that miR-199-5p and miR-34a were upregulated in lung tissue samples from COPD patients with moderate to severe COPD compared with nonsmoking individuals with normal lung function, and this increased expression was clearly associated with impaired lung function. This suggests that these changes in miRNA profile might not only be related to cigarette smoke exposure, but are in fact changes associated with pathogenesis of disease and reflected by the reduced lung function present [32].
Involvement of miRNAs in inflammatory responses
Although different miRNAs have been implicated in different aspects of the regulation of the inflammatory response, there is one miRNA in particular that has been directly linked to COPD pathogenesis, miR-146a. Sato et al. [33] found that lung fibroblasts from COPD patients have a lower induction of miR-146a expression upon IL-1β/TNF-α stimulation in comparison with fibroblasts from healthy controls. In addition, they showed that downregulation of miR-146a expression increases the levels of its target, cyclooxygenase 2, subsequently leading to higher prostaglandin (PG)E2 production in lung fibroblasts. These findings are in line with the high levels of PGE2 in sputum of COPD patients and the association of higher PGE2 levels with more severe airflow limitation during progression of COPD [48]. Of note, cigarette smoke exposure has been shown to increase miR-146a expression in murine pulmonary fibroblasts by a mechanism involving the nuclear factor-κB member RelB [49]. Various other studies have linked miR-146a to the regulation of inflammatory responses, showing that miR-146a targets IRAK1 and TRAF6, two adapter proteins involved in cytokine receptor signalling [50], and IL-1β-induced IL-8 production in human lung epithelial cell lines [51]. Furthermore, miR-146a negatively modulates the inflammatory mediators IL-6 and IL-8 in human skin fibroblasts [52].
There are other miRNAs that, although not directly related to the inflammatory response in COPD, have been linked to inflammation and to COPD separately. Among the most downregulated miRNAs in COPD found by Van Pottelberge et al. [22] there are several miRNAs that are known to regulate inflammatory responses, including miR-125b, miR-150 and miR-203. MiR-125b was found to significantly reduce lipopolysaccharide (LPS)-induced pulmonary inflammation in a mouse model, as reflected by reductions in total and neutrophil cell counts [53]. Furthermore, higher miR-150 expression correlated with lower CXCL1 expression in mice, while miR-203 was found to target MyD88 in LPS-stimulated monocytes [54]. In addition, miR-181a and miR-20a, which are also downregulated in COPD [21], have been linked to inflammatory processes. MiR-181a regulates inflammatory responses associated with IL-1β levels in LPS-stimulated monocytes [55] and IL-8 levels in human fibroblasts [56]. Transfection of miR-20a mimics in human monocytes decreased the release of IL-1β and TNF-α, and injection of miR-20a mimics in mice led to a global decrease in IL-6 secretion in macrophages that were stimulated with LPS in vitro [57]. Considering that miR-125b, miR-150, miR-203, miR-181a and miR-20a are downregulated in COPD patients and that they all regulate inflammatory processes, it is possible that their alteration is involved in the pro-inflammatory phenotype observed in COPD.
Involvement of miRNAs in emphysematous lesions
Emphysema is induced by chronic inflammation upon cigarette smoking and characterised by loss of alveolar tissue and a decrease in lung elastic recoil. In collaboration with the groups of Spira and Hogg, we were involved in the first study investigating the direct relationship between mRNA [58] and miRNA expression changes, both measured by microarrays, and emphysema severity as measured by micro-computed tomography scans [34]. Increased emphysema severity was associated with increased expression of 35 miRNAs, including miR-520e, miR-302d, miR-92a, miR-638, miR-211 and miR-150. Increased emphysema severity was associated with decreased expression of 28 miRNAs, including let-7c, let-7d, let-7e, let-7f, miR-181c, miR-181d, miR-30a- 3p, miR-30c, miR-30e-5p and miR-30e-3p. The expression of five miRNAs (miR-638, miR-18a-3p, miR-483-3p, miR-181d and miR-30c) was significantly correlated with more than 50 of their predicted target genes. Of those, miR-638, miR-30c and miR-181d had the most anti-correlated predicted target genes, and these target genes were enriched in pathways associated with emphysema, suggesting that these miRNAs may be important regulators of the gene expression changes associated with emphysema severity. Subsequent inhibition of miR-638 in primary lung fibroblasts led to the modulation of pathways that were previously implicated in oxidative stress and accelerated lung ageing responses.
In a study by Ezzie et al. [29], in situ hybridisation showed miR-15b localisation in areas of emphysema as well as in areas of fibrosis, with increased expression in COPD patients compared with controls. Although the authors did not further investigate the role of miR-15b in emphysema, they found that miR-15b overexpression in bronchial epithelial BEAS-2B cells resulted in decreased SMAD7, decorin and SMURF2 protein expression. Our group has shown that stimulation with TGF-β1 and basic fibroblast growth factor induces a significantly more pronounced downregulation of decorin in cultured fibroblasts from patients with severe emphysema than from those with mild emphysema [59], and miR-15b may thus be related to the severity of emphysema in COPD.
