





Abstract
Reduced vascular endothelial growth factor (VEGF) has been reported in bronchoalveolar lavage fluid and lungs of severe emphysema patients. Airway epithelial cells (AEC) are exposed to various environmental insults like cigarette smoke and bacterial infections, but their direct effect on VEGF production in well-differentiated primary human AEC remains unclear.
The current authors determined the effect of cigarette smoke extract (CSE) alone and in combination with Mycoplasma pneumoniae (Mp) on VEGF production in well-differentiated primary normal human bronchial epithelial (NHBE) and small airway epithelial cells (SAEC) in air–liquid interface cultures. Secretion and expression of VEGF were determined by ELISA and real-time RT-PCR, respectively. Cell growth, apoptosis, extracellular signal-regulated kinase (ERK)1/2 and protein kinase (PK)C signalling pathways were evaluated to further dissect VEGF regulation under CSE treatment.
CSE significantly reduced VEGF secretion in NHBE and SAEC. In SAEC, Mp alone significantly increased the VEGF, while the presence of CSE attenuated Mp-induced VEGF production. While ERK inhibitor reduced VEGF secretion only in NHBE, a PKC inhibitor significantly decreased VEGF secretion in both NHBE and SAEC.
In conclusion, direct cigarette smoke extract exposure significantly reduced vascular endothelial growth factor production in well-differentiated primary human airway epithelial cells, in part through modifying extracellular signal-regulated kinase 1/2 and protein kinase C signalling pathways.
- Airway epithelial cells
- cigarette smoke extract
- extracellular signal-regulated kinase 1/2
- protein kinase C
- vascular endothelial growth factor
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death worldwide 1, 2. Emphysema is one of the important pathological features in severe COPD, which is associated with irreversible loss of lung function largely due to destruction of alveolar structure including microvasculature 3. Cigarette smoke (CS) is one of the major contributing factors in the development of emphysema.
Vascular endothelial growth factor (VEGF)-A (also known as VEGF) is a critical angiogenic factor in maintaining normal tissue vasculature and endothelial cell growth and survival 4. CS exposure has been associated with reduced VEGF and VEGF receptor (VEGFR)-2 expression and VEGFR-2 activation in severe emphysema patient lungs and rodent lungs 5, 6. Moreover, reduced VEGF has been clearly implicated in the destruction of alveolar wall components including microvasculature 6–9. As the first line of lung host defence mechanism, airway epithelial cells (AEC) are exposed to the highest level of CS and other environmental insults such as pathogens. However, the direct impact of CS on well-differentiated human primary AEC with relation to VEGF production has not been well studied.
Most previous studies aimed at determining the effects of CS on cell proliferation, apoptosis, inflammation and senescence have been performed using either immortal lung epithelial cell lines or primary large AEC cultured under submerged conditions 10–14. AEC grown under submerged culture conditions are poorly differentiated and do not mimic major in vivo features such as mucociliary differentiation. Boussat et al. 15 found that well-differentiated normal human bronchial epithelial (NHBE) cells produced significantly higher amounts of VEGF compared with transformed epithelial cell lines. However, it remains unclear whether CS exposure can directly decrease VEGF levels in well-differentiated primary human AEC. As small airways and lung parenchyma are the major affected sites in COPD, it is critical to evaluate the direct effect of CS on VEGF in well-differentiated small airway epithelial cells (SAEC). Findings from the SAEC will further reveal how CS affects distal lungs in COPD and illustrate whether SAEC and NHBE respond differently to CS exposure with regard to VEGF production.
Increased prevalence of bacterial colonisation has been reported in the airways of smokers with and without COPD 16, 17. Mycoplasma pneumoniae (Mp), an atypical respiratory bacterium, is associated with upper as well as lower respiratory tract infections in smokers suffering from COPD 18–20. The impact of bacterial infection on tissue remodelling (e.g. angiogenesis) in COPD patients is poorly understood. Therefore, the current authors also examined whether Mp can modify VEGF production in CS-exposed AEC.
Signalling pathways such as mitogen-activated protein kinase (MAPK) and protein kinase (PK)C have been shown to regulate VEGF expression in various cell types, including smooth muscle cells, human umbilical vein endothelial cells, retinal pigment epithelial cells, macrophages and renal glomerular epithelial cells 21–27. CS and CS extract (CSE) have been shown to activate MAPK and PKC signalling pathways in lung epithelial cell lines (e.g. A549) and primary cells (e.g. NHBE) cultured under submerged conditions 28–32. Although reduced VEGF expression is observed in severe emphysematous lungs, the molecular mechanisms involved in CS-mediated VEGF reduction remain unclear. In the present study, the role of MAPK and PKC signalling pathways in VEGF production was determined in well-differentiated human primary AEC that were treated with CSE.
The current results demonstrated that CSE significantly reduced VEGF expression in well-differentiated NHBE and SAEC. The inhibitory effect of CSE on VEGF was more pronounced in SAEC in the presence of Mp infection.
