





Abstract
We investigated the role of granulocyte-macrophage colony-stimulating factor (GM-CSF) in a subchronic exposure model of cigarette smoke (CS)-induced inflammation using antibodies directed against GM-CSF or the GM-CSF receptor (GM-CSFR) α-chain.
CS-induced mononuclear and neutrophilic inflammation following 4 days of CS exposure in BALB/c mice was assessed in bronchoalveolar lavage (BAL) fluid. An increase in mature dendritic cells (DCs) (CD11c+ and major histocompatibility complex II+) and Gr-1-high neutrophils was also observed by flow cytometric analysis of whole-lung tissue.
Daily i.p. injection of 400 μg GM-CSF or GM-CSFR antibody prior to daily smoke exposure attenuated the accumulation of neutrophils within the BAL by 60%. A reduction in mature DCs was also observed. Anti-GM-CSFR antibody administration did not have an effect on the percentage of lung T-cells; however, a significant decrease in activated CD69+ CD8+ T-cells was observed. Anti-GM-CSFR antibody administration decreased the mRNA and protein expression of interleukin-12 p40 and matrix metalloproteinase 12.
Taken together, intervention with this receptor antibody implicates the GM-CSF pathway as an important mediator of smoke-induced inflammation.
- Antibody neutralisation
- cigarette smoke
- granulocyte-macrophage colony-stimulating factor
- inflammation
- neutrophils
Whilst lung exposure to cigarette smoke (CS) has been identified as the leading cause of chronic obstructive pulmonary disease (COPD), the exact pathogenic mechanisms of the disease are not understood [1]. COPD is characterised by irreversible airflow obstruction and progressive lung inflammation that correlates with disease severity [2]. Whilst asthma is more associated with eosinophilic inflammation, in COPD, neutrophils and macrophages predominate [3].
As a key regulator of myeloid cell survival and activation [4], the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) has been shown to have a central role in maintaining the innate immune response in the healthy lung [5, 6]. It is released by a range of structural and inflammatory cells, such as the airway epithelium, smooth muscle cells, T-cells and macrophages. It has also been shown that GM-CSF is elevated in the lungs of patients with COPD [7–9], suggesting that this cytokine may also play a role in the exaggerated inflammatory response in the disease. The contribution of GM-CSF is unclear; however, it has been shown that GM-CSF not only activates macrophages and neutrophils in concert with stimuli, such as lipopolysaccharide, that may be present in the COPD lung, but also promotes cytokine release and cell survival [2]. Recently, it has also been demonstrated that intranasal delivery of an anti-GM-CSF antibody in a mouse model of CS-induced inflammation attenuates lung neutrophilia [10].
GM-CSF mediates its effects via a GM-CSF receptor (GM-CSFR) that consists of two subunits, an α-subunit, which binds the cytokine with low affinity, and a larger β-subunit (beta common; βc), responsible for signalling, forming a ternary receptor complex [11]. Signal transduction in response to the cytokines interleukin (IL)-3 and IL-5 is also mediated by βc; therefore, receptor specificity is due to GM-CSFRα [12]. Because CS is one of the main causative agents of COPD and a number of CS-exposure models have been developed that present with phenotypes analogous to COPD, such as emphysema and lung inflammation [13–15], these models provide platforms to evaluate the efficacy and mechanisms of new therapies. Biologics are increasingly being developed for chronic diseases