





Abstract
Bronchopulmonary dysplasia (BPD) is characterised by impaired alveolarisation, inflammation and aberrant vascular development. Phosphodiesterase (PDE) inhibitors can influence cell proliferation, antagonise inflammation and restore vascular development and homeostasis, suggesting a therapeutic potential in BPD.
The aim of the present study was to investigate PDE expression in the lung of hyperoxia-exposed mice, and to assess the viability of PDE4 as a therapeutic target in BPD.
Newborn C57BL/6N mice were exposed to normoxia or 85% oxygen for 28 days. Animal growth and dynamic respiratory compliance were reduced in animals exposed to hyperoxia, paralleled by decreased septation, airspace enlargement and increased septal wall thickness. Changes were evident after 14 days and were more pronounced after 28 days of hyperoxic exposure. At the mRNA level, PDE1A and PDE4A were upregulated while PDE5A was downregulated under hyperoxia. Immunoblotting confirmed these trends in PDE4A and PDE5A at the protein expression level. Treatment with cilomilast (PDE4 inhibitor, 5 mg·kg−1·day−1) between days 14 and 28 significantly decreased the mean intra-alveolar distance, septal wall thickness and total airspace area and improved dynamic lung compliance.
Pharmacological inhibition of phosphodiesterase improved lung alveolarisation in hyperoxia-induced bronchopulmonary dysplasia, and thus may offer a new therapeutic modality in the clinical management of bronchopulmonary dysplasia.
Bronchopulmonary dysplasia (BPD) is a chronic lung disease of premature infants exposed to high oxygen levels and mechanical ventilation 1, 2 and is characterised by a pronounced arrest of alveolarisation. The overall incidence of BPD has not changed over the past decade 3. In total, 75% of infants born weighing ≤1,000 g and earlier than 32 weeks gestational age present with BPD, which remains a major cause of pulmonary morbidity that persists into adulthood 4. As a consequence of improved therapeutic strategies, the pathological picture of BPD has changed. The “old” BPD with prominent fibroproliferation is now less frequent 5. The “new” BPD is characterised primarily by the disruption of distal lung growth with large, simplified alveolar structures, a dysmorphic capillary configuration and variable interstitial cellularity 5. This leads to a reduction in the number of alveoli and a reduced gas exchange surface area 6.
BPD is a disease with a multifactorial aetiology 5. The pathogenesis of BPD involves extreme lung immaturity, treatment induced oxygen, volutrauma and barotrauma injury, an inflammatory response that elicits lung injury and varying degrees of disorganised repair 6. Impaired microvascular development is also observed 7, 8, and the disease can be complicated by pulmonary hypertension (PH) 9. The pathophysiological mechanisms include aberrant cell proliferation, inflammation, oxidative stress, infection and impaired microvascular development 10.
The secondary messengers, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), play a key role in signal transduction 11 and, thus, the regulation of physiological processes central to the pathogenesis of BPD. These cyclic nucleotides are metabolised intracellularly by cyclic nucleotide phosphodiesterases (PDEs) 12. The PDE superfamily currently consists of 11 family members with differing selectivity for cAMP and/or cGMP 11. Several PDEs are abundant in neonatal and adult lung tissue 13, 14, including PDE1, PDE4 and PDE5 isoforms 11. The documented roles for PDE5 in vascular homeostasis resulted in the evaluation of PDE5 inhibitors for the treatment of disorders with a vascular component, including PH 15, 16 and BPD 17. In the context of BPD, treatment of hyperoxia-exposed rat pups with sildenafil, a selective PDE5 inhibitor, preserved alveolar growth and angiogenesis, and decreased the structural evidence of PH 17. These data suggested that PDE-regulated cyclic nucleotide levels do play a role in hyperoxic lung injury in mice.