METHODS
Primary human AEC air–liquid interface culture
Primary human AEC (Clonetics NHBE and SAEC) were obtained from different healthy donors (Cambrex Bio Science, Walkersville, MD, USA). They were cultured in serum-free medium (Dulbecco’s modified Eagle medium and bronchial epithelial cell basal medium at 1:1) supplemented with various growth factors, including insulin (0.4 μg·mL−1), transferrin (5 μg·mL−1), hydrocortisone (0.5 μg·mL−1), epinephrine (0.5 μg·mL−1), epidermal growth factor (10 ng·mL−1), bovine pituitary extract (52 μg·mL−1), retinoic acid (30 ng·mL−1), bovine serum albumin (50 µg·mL−1), gentamycin (50 μg·mL−1) and amphoterisin B (0.05 μg·mL−1). Cells between passages 2 and 4 were plated onto 12-mm collagen-coated polyester transwell inserts (pore size 0.4 µm; Corning Inc., Corning, NY, USA) at 4×104 cells·cm−2. Within a week of submerged culture conditions these epithelial cells attained 100% confluence, and were then transferred into an air–liquid interface (ALI) condition by reducing apical surface medium volume to 50 μL. Epithelial cells were maintained in an ALI culture condition for a week to induce mucociliary differentiation 33, 34. On day 7 of ALI culture, CSE treatment was started and refreshed daily for up to 14 days. CSE was applied to the apical side of epithelial cells to mimic the in vivo pattern of CS exposure. At days 1, 7 and 14 after CSE treatments, basolateral supernatants were collected for VEGF ELISA, while cells were collected to evaluate VEGF mRNA expression, cell morphology, DNA content and activation of signalling pathways.
CSE preparation
CSE was prepared fresh daily, as previously described but with slight modification 35–38. Unfiltered research grade cigarettes (2R1) were purchased from Kentucky Tobacco Research and Development Center at the University of Kentucky (Louisville, KY, USA). Briefly, CSE was prepared by combusting one cigarette and bubbling mainstream cigarette smoke into 25 mL of sterile serum-free cell culture medium with the help of a peristaltic pump. This preparation was sterilised through a 0.22-µm syringe filter and considered as 100% CSE. Complete cell culture medium was used to dilute 100% CSE to the required CSE concentrations, which were used within 30 min of CSE preparation.
Mp infection in AEC
Mp strain 15531 was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and was grown at 37°C in SP4 glucose broth. Adherent Mp was collected, then frozen at -80 °C for infection experiments 34. Frozen Mp stock (passage 4) was resurrected in SP4 broth at 37°C for 2 h, and then used for infecting the epithelial cells at a concentration of 1 colony-forming unit (cfu)·cell−1 (a physiological dose) or 4×104 cfu·transwell−1 at the apical side in the presence or absence of 20% CSE. Epithelial cells were infected with Mp once only at the beginning of CSE treatment (day 7 of ALI culture). On days 1, 7 and 14 post Mp infection, cells and supernatants were harvested for VEGF detection, and also cultured to verify Mp infection.
ELISA for VEGF
VEGF secretion in basolateral supernatants from AEC was measured using a VEGF ELISA kit (ELISA Tech., Aurora, CO, USA) according to the manufacturer’s instructions. A standard curve was generated using recombinant VEGF (0.05–2 ng·mL−1) to calculate VEGF concentrations in AEC supernatants.
Quantitative real-time RT-PCR
Expression of human airway epithelial VEGF mRNA was determined by quantitative real-time RT-PCR. Total RNA was extracted using the Trizol reagent (Gibco BRL, Rockville, MD, USA). Reverse transcription was performed using 1 µg of total RNA as described previously 34. Human VEGF ( Genbank accession number M27281. 1) primers (forward 5′-CTTGCCTTGCTGCTCTACC-3′ and reverse 5′-CACACAGGATGGCTTGAAG-3′) and probe (5′-AGTTCATGGATGTCTATCAGCGCAGCT-3′) were designed using the Primer Express software (Applied Biosystems Inc., Foster City, CA, USA). Primers and probes for human glyceraldehyde-3-phosphate dehydrogenase (4326317E) and B-cell lymphoma (Bcl)-2 (assay ID HS00153350_m1) were purchased as premixed solution from Applied Biosystems. Real-time PCR was performed using 25 µL reaction mixture containing 30 ng cDNA, fluorogenic probe, primers and other components in the TaqMan RT-PCR kit. The comparative threshold cycle method was used to determine the relative gene expression levels in AEC.
Total DNA quantification
To monitor cell growth of well-differentiated epithelial cells, total DNA content, which directly reflects the total number of cells, was determined using the Quant-iT dsDNA broad range (BR) kit from Molecular Probes (Eugene, OR, USA), according to the manufacturer’s instructions. In brief, whole cell lysates were prepared from each transwell using 200 µL of cell lysis solution (0.2% TritonX-100). Cell lysates (20 µL) were added directly to 200 µL of working Quant-iT dsDNA BR reagent. DNA content was determined by measuring fluorescence intensity using 510-nm/527-nm (excitation/emission) filters in a fluorescence plate reader. A standard curve was prepared using the λDNA standards (50–1,000 ng·well−1) provided with the kit and used to determine the DNA content of cell lysates.