Savarimuthu Francis et al. [35] evaluated miRNA expression by miRNA microarray in lung tissue samples of COPD patients with moderate and mild emphysema defined by gas transfer measurements. They found five miRNAs that were significantly downregulated in lung tissue from patients with moderate emphysema compared with mild emphysema: miR-34c, miR-34b, miR-149, miR-133a and miR-133b. MiR-34c exhibited the largest difference. Although it is not clear if and how miR-34c may contribute to the severity of emphysema, the authors found that higher miR-34c expression correlated with lower mRNA expression of five of its predicted targets: MAP4K4, SERPINE1, ALDOA, HNF4A and ZNF3. Among these, SERPINE1 had the strongest correlation with the miR-34c expression. Since SERPINE1 is a protease inhibitor, it is possible that miR-34c expression in emphysema is involved in the disturbed protease–antiprotease imbalance that is characteristic of the pathogenesis of emphysema [35].
Involvement of miRNAs in tissue repair
There are several miRNAs that have been linked to the development of pulmonary fibrosis, but not many have been described as regulators of hampered tissue repair, airway remodelling and fibrosis in COPD. As mentioned, Ezzie et al. [29] reported increased expression of miR-15b in areas of fibrosis in lung tissue samples from COPD patients and found that miR-15b targets several genes from the TGF-β superfamily, including SMAD7.
Since tissue repair and the fibrotic response have common initiating events, it is possible that the same miRNAs are involved in their regulation. Therefore, it would be of interest to investigate whether miRNAs that have been associated with fibrosis are also involved in tissue repair and airway remodelling in COPD. Potential candidates are miR-21 [21] and let-7 [19, 22, 34], since these were found to be involved in repair of damaged lungs after influenza infection [60], while miR-21 also mediates the fibrogenic activation of pulmonary fibroblasts [61]. Huleihel et al. [62] showed that the overexpression of let-7d in lung fibroblasts induced a delay in tissue repair and reduced motility and proliferation of fibroblasts. Furthermore, let-7d attenuated the expression of TGF-β downstream targets such as the high-mobility group AT-hook 2 (HMGA2) and α-smooth muscle actin (α-SMA) [62]. Several members of the let-7 family, including let-7d and let-7c, were associated with emphysema severity in lung tissue samples [34], and let-7c was found to be decreased in sputum of COPD patients and correlated positively with FEV1 [22].
Other miRNAs that could be of importance for tissue repair in COPD are miR-29b (downregulated in COPD) and miR-145 (upregulated in COPD) [28, 63]. MiR-29b has been found to mediate TGF-β1-induced extracellular matrix synthesis through activation of the PI3K-AKT pathway in human lung fibroblasts [64]. MiR-145 has been shown to regulate myofibroblast differentiation and lung fibrosis [63]. Yang et al. [63] found that miR-145 expression is upregulated in TGF-β1-treated lung fibroblasts and that its expression is also higher in the lungs of patients with idiopathic pulmonary fibrosis compared with normal human lungs. Overexpression of miR-145 in lung fibroblasts increased α-SMA expression, enhanced contractility and promoted formation of focal and fibrillar adhesions [63]. One of the most interesting results of this study is the protection of miR-145−/− mice from bleomycin-induced pulmonary fibrosis, suggesting that miR-145 may be a potential target in the development of novel therapies to treat pathological fibrotic processes, as observed in the airway wall in COPD.
MiRNAs as targets for future therapeutic strategies in COPD
The first successful miRNA-based therapy involved treating hepatitis C virus (HCV) infections. Miravirsen is a locked nucleic acid-modified DNA phosphorothioate antisense oligonucleotide that sequesters mature miR-122 in a highly stable heteroduplex. This inhibits the function of miR-122, which is required for the propagation of HCV in the liver [15]. The success of the phase 1 and 2 clinical trials of this new drug represented a major breakthrough for miRNA therapy in humans.
Depending on how a particular miRNA is dysregulated in disease, two different therapeutic approaches can be used. The first approach involves the use of miRNA antagonists, including anti-miRNAs, locked nucleic acids and antagomiRs, to inhibit the activity of miRNAs. These molecules are complementary oligonucleotides that either degrade the endogenous miRNA or trap it in a specific configuration to prevent its processing by the RNA-induced silencing complex (RISC) [65]. In the second approach, oligonucleotide miRNA mimics or gene vectors are used to increase the expression levels of miRNAs. This approach has been studied extensively in various cancers, where miRNA mimics are introduced into cells to restore miRNA function where they are downregulated [6, 65, 66]. An important advantage of miRNA therapy is the potential to affect and regulate several pathways that are dysregulated in disease [65].