The bulk of cAMP-hydrolysing activity of a cell is attributed to the PDE4 family 11, which consists of four distinct genes that are ubiquitously expressed 18 and are already under evaluation as therapeutic targets in inflammatory diseases, such as chronic obstructive pulmonary disease (COPD), with the PDE4-specific inhibitor cilomilast 19. Pentoxifylline, a nonselective PDE inhibitor has been shown to reduce fibrin deposition and to prolong survival in hyperoxic lung injury 20. A pilot clinical study has demonstrated the therapeutic benefits of pentoxifylline in the prevention of BPD in very low birth weight infants 21. The anti-inflammatory effects of selective PDE4 inhibitors have also been assessed in hyperoxic lung injury in rats. In this model, rolipram and piclamilast reduced inflammation and fibrin deposition and prolonged survival 22. Together, these data support a role for PDEs in BPD and the therapeutic potential of targeting PDE4 in particular in BPD. To this end, the aim of the present study was to investigate the expression profile of PDEs in lung tissue from mice with hyperoxia-induced BPD, and to assess the potential of PDE4 inhibition by cilomilast as a novel therapeutic approach.
METHODS
Experimental animals and exposure to hyperoxia
Pregnant C57Bl/6N mice from Charles River Laboratories (Sulzfeld, Germany) were maintained on a 12/12-h light/dark cycle. Experiments were performed according to institutional guidelines that comply with national and international regulations. Litters of pups were placed in either 85% oxygen (hyperoxia) or room air (normoxia) 12 h after birth for 28 days. Dams were rotated between hyperoxia and normoxia cages every 24 h to avoid oxygen toxicity. Body weights were recorded at post-natal (P) days P7, P14, P21 and P28.
Drug administration
Hyperoxia-exposed animals were treated with the PDE4 inhibitor, cilomilast, dissolved in dimethylsulfoxide (DMSO) and subcutaneously injected in the neck at 5 mg·kg−1 body weight once daily between days P14 and P28. The control group received DMSO alone. Cilomilast was initially injected at a dose of 30 mg·kg−1·day−1, a treatment protocol that has previously been reported in an in vivo transplantation model in rats 23; however, all animals died within 5 days. Therefore, the experimental dose was set at 5 mg·kg−1·day−1, which was well tolerated (survival of the cilomilast-treated group was 100%, 14 days post-administration).
Tissue preparation, lung morphometric analysis and compliance measurements
At days P7, P14, P21 and P28, pups were euthanised by an intraperitoneal injection of sodium pentobarbital. Lungs were processed for protein or RNA isolation as previously described 24. For histological analysis, fixation of the lungs was performed as follows. After mid-sternal thoracotomy the trachea was cannulated and lungs were fixed in situ with 4.5% formaldehyde in phosphate-buffered saline (pH 7.0) at a pressure of 20 cmH2O (1.96 kPa). After overnight fixation in 4.5% formaldehyde, the lungs were embedded in paraffin. The mean linear intercept, mean septal wall thickness and total airspace area were measured in 3-µm lung sections, as previously described 24, 25. Dynamic lung compliance (CL,dyn) was assessed by a volume-controlled pressure-limited compliance method, where mice were ventilated with a tidal volume of 6 mL·kg-1 for 10 min 24.
Semi-quantitative RT-PCR
Total RNA was isolated from lung and kidney homogenates using TRIzol (Invitrogen, Karlsruhe, Germany). The reverse transcription (RT) reactions were performed with the ImProm-II RT system (Promega, Mannheim, Germany) as previously described 26. For cDNA synthesis, 2 µg of total RNA was used. The forward and reverse PCR primers were: PDE1A 5′-GCTTCCATTTTCTCCACTTTGTG-3′ and 5′-AGCCAACTCTTTCCACCTCTCT-3′ (361 bp fragment); PDE4A 5′-AGGAGCGGGACTTACTGAAGA-3′ and 5′-CCTTTGACCTTTGACTGTTCCAT-3′ (1,057 bp fragment); PDE5A 5′-CCTGACTCTTTCTCTCGTGTGTT-3′ and 5′-CTGCTCATCATTTTGGTTGA-3′ (342 bp fragment); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 5′-GCAGTGGCAAAGTGGAGATT-3 and 5′-ACAGTCTTCTGGGTGGCAGT-3′ (493 bp fragment). After resolving the PCR products in an agarose gel, the band intensity of the amplicons were quantified using BioDoc Analyzer software (Biometra, Goettingen, Germany), and band intensities for PDE isoforms were normalised for the band intensity of the GAPDH amplicon from the same sample.