AEC morphology
For cell morphology analysis, cells were fixed in ice-cold acetone and embedded in JB-4 polymer 33. Cell sections of 2 µm thickness were cut with a microtome and stained with haematoxylin and eosin to examine the cell morphology, including cell integrity, nuclear shape and mucociliary differentiation, under a light microscope.
Western blot analysis
To prepare whole cell lysates, NHBE or SAEC were lysed in Western blot lysis buffer containing 50 mM Tris-HCl (pH7.4), 0.15 M NaCl, 0.1% SDS, 1.0% Nonidet P-40 and 10 mM EDTA with 1% protease inhibitors and 1% phosphatase inhibitor cocktails. Equal amounts of protein samples were resolved on 10% or 12% SDS-PAGE gels, and transferred onto nitrocellulose membranes as described previously 33. Antibodies against extracellular signal-regulated kinase (ERK)1/2, phospho-ERK1/2, c-Jun N-terminal kinase (JNK), phospho-JNK, p38MAPK and phospho-p38MAPK (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA, and Cell Signaling Technology Inc., Danvers, MA, USA) were applied to the nitrocellulose membranes to detect phosphorylated and total ERK1/2, JNK and p38MAPK. Densitometry was performed using the NIH Image-J software, and the ratio of phospho-MAPK/total MAPK was used to determine MAPK activation levels.
Statistical analysis
Statistical analyses were performed using Graphpad PRISM 4.0 software and data expressed as mean±sem. Statistical significance between different groups was analysed using paired t-tests and ANOVA. A p-value of <0.05 was considered statistically significant.
RESULTS
Effect of CSE on VEGF production in human primary AEC
Initial experiments using different doses of CSE (1–20%) showed a significant decrease of VEGF protein secretion in NHBE after 7 days of CSE treatment compared with the untreated group (fig. 1a⇓). CSE tended to decrease VEGF mRNA expression, but the differences between CSE-treated and -untreated cells were not statistically significant (p>0.05; fig. 1b⇓). The current authors decided to use 20% CSE for subsequent experiments, as 20% CSE produced a maximal decrease in VEGF secretion without apparent cell damage (e.g. cell death or cell detachment from the transwell membrane).

Dose response of cigarette smoke extract (CSE; 1–20%) on vascular endothelial growth factor (VEGF) a) secretion and b) mRNA expression in well-differentiated normal human bronchial epithelial (NHBE) cells. CSE was applied to the apical side of NHBE cells for 7 days. a) VEGF levels in basolateral supernatants were determined using an ELISA. CSE at 5 and 20% significantly reduced VEGF secretion compared with untreated cells. b) VEGF mRNA relative expression levels were determined using quantitative real-time RT-PCR. Data are expressed as mean±sem (n = 6). ns: nonsignificant (one-way ANOVA). *: p<0.05; **: p<0.01.
AEC were exposed to media alone or 20% CSE for 1, 7 and 14 days. Repeated (day 7 and 14) but not a single (day 1) CSE exposure significantly reduced VEGF in NHBE cells (fig. 2a⇓). Unlike NHBE, both a single (day 1) and repeated (day 14) CSE exposure significantly reduced VEGF secretion in SAEC compared with untreated cells (fig. 2b⇓).