The various miRNAs that are deregulated in COPD pathogenesis could be candidates for miRNA-based therapy. We have described studies in this review that have highlighted this dysregulation and may serve as the basis for miRNA therapy studies. Although there is little overlap in the miRNAs reported, common miRNAs to various features of COPD, such as let-7c, miR-15b, miR-21 and miR-34b (table 2), could serve as a basis for future therapeutic investigations.
As described, we have also shown that inhibiting miR-638 in lung fibroblasts modulates genes that may be important in emphysema [34]. This could serve as the beginning of studies that investigate the use of miRNA therapy to reverse the cellular damage and aberrant repair in emphysema. Another example is the use of a miRNA antagonist to inhibit miR-15b, which has been shown to be upregulated in lung tissue from COPD patients [29]. Studies may also focus on the use of mimics to increase the expression of miR-146a, which has been shown to have anti-inflammatory effects on both pulmonary fibroblasts [33] and bronchial epithelium [24].
Due to the fact that remodelling processes are opposite in COPD airways (fibrosis) compared with parenchyma (emphysema), when considering these approaches, attention must also be paid to targeting strategies to direct the miRNA modulators to the correct lung compartment.
MiRNA antagonists and mimics have been studied in experimental models, providing promising outcomes. Although these studies were mainly performed with other lung disease models, they can serve as good examples for investigating miRNA therapy in animal models of COPD. For example, in a mouse model of lung cancer, let-7 mimics were successfully used to inhibit tumour growth after intravenous administration of a neutral lipid emulsion that contained the mimic [67]. A synthetic miR-34a mimic delivered to another mouse model of lung cancer inhibited lung tumour growth and suppressed known cancer-promoting genes [68]. The knock-down of miR-155 in a mouse model of allergic asthma led to an attenuated T-helper cell (Th)2 response, with a reduction in Th2 cytokines compared with wildtype mice [69]. These studies show the potential of bringing in vitro miRNA studies in COPD to animal models and, in due course, to clinical trials.
It is important to realise that there may also be limitations to a therapeutic approach involving miRNAs. There is the possibility of toxicity as a result of off-target miRNAs or unintended effects of miRNA mimics or antagonists, which could be due to nonspecific delivery. Furthermore, the pathways affected by miRNAs could be adversely deregulated into hyper- or hypo-activation, which may have deleterious consequences for normal cells [65]. In this respect, it is worth noting that most manuscripts describing the use of miRNA mimics and antagonists have not adequately addressed the quantitative effects and physiological relevance of the increased or decreased miRNA levels. These concerns especially apply to in vitro studies, where miRNA levels may be increased far beyond levels of physiological relevance. When making the translation of experimental in vitro and in vivo models to clinical trials in humans, as was done for miravirsen, dose-finding experiments are mandatory. In the case of miravirsen, no dose-limiting toxic effects or binding-site escape mutations were observed after delivery of 3, 5 or 7 mg·kg−1 of body weight for 5 weeks [15]. Notwithstanding this, the toxicity of exogenous miRNAs still remains an important area to investigate in miRNA therapeutics, in which specific cell or tissue compartment targeting may also play an important role [65].
Conclusions
In this review, we have provided an up-to-date overview of the current knowledge on differential miRNA expression in COPD and discussed how the dysregulation of miRNAs may contribute to different aspects of COPD pathogenesis. Increasing evidence supports a role for miRNA deregulation in persistent inflammation, tissue repair and tissue remodelling, leading to large and small airways pathology and emphysema in COPD. Although there are several publications on miRNAs and their dysregulation in COPD, there is relatively limited overlap in the observed miRNAs and few studies have directly related the observed dysregulation in miRNAs to their biological function in COPD. This limited overlap might be due to the fact that miRNAs are cell type specific and the discrepancy between studies in terms of the use of different samples (e.g. cell type, type of tissue, serum versus plasma) and different experimental approaches. However, several miRNAs have been shown to be involved in more than one feature of COPD (table 2) and those could serve as a starting point for the functional studies that are needed to make the translation to new therapeutics in COPD that are directed towards inflammation and remodelling in COPD. This will improve our insights into the various roles of miRNAs and their potential as novel therapeutic targets in COPD.
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received November 20, 2014.
- Accepted June 15, 2015.
- Copyright ©ERS 2015
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- The effects of cigarette smoke exposure on miRNA expression
- Differential miRNA expression in COPD
- Involvement of miRNAs in inflammatory responses
- Involvement of miRNAs in emphysematous lesions
- Involvement of miRNAs in tissue repair
- MiRNAs as targets for future therapeutic strategies in COPD
- Conclusions
- Footnotes
- References
- Figures & Data
- Info & Metrics