Immunoblotting
After normalisation of the protein lysates, 50–60 µg of total protein was loaded onto a 10% polyacrylamide gel. Following electrophoresis, the proteins were transferred to a nitrocellulose membrane, blocked and probed overnight with one of the following antibodies: anti-PDE1A (Abcam, Cambridge, MA, USA), anti-PDE4A (Abcam), anti-PDE5A (Cell Signalling, Danvers, MA, USA) or anti-GAPDH (Abcam). Membranes were incubated for 1 h in horseradish peroxidase-labelled secondary antibody (Sigma Aldrich, Munich, Germany) and immune complexes were detected by enhanced chemiluminescence using the ECL Plus Western Blotting Detection System (Amersham Biosciences, Munich, Germany).
Statistical analysis
All data are presented as mean±sem. Differences between the groups were assessed by an unpaired t-test with a p-value of ≤0.05 regarded as significant.
RESULTS
PDE1A expression analysis
At days P14, P21 and P28, mRNA levels for PDE1A were consistently higher in the lungs of hyperoxia-treated mice compared with normoxic controls (fig. 1⇓). This effect was confined to the lung, since no change in the levels of mRNA encoding PDE1 were observed in the kidney over the same time-course of hyperoxia exposure (fig. 1a⇓ and b). The PDE1A protein was detected as a single band of 72 kDa by an immunoblot (fig. 2a⇓). After an initial upregulation on day P7 and P14, the expression of PDE1A protein was downregulated at days P21 and P28 in the lungs of hyperoxia-treated mice compared with normoxia-treated mice (fig. 2⇓).

Effects of hyperoxia on phosphodiesterase (PDE)1A mRNA. a) The lungs and kidneys from neonatal mice (more than eight per condition) at post-natal (P) days P7, P14, P21 or P28 after exposure to hyperoxia (85% O2) or normoxia (21% O2) were screened for PDE1A mRNA expression by RT-PCR. Band intensities were compared using densitometry, after normalisation for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity in the b) lung and c) kidney. ▪: normoxia; ░: hyperoxia. *: p<0.05 comparing hypoxia with normoxia.

a) Lung homogenates from neonatal mice at post-natal (P) days P7, P14, P21 or P28 after exposure to hyperoxia (85% O2) or normoxia (21% O2) were screened for phosphodiesterase (PDE)1A protein expression by immunoblot. b) Band intensities were compared using densitometry, after normalisation for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity. ▪: normoxia; ░: hyperoxia.
PDE4A expression analysis
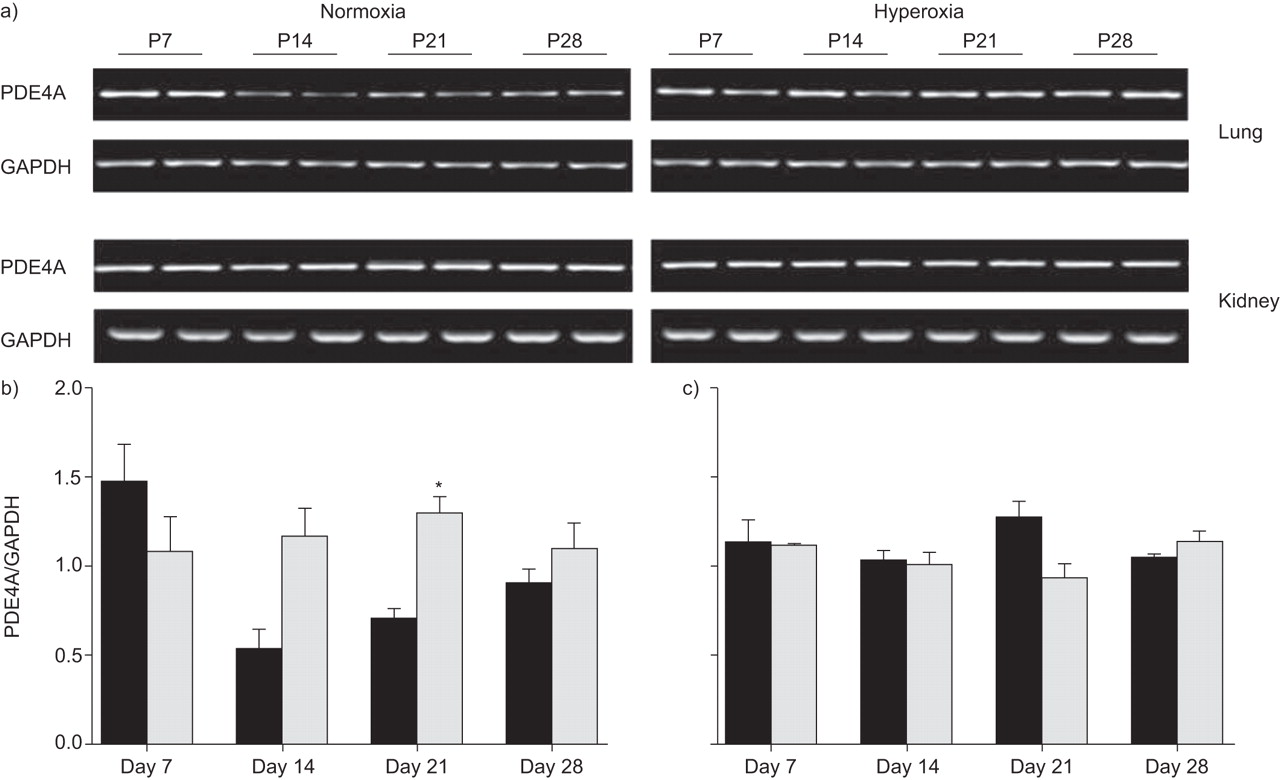