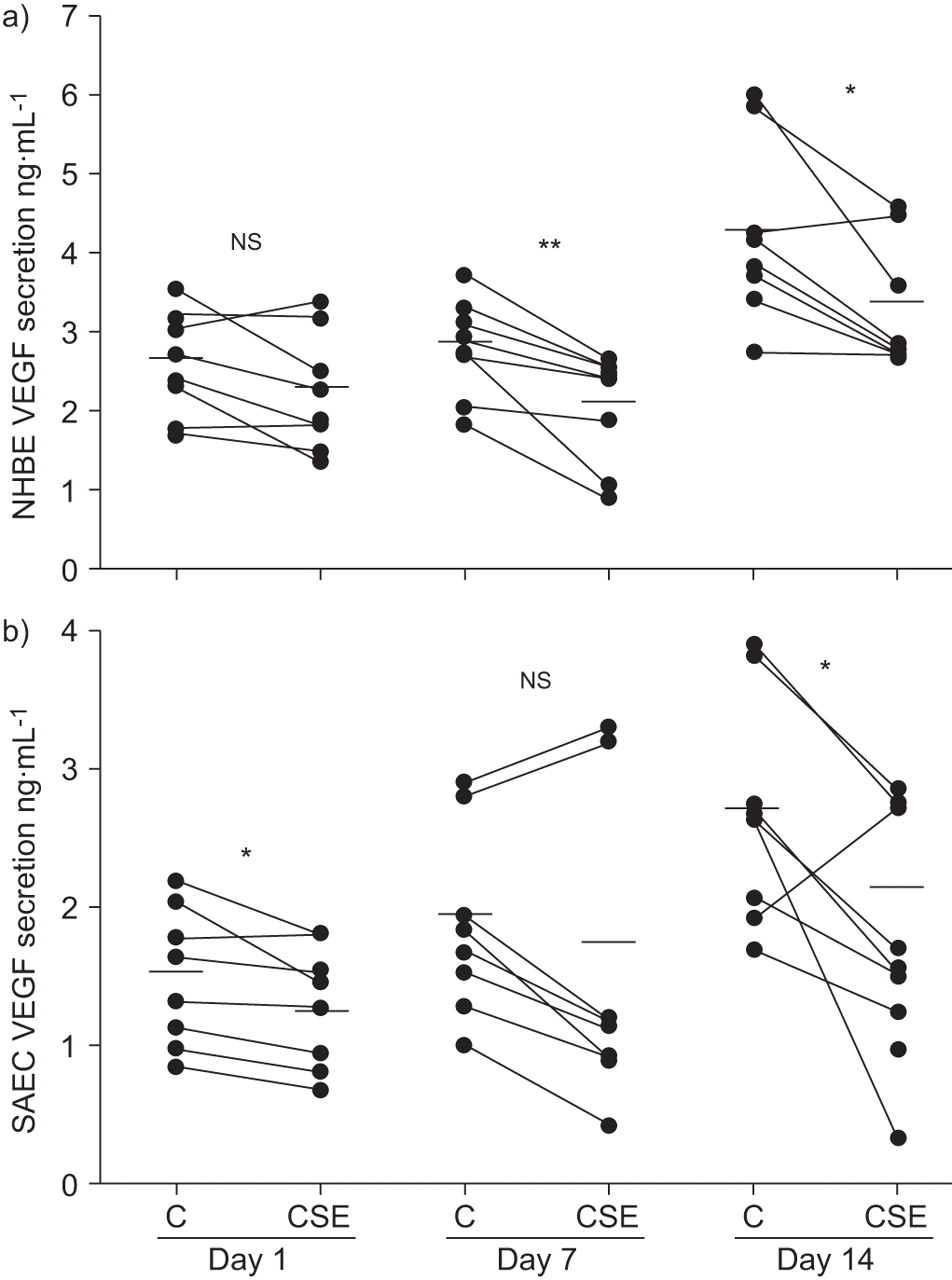
Cigarette smoke extract (CSE) exposure reduced vascular endothelial growth factor (VEGF) secretion in well-differentiated a) normal human bronchial epithelial (NHBE) cells and b) small airway epithelial cells (SAEC), compared with the respective untreated controls (C), after 1, 7 and 14 days of CSE treatments. Data are expressed as individual values (•) and means (horizontal bars); n = 8. ns: nonsignificant (paired t-test). *: p<0.05; **: p<0.01.
Effect of CSE on VEGF production in the presence of Mp infection in human primary AEC
Mp alone significantly increased VEGF secretion in SAEC but not in NHBE (table 1⇓; fig. 3a⇓ and b). The presence of CSE significantly attenuated Mp-induced VEGF secretion in SAEC (fig. 3b⇓). The inhibitory effect of CSE in combination with Mp on VEGF production occurred at earlier time-points (days 1 and 7) and was more pronounced in SAEC compared with NHBE (table 1⇓). Furthermore, in response to simultaneous CSE and Mp exposure, the degree of VEGF reduction in SAEC was greater than that in NHBE (table 1⇓).

Effects of cigarette smoke extract (CSE) on vascular endothelial growth factor (VEGF) secretion in Mycoplasma pneumoniae (Mp)-infected well-differentiated a) normal human bronchial epithelial (NHBE) cells and b) small airway epithelial cells (SAEC). Epithelial cells were infected with Mp in the presence or absence of CSE for 14 days. Data are expressed as mean±sem (n = 8). *: p<0.05 (paired t-test).
Change in vascular endothelial growth factor secretion following exposure to cigarette smoke extract(CSE), Mycoplasma pneumoniae (Mp) or both
Effect of CSE on cell growth and apoptosis in human primary AEC
To reveal the mechanisms underlying CSE-mediated VEGF reduction, the effect of 20% CSE on NHBE and SAEC growth or apoptosis was determined by examining cell morphology, total DNA content and Bcl-2 mRNA expression, respectively (fig. 4⇓). No apparent changes in the cell morphology were observed in NHBE or SAEC following CSE treatment, in comparison with their respective controls (fig. 4a–d⇓). Similarly, no significant differences existed in the total DNA content of CSE-untreated and -treated cells, suggesting that 20% CSE treatment did not increase cell growth or induce cell death in either NHBE or SAEC (fig. 4e⇓). Bcl-2 is an anti-apoptotic protein, and overexpression of Bcl-2 has been shown to protect human airway epithelial cell line 1HAE0 and lung adenocarcinoma cell line A549 from corticosteroid- and hydrogen peroxide-induced apoptosis, respectively 39, 40. In the present study, CSE did not significantly reduce or alter Bcl-2 mRNA expression compared with untreated cells (fig. 4f⇓). Collectively, these data indicate that 20% CSE treatment did not have any noticeable effects on NHBE or SAEC growth and survival.