PDE4A mRNA expression was elevated in mouse lungs and kidneys over the time-course of hyperoxic exposure (fig. 3⇓). The most pronounced changes in PDE4 mRNA expression were observed at days P14 and P21, where expression levels were two-fold elevated in the lungs of the hyperoxia-treated group (fig. 3⇓). Normoxic controls exhibited an initial peak in PDE4A expression on day P7, and then a three-fold downregulation at day P14 (compared with day P7 in the normoxia group), which increased by day P28. No change in PDE4A mRNA expression levels were observed in the kidney (fig. 3a and c⇓) in either group. The different splice-isoforms of PDE4A were detected by immunoblot: PDE4A1 (66 kDa) and PDE4Ax (at 76 kDa and 102 kDa; fig. 4⇓). In general, PDE4A protein expression in mouse lungs was elevated between days P7 and P28 under hyperoxic conditions.

Effects of hyperoxia on the phosphodiesterase (PDE)4A mRNA. a) The lungs and kidneys from neonatal mice (more than eight per condition) at post-natal (P) days P7, P14, P21 or P28 after exposure to hyperoxia (85% O2) or normoxia (21% O2) were screened for PDE4A mRNA expression by RT-PCR. Band intensities were compared using densitometry, after normalisation for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity in the b) lung and c) kidney. ▪: normoxia; ░: hyperoxia. *: p<0.05 comparing hyperoxia with normoxia.

a) Lung homogenates from neonatal mice at post-natal (P) days P7, P14, P21 or P28 after exposure to hyperoxia (85% O2) or normoxia (21% O2) were screened for phosphodiesterase (PDE)4A protein expression by immunoblot. b) Band intensities of the 106 kDa band were compared using densitometry, after normalisation for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity. ▪: normoxia; ░: hyperoxia. *: p<0.05 comparing hyperoxia with normoxia.
PDE5A expression analysis
The expression of PDE5A at the mRNA level was progressively downregulated between day P7 and P28 in both the lungs and the kidneys (fig. 5⇑) of normoxia- and hyperoxia-exposed neonatal mice. Except for day P28, where expression of PDE5A mRNA was downregulated in the lungs of hyperoxia-exposed mice (fig. 5⇓), no differences between normoxia- and hyperoxia-treated groups were evident. In contrast, PDE5A protein expression was consistently downregulated in the lungs of the hyperoxia-treated group (fig. 6⇓).

Effects of hyperoxia on the phosphodiesterase (PDE)5A mRNA. a) The lungs and kidneys from neonatal mice (more than eight per condition) at post-natal (P) days P7, P14, P21 or P28 after exposure to hyperoxia (85% O2) or normoxia (21% O2) were screened for PDE5A mRNA expression by RT-PCR. Band intensities were compared using densitometry, after normalisation for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity in the b) lung and c) kidney. ▪: normoxia; ░: hyperoxia. *: p<0.05 comparing hyperoxia with normoxia.