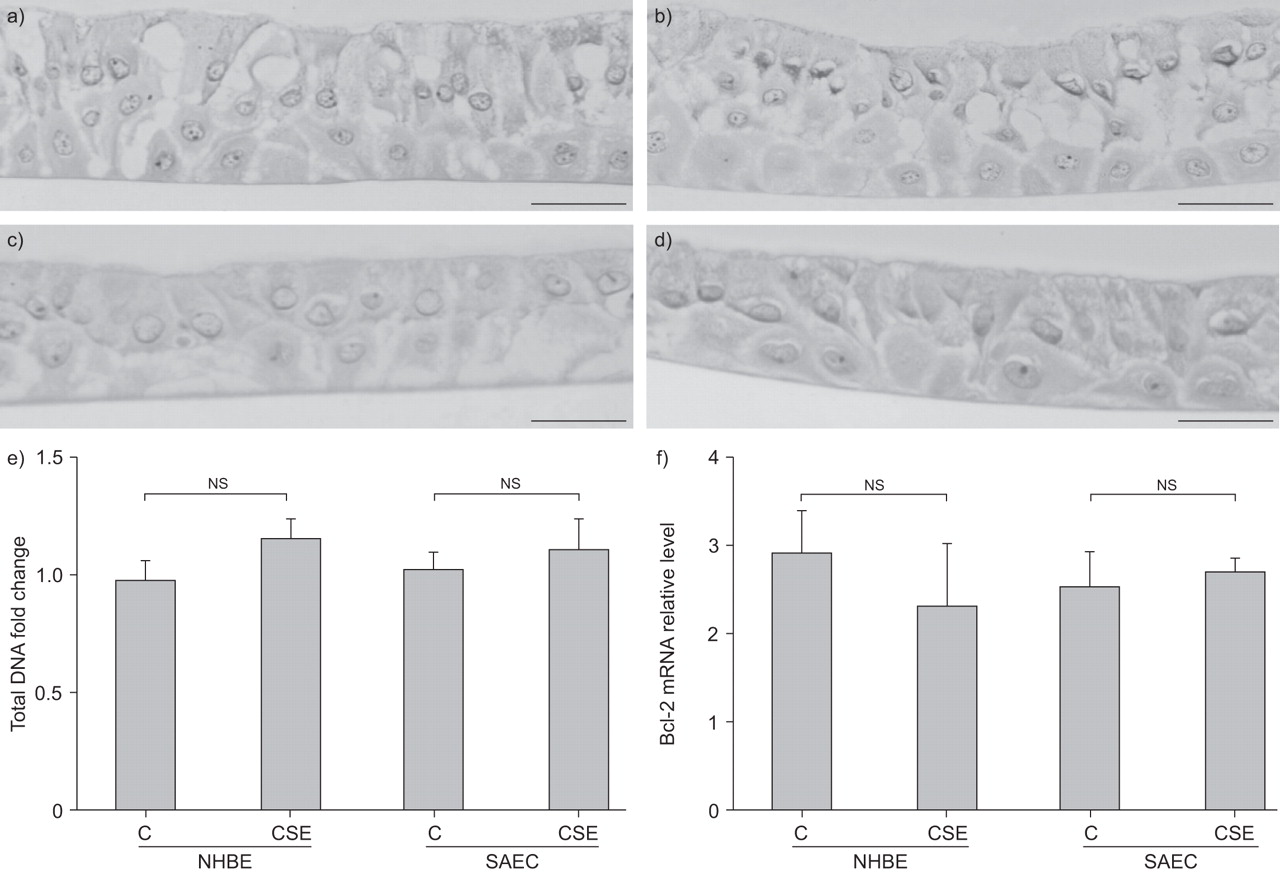
Effects of cigarette smoke extract (CSE) on cell growth and apoptosis in normal human bronchial epithelial (NHBE) cells and small airway epithelial cells (SAEC). Cells were exposed to medium alone or 20% CSE for 14 days, and were harvested to examine cell morphology using haematoxylin and eosin staining. a) Untreated NHBE cells, b) CSE-treated NHBE cells, c) untreated SAEC and d) CSE-treated SAEC. e) Total DNA content was determined using whole cell lysates and expressed as fold change over untreated cells (C). f) B-cell lymphoma (Bcl)-2 mRNA relative expression levels in NHBE cells and SAEC were determined by quantitative real-time RT-PCR. Data are expressed as mean±sem (n = 4 to n = 6). ns: nonsignificant (paired t-test). Scale bars = 100 μm.
Role of MAPK signalling in VEGF expression in human primary AEC
To elucidate the role of individual MAPK signalling pathways in regulating VEGF expression in NHBE and SAEC, MAPK activation was examined at 5 min, 15 min, 2 h and 24 h following 20% CSE exposure. In NHBE cells, CSE exposure caused acute increase (after 15 min) but subsequent decrease (at 2 h and 24 h) in ERK1/2 phosphorylation, compared with untreated cells (at 24 h, p = 0.066). In the same time-frame, no changes were observed in p38MAPK or JNK phosphorylation following CSE exposure (fig. 5a⇓ and b). Acute CSE exposure also increased ERK1/2 activation in SAEC but, unlike NHBE, ERK1/2 remained activated at 24 h compared with untreated cells (fig. 5c⇓). Similar to the findings in NHBE, CSE did not alter p38MAPK and JNK activation in SAEC (data not shown).

Role of mitogen-activated protein kinase (MAPK) in cigarette smoke extract (CSE)-mediated vascular endothelial growth factor (VEGF) reduction in normal human bronchial epithelial (NHBE) cells and small airway epithelial cells (SAEC). Cells were treated with either medium alone (□) or 20% CSE (▓). Activation of three MAPK pathways (phosphorylated (p)-MAPK/total MAPK ratio) was determined at the indicated time-points by a) Western blot and b) densitometry in NHBE cells and c) in SAEC. CSE exposure acutely (at 15 min) increased extracellular signal-regulated kinase (ERK)1/2 phosphorylation in both NHBE cells and SAEC, but at 24 h it decreased ERK1/2 phosphorylation in NHBE cells. CSE did not alter p38MAPK or c-Jun N-terminal kinase (JNK) phosphorylation in NHBE cells or SAEC. d and e) Cells were treated with 5 μM PD98059, a specific inhibitor of MEK1/2 (the kinase upstream of ERK1/2), for 2 and 24 h, or with 0.1% dimethyl sulphoxide (DMSO) as a vehicle control. d) Inhibition of ERK1/2 phosphorylation was verified by Western blot at 2 h. e) VEGF secretion was determined by ELISA at 24 h in NHBE cells (▪) and SAEC (░). Data are expressed as mean±sem (n = 6). AU: arbitrary units. **: p<0.01; ns: nonsignificant (paired t-test).
To confirm a role of ERK1/2 signalling in the regulation of airway epithelial VEGF production, PD98059 (5 μM; Calbiochem, San Diego, CA, USA), a specific inhibitor of MEK1/2 (the kinase upstream of ERK1/2), was applied to the cells 2 h prior to the CSE treatment. VEGF secretion and mRNA expression were measured after 24 h of CSE treatment. PD98059 pretreatment significantly reduced basal ERK1/2 phosphorylation in both NHBE and SAEC (fig. 5d⇑). Treatment with PD98059 alone for 24 h significantly reduced VEGF secretion in NHBE cells, but not in SAEC (fig. 5e⇑). PD98059 treatment together with 20% CSE further reduced VEGF in NHBE compared with either treatment alone, but the difference was not statistically significant compared with PD98059 alone (data not shown). PD98059 did not significantly reduce VEGF mRNA expression in NHBE at 24 h post CSE treatment (data not shown).
Role of PKC signalling in VEGF expression in human primary AEC
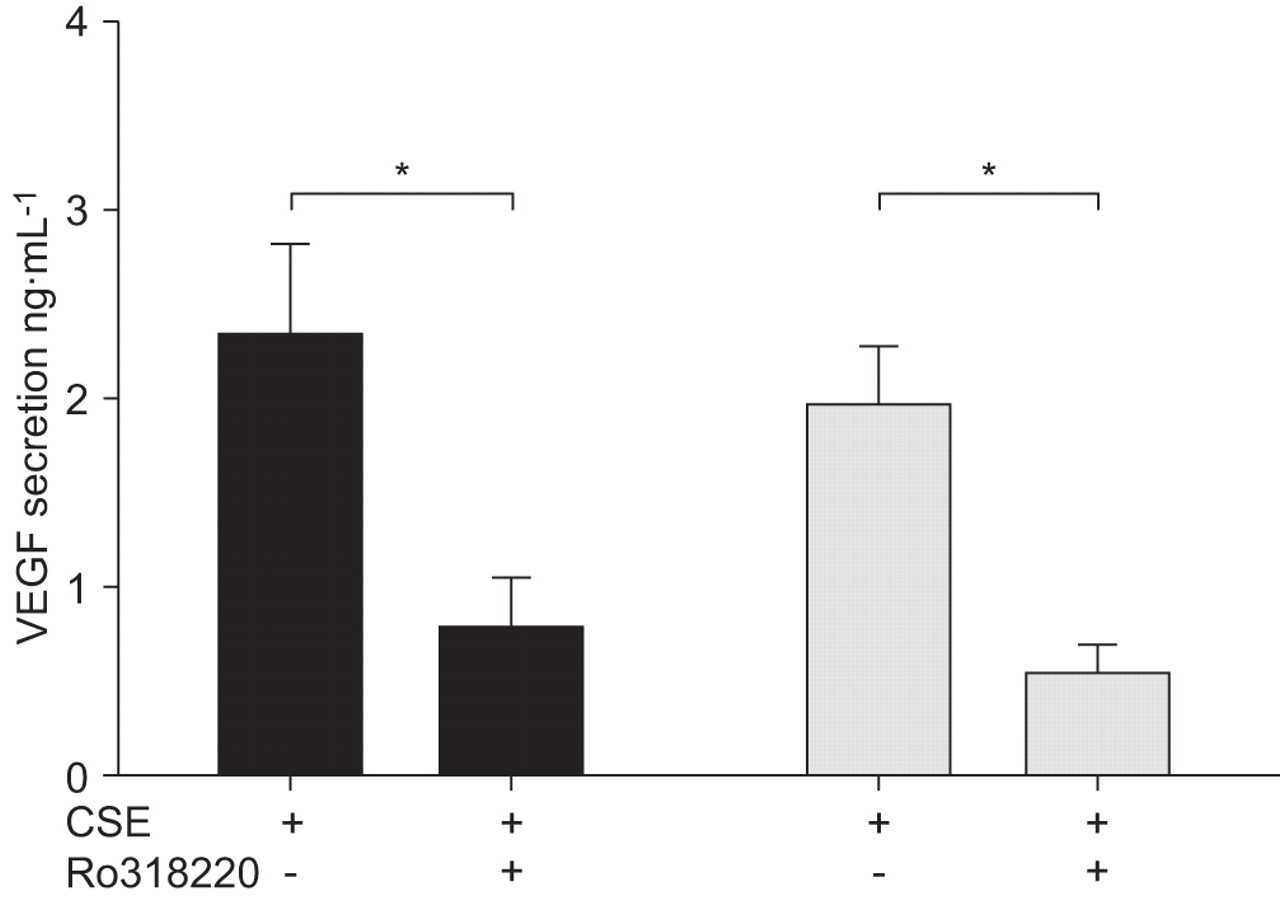
To understand the role of PKC signalling in regulating VEGF expression in NHBE and SAEC, cells were pretreated with the pan-PKC inhibitor Ro318220 (10 µM; Calbiochem) for 2 h, followed by 20% CSE treatment for 24 h. VEGF secretion and relative mRNA expression were determined at 24 h post CSE treatment. Pharmacological inhibition of PKC significantly reduced VEGF secretion in NHBE and SAEC, suggesting that this pathway is required for VEGF production in both cell types (fig. 6⇓). PKC inhibition also tended to reduce VEGF mRNA expression (up to 2.5-fold).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Role of protein kinase (PK)C in cigarette smoke extract (CSE)-mediated vascular endothelial growth factor (VEGF) reduction in normal human bronchial epithelial cells (▪) and small airway epithelial cells (░). Cells were pretreated with a PKC inhibitor, Ro318220 (10 µM), for 2 h, followed by 20% CSE treatment for 24 h. VEGF secretion was determined by an ELISA at 24 h post CSE treatment. Data are expressed as mean±sem (n = 3). *: p<0.05.
DISCUSSION