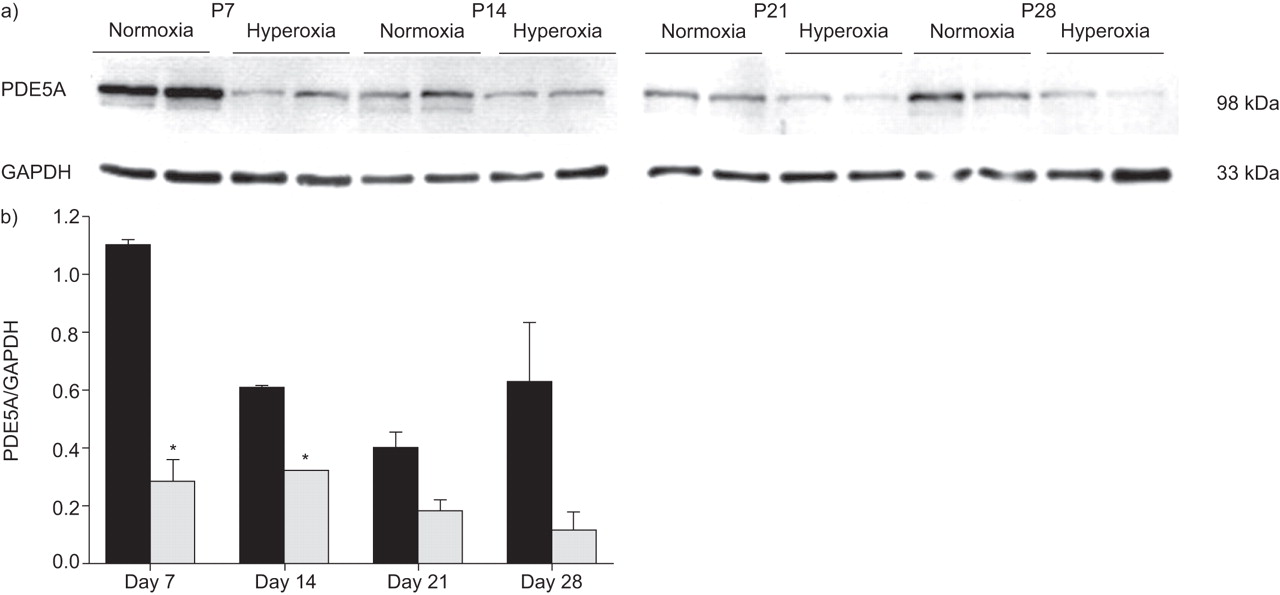
a) Lung homogenates from neonatal mice at post-natal (P) days P7, P14, P21 or P28 after exposure to hyperoxia (85% O2) or normoxia (21% O2) were screened for phosphodiesterase (PDE)5A protein expression by immunoblot. b) Band intensities were compared using densitometry, after normalisation for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) band intensity. ▪: normoxia; ░: hyperoxia. *: p<0.05 comparing hyperoxia with normoxia.
Effects of cilomilast treatment
The expression of PDE4 in hyperoxia-exposed animals was elevated most prominently between days P14 and P28, which suggested a potential therapeutic benefit of PDE4 inhibition in hyperoxia-induced BPD. Thus, the PDE4-specific inhibitor cilomilast (or vehicle alone) was administered to pups between days P14 and P28 during hyperoxic exposure. All animals in the normoxia- and cilomilast-treated groups exposed to hyperoxia survived, while the mean survival of the vehicle-treated group exposed to hyperoxia was 98%. As reported previously 24, continuous exposure of neonates to 85% oxygen adversely affected growth, where hyperoxia-exposed pups weighed ∼50% less at P28 than their normoxia-exposed littermates (fig. 7⇓). Cilomilast treatment did not alter the body mass of pups exposed to hyperoxia (fig. 7⇓). No other side-effects of cilomilast could be observed.