The present study demonstrated that CSE exposure reduced VEGF expression in well-differentiated primary human large and small AEC. Inhibitory effects of CSE on VEGF secretion were more pronounced in SAEC in the presence of a bacterial infection. Furthermore, the results revealed that ERK1/2 and PKC inhibition significantly reduced VEGF production, and might be in part responsible for CSE-mediated reduction of VEGF secretion in NHBE cells and/or SAEC.
CS or CS-related component exposure has been shown to induce a variety of biological responses, such as increased pro-inflammatory cytokine release, oxidative stress and apoptosis in lung epithelial cell lines or primary AEC under submerged culture conditions 10–14. A comparative study among various transformed lung epithelial cell lines and primary epithelial cells cultured under submerged culture conditions suggested that primary AEC were more responsive to CSE than transformed cell lines with regard to the release of inflammatory cytokines such as interleukin (IL)-8 and IL-6 11. Most of the previous studies were performed under submerged culture conditions, which do not mimic many of the major in vivo features of AEC such as mucociliary differentiation. Unlike previous studies, the current study utilised well-differentiated AEC to unravel the impact of CSE on airway epithelial biology such as VEGF production. The present results provide direct evidence that CSE exposure significantly reduced VEGF secretion in both large and small AEC, and thus have extended previous studies in whole lung tissues of severe emphysema patients as well as rodents 5, 6, 41. The maximum inhibition of CSE on VEGF secretion in the current study was 27% for NHBE and 34% for SAEC. Since the absolute VEGF protein levels (up to 6 ng·mL−1) in cultured AEC were very high, a reduction of 27% or 34% in VEGF should be significant. In support of this notion, previous studies demonstrated 20–38% VEGF reduction in human emphysematous lungs compared with normal lungs 5, 6, 42. Although the direct consequences of reduced VEGF from AEC with CS exposure were not explored in the current study, decreased VEGF has been clearly implicated in the destruction of lung parenchyma, including microvasculature, in severe emphysematous lungs 6, 8, 9.
Large AEC are exposed to the highest concentration of CS, but major pathological lesions in COPD patients exist in small airways and lung parenchyma. Hence, it is important to understand the effect of CS exposure on SAEC and to compare it with large AEC (e.g. NHBE cells). The current study is the first to determine the impact of CSE on VEGF production in both well-differentiated SAEC and NHBE cells. The results demonstrated that SAEC were more responsive to acute CSE exposure than NHBE cells, with regard to VEGF reduction. Although some reports have proposed certain differences of cellular population or gene expression, such as galectin-3 and Ki-67, in small and large AEC in COPD 43, no previous studies have addressed the molecular mechanisms responsible for the different responses to CS exposure between these two types of AEC. Further studies are needed to demonstrate the molecular basis upon which SAEC are more responsive to CSE exposure than NHBE cells with regard to VEGF reduction. Since both types of epithelial cells express different repertoires of nicotine receptors 44, it may be speculated that differing levels of nicotine receptors on SAEC and NHBE cells may be involved, but this needs to be explored in future experiments.