Effects of normoxia, hyperoxia, and hyperoxia and cilomilast treatment on the body weight of neonatal mice at post-natal day 28 (n = 8, n = 8 and n = 6, respectively). *: p<0.05 comparing hyperoxia with normoxia.
Exposure of neonatal mice to hyperoxia caused airspace enlargement that was evident upon microscopic analysis of lung tissue sections (fig. 8⇓). These morphological changes were quantified by assessment of the mean linear intercept (MLI), which describes the mean intra-alveolar distance (fig. 9a⇓), assessment of the thickness of the septae (fig. 9b⇓) and estimation of the total airspace area (fig. 9c⇓). Exposure of neonatal mice to hyperoxia generated a two-fold increase in the MLI compared with normoxia-treated pups. Cilomilast treatment significantly reduced the MLI (fig. 9a⇓), although it was not restored to normal baseline values observed in the normoxia-treated group. Similarly, hyperoxia-exposed pups exhibited a significant increase in septal wall thickness compared with normoxia-treated pups (fig. 9b⇓), and cilomilast treatment significantly reduced septal wall thickness in the hyperoxia-treated group, bringing septal wall thickness back into the range observed in the normoxia-treated group. Cilomilast treatment under hyperoxic conditions also significantly decreased airspace volume in the lungs of hyperoxia-exposed pups (fig. 9c⇓) when compared with the hyperoxia-treated pups that did not receive cilomilast. These data indicate that cilomilast was able to either partially correct, or prevent additional damage to lung alveolar structure induced by hyperoxia.

Effect of cilomilast treatment on architectural changes in the lungs of neonatal mice exposed to 85% oxygen. Lung sections (obtained at post-natal day 28) were stained with haematoxylin and eosin. Lung sections were obtained from the right lower lobe of a) normoxia-exposed, b) hyperoxia-exposed mice and c) cilomilast-treated hyperoxia-exposed mice. Scale bars = 20 μm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Quantification of the effect of cilomilast on lung architectural changes and lung function of neonatal mice exposed to 85% oxygen. Lung sections were obtained from the right lower lobe from 10 hyperoxia-exposed mice (for dynamic lung compliance (CL,dyn) n = 6), eight cilomilast-treated, hyperoxia-exposed mice (CL,dyn n = 5) and eight normoxia-exposed mice (CL,dyn n = 8) at post-natal day 28, and were stained with haematoxylin and eosin. Sections were quantified for a) the mean linear intercept (MLI), b) septal wall thickness and c) total airspace area. d) Additionally, CL,dyn was assessed in pups from all three groups. #: p<0.05 comparing hyperoxia with normoxia groups; ¶: p<0.05 comparing hyperoxia with hyperoxia/cilomilast groups.
Cilomilast treatment improved the mean CL,dyn of hyperoxia-treated pups, restoring the CL,dyn to normal levels, as assessed by whole body plethysmography in a volume-adapted, pressure-regulated protocol (fig. 9d⇑). Variability in CL,dyn between individual animals yielded mean values for experimental groups that were not statistically different, irrespective of the groups compared. Hyperoxia-exposed pups tended towards decreased CL,dyn versus CL,dyn of normoxia-exposed pups. When hyperoxia-exposed pups were treated with cilomilast between days P14 and P28, CL,dyn tended towards an increase, where the mean value was restored to that of the normal CL,dyn of normoxia-treated pups (fig. 9d⇑). Taken together, these data indicate that administration of cilomilast blunted the deleterious effects of hyperoxia on the architectural changes and function in neonatal lungs.
DISCUSSION
Exposure of neonatal mice to hyperoxia resulted in disturbed alveolar development, retarded lung growth and adverse growth. Expression analysis of cyclic nucleotide PDEs revealed changes in the expression of several PDE isoforms during hyperoxia-induced lung injury in mice. With the exception of day 7, PDE1 was upregulated at the mRNA level and downregulated at the protein level in the lungs of hyperoxia-exposed mouse pups. These differences in gene expression in the first week of hyperoxic exposure were most likely due to an adaptation of the lung to higher oxygen concentrations and remained at the same level until the end of the exposure period. The downregulation of the PDE1A protein, which was reported to be present primarily in vascular smooth muscle cells 27, could occur either by negatively influenced protein synthesis, or through an overall decrease in vessel formation, which is one pathological aspect of hyperoxic lung injury. The expression of PDE5A was downregulated by hyperoxia at both the mRNA and protein levels, which is consistent with the microarray observations of Wagenaar et al. 28. In contrast, the expression of type four PDEs in the lungs of neonatal mice was dramatically upregulated over the time-course of exposure to hyperoxia. These data suggested a possible therapeutic benefit of PDE4 inhibition.
Nonselective PDE inhibitors, such as pentoxifylline, reduced fibrin deposition and prolonged survival in neonatal rats, in which lung injury was induced by hyperoxia 20. A pilot clinical study has also demonstrated the therapeutic benefit of pentoxifylline in the prevention of BPD in very low birth weight infants, suggesting that PDE inhibition may represent an alternative to steroid use for the management of BPD 21. Specific inhibition of PDE5 (and thus, elevation of the cGMP levels) by sildenafil improved lung growth and vascularisation, and attenuated pulmonary hypertension in rat pups exposed to hyperoxia 17, which is consistent with the therapeutic use of sildenafil for the treatment of adult pulmonary arterial hypertension 29. Inhibitors of PDE4, including rolipram and piclamilast, have also recently been demonstrated to improve survival and attenuate pulmonary inflammation in hyperoxia-induced lung injury in neonatal rats 22, suggesting a role for both PDEs in the development of BPD and the possibility of targeting PDEs in the treatment of BPD.
Cilomilast (Ariflo; SB-207499), a second generation PDE4 inhibitor, has generated renewed interest in the therapeutic applications of PDE4 inhibition and has the added advantage of being orally active. It exhibits a lower emetic potential compared with rolipram and is negatively charged at physiological pH, limiting its penetration in the central nervous system, leading to improved tolerance and fewer side-effects 30. Cilomilast has now been evaluated in phase I, II and III trials, and dose–response studies have suggested clinical benefits when cilomilast was employed in COPD patients, but not in asthma patients 31.
In the present study, cilomilast was administered to neonatal mice for 2 weeks over the period P14–P28, after an initial 2-week exposure to hyperoxia over P1–P14. The onset of cilomilast therapy was timed to correlate with changes in PDE gene expression, which were most pronounced at day P14. Since murine alveolarisation approximately starts at day P3 and finishes approximately at day P14 32, therapy with cilomilast could not directly influence alveolarisation, but rather would impact the later maturation and growth process of the lung. Earlier treatments starting from day P1, by virtue of suppression of inflammation would probably have further improved lung structural development. Indeed, one of the weaknesses of the present study is the late application of cilomilast (at day P14); however, at the doses required, application of cilomilast to mice prior to day P14 was both traumatic and technically challenging. However, cilomilast applied from day P14 (which was well tolerated) was able to limit damage to lung architecture suggesting the beneficial effects of cilomilast in the current model.
While without any effect on body mass of hyperoxia-treated mouse pups, cilomilast had a pronounced effect on lung structure and architecture. After treatment with cilomilast, the lungs of hyperoxia-treated pups exhibited a significantly reduced mean intra-alveolar distance (measured by mean linear intercept), a septal wall thickness that was restored to normal values and a significantly reduced total airspace area, when compared with lungs from age-matched hyperoxia-exposed pups that received vehicle alone. These structural changes translated to improved dynamic respiratory compliance, which tended towards normal values. Together, these data indicate that cilomilast limited structural and architectural damage to the developing lung caused by chronic exposure to normobaric hyperoxia.
The beneficial effects of PDE4 inhibition in the present model are most likely attributable, at least in part, to the well recognised anti-inflammatory properties of PDE4 inhibitors 30, 31, 33, since the exposure of neonates to normobaric hyperoxia promotes lung inflammation 34. Indeed, in one study it was demonstrated that PDE4 inhibition can impact on the production of inflammatory cytokines, such as tumour necrosis factor-α 35. In another study, both PDE4 inhibitors, rolipram and piclamilast, significantly attenuated the induction of interleukin-6, chemokine-induced neutrophilic chemoattractant-1, monocyte chemoattractant-1 and amphiregulin by hyperoxia in rat pups 22. These data suggest that the protective effects of cilomilast may be attributed, at least in part, to promoting increased expression of anti-inflammatory cytokines in hyperoxia-induced BPD in mice.