Increased bacterial colonisation has been reported in the airways of asymptomatic smokers as well as smokers with COPD. Among various bacteria, Mp, an atypical respiratory pathogen, was detected in 6.7% of COPD patients 19. The significance of Mp colonisation/infection in COPD is largely unknown. The current authors found that Mp alone significantly increased VEGF secretion in SAEC, but not in NHBE cells. Moreover, the presence of CSE abrogated Mp-induced VEGF secretion in SAEC, suggesting that persistent bacterial infection in a CS exposure milieu can significantly reduce VEGF secretion in lower airways, and may accelerate the loss of microvasculature, a pathological feature highly relevant to the development of emphysema in human lungs.
CS exposure has been shown to induce apoptosis, senescence, cell proliferation and inflammatory response in different cell types, including AEC cultured under submerged conditions 10–14. In contrast to previous findings, the well-differentiated primary human AEC in the current experiments did not increase cell growth, death or apoptosis upon CSE exposure. Therefore, the current findings suggest that CSE-mediated reduction in VEGF secretion is an active process and might be occurring at the transcriptional and/or translational level. Although there was a significant decrease in the VEGF protein secretion in CSE-treated cells compared with untreated cells at different time-points, the decrease in VEGF mRNA levels did not attain a significant difference between CSE-treated and -untreated cells. The inconsistency of VEGF mRNA and protein data may reflect the timing effect after CSE treatment. In a study by Koyama et al. 45, VEGF protein, but not VEGF mRNA, was increased in lung epithelial A549 cells at 72 h following IL-1β stimulation. The VEGF mRNA level increased during the first 12 h of IL-1β stimulation when VEGF protein was not increased 45. Therefore, it is not surprising that, after 24 h of CSE treatment, VEGF protein decreased in AEC but VEGF mRNA did not change significantly.
Boussat et al. 15 showed that well-differentiated NHBE cells produced a noticeable amount of VEGF at the baseline, which increased over the culture time. However, the molecular mechanisms governing the regulation of VEGF expression in well-differentiated primary human AEC are still not clear. Signalling pathways, including MAPK and PKC, have been shown to regulate VEGF expression in different cell types 21–27. In the present study, the role of MAPK pathways, including ERK1/2, JNK and p38MAPK, in regulating CSE-mediated decrease of VEGF secretion in well-differentiated AEC was examined. It was found that CSE exposure did not change JNK or p38MAPK phosphorylation/activation in either cell type, but an acute (15 min) increase followed by a decrease (by 24 h) in phospho-ERK/total ERK ratio was seen in CSE-treated NHBE cells compared with CSE-untreated cells. The current authors note that the Western blot data plus ERK inhibitor studies did not fully explain the significant difference in VEGF levels between CSE-treated and -untreated NHBE cells. Also, the ERK inhibition data did not explain the CSE-mediated decrease of VEGF in SAEC. Together, the data suggest that additional signalling pathways could also be involved in CSE-induced reduction of VEGF. Indeed, it was confirmed that the PKC pathway may also contribute to regulation of VEGF production in NHBE and SAEC, by use of a pan-PKC inhibitor (fig. 6⇑). Nonetheless, due to the complexity of VEGF regulation, further studies are warranted to investigate the role of additional signalling pathways, in order to better reveal the process of VEGF regulation following CS exposure.
The present study demonstrated that NHBE and SAEC differed in their responses to CSE with regard to VEGF production, and showed a role of the ERK1/2 pathway in the regulation of VEGF expression. Future experiments may need to focus on signalling molecules or pathways that are uniquely utilised by either type of epithelial cells to regulate VEGF synthesis. For example, the prostaglandin (PG)E2 pathway, which is clearly involved in tumour angiogenesis 46, 47, may play an important role in airway epithelial VEGF production. The current authors observed an induction (1.9-fold increase) of mitochondrial PGE synthase-1 (mPGES-1; a key enzyme in PGE2 synthesis) protein expression in SAEC, but not in NHBE, at day 7 post Mp infection (data not shown). This increased PGE2 pathway activity could be responsible for the significant increase in VEGF production in SAEC following Mp infection. Mp-induced mPGES-1 expression in SAEC was reduced (2.7-fold) in the presence of CSE. This observation needs to be further explored.
The current study demonstrated that cigarette smoke extract exposure significantly reduced vascular endothelial growth factor secretion in well-differentiated primary human large and small airway epithelial cells. Inhibitory effects of cigarette smoke extract on vascular endothelial growth factor secretion were more pronounced in small airway epithelial cells in the presence of a bacterial infection. Greater reduction of vascular endothelial growth factor secretion from small airway epithelial cells following cigarette smoke extract exposure could contribute to the loss of microvasculature in lung parenchyma of chronic obstructive pulmonary disease patients. Persistent bacterial infections or colonisation in cigarette smoke-exposed lungs may further enhance the loss of microvasculature and subsequent development of emphysema.
Support statement
This study was supported by the Flight Attendant Medical Research Institute (FAMRI; Miami, FL, USA).
Statement of interest
None declared.
- Received May 30, 2008.
- Accepted December 3, 2008.
- © ERS Journals Ltd
References