The beneficial effects of PDE4 inhibition in hyperoxia-induced BPD may also extend beyond anti-inflammatory effects of PDE4 inhibitors. Transforming growth factor (TGF)-β has a well documented role in late lung development 36; however, TGF-β signalling is dysregulated in BPD, both in human patients 37, as well as in hyperoxia-induced BPD in rodents 24. Interestingly, PDE4 inhibitors are potent antagonists of TGF-β signalling, since roflumilast antagonises the induction of connective tissue growth factor, collagen I and fibronectin by TGF-β in human airway smooth muscle cells 38. The increased septal wall thickness observed in hyperoxia-induced BPD is thought to originate, at least in part, from over-deposition of matrix molecules and improper remodelling of the extracellular matrix 6, 39. In the present study, cilomilast administration restored normal septal wall thickness to hyperoxia-exposed mouse pups. It is tempting to speculate that this may have been achieved, in part, by the antagonism of aberrant matrix production produced in response to excessive (abnormal) TGF-β signalling. Along these lines, excessive matrix production in BPD has also been attributed to an over-abundance of myofibroblasts, which produce and deposit excessive matrix, as well as matrix metabolising enzymes such as matrix metalloproteinases (MMPs), the over-production of which contributes to alveolar destruction 40. The PDE4 inhibitors may blunt this process, since piclamilast has been shown to block lung fibroblast to myofibroblast conversion driven by TGF-β 41. Thus, apart from anti-inflammatory effects, cilomilast may directly antagonise aberrant growth factor signalling, which drives the pathological changes in alveolar architecture observed in BPD.
A PDE4 inhibitor has also been employed in a mouse model of cigarette-smoke induced emphysema 42 a pathology that, much like BPD, is characterised by a pronounced loss of alveolar architecture. In cigarette smoke-induced emphysema, the selective PDE4 inhibitor, GDP-1116, attenuated alveolar destruction by inhibiting smoke-induced MMP-12 production and protected against type II pneumocyte apoptosis 42. The PDE4 inhibitors rolipram and cilomilast can also directly block secretion of pro-MMP-1 and pro-MMP-2 secretion by lung fibroblasts 43. Although emphysema and BPD both exhibit loss of alveolar architecture, the aetiologies of these two diseases are vastly different. Emphysema arises from the loss of alveolar structure due to insult to the adult lung, while BPD arises from impaired development of the immature (neonatal) lung. Thus, PDE4 inhibitors most likely promote lung repair in the case of emphysema, and conversely, limit hyperoxic damage to the developing lung, in the case of the present BPD model, and thereby facilitate improved lung development. Nevertheless, the aberrant production of MMPs 6 and increased type II cell apoptosis 44 are also consequences of hyperoxia-induced lung injury, and the protective effects of cilomilast might also be partly attributed to the prevention of excessive MMP production and protection against hyperoxia-induced epithelial cell apoptosis.
In summary, the data presented in the present study indicate that phosphodiesterase expression is impacted in hyperoxia-induced bronchopulmonary dysplasia, and the upregulation of phosphodiesterase, in particular, suggests a pathological role for phosphodiesterase isoforms in the current model. The ability of cilomilast to protect against or reverse the aberrant remodelling of the alveolar architecture in response to hyperoxic insult supports the further development of phosphodiesterase inhibitors for the treatment of bronchopulmonary dysplasia.
Support statement
The study was supported, in part, by grants from the German Research Foundation (grants SFB547 and KFO118; Bonn, Germany), the Excellence Cluster Cardio-Pulmonary System (Hessen, Germany); and the European Commission (Brussels, Belgium) under the Sixth Framework Program (contract no. LSHM-CT-2005-018725, PULMOTENSION).
Statement of interest
Statements of interest for K. Woyda and F. Grimminger can be found at www.erj.ersjournals.com/misc/statements.dtl
Acknowledgments
The present authors acknowledge the excellent technical support offered by M. Zoremba (Dept of Paediatrics, University Hospital of Giessen, Giessen, Germany) for the current study.
- Received July 18, 2008.
- Accepted October 9, 2008.
- © ERS Journals Ltd
References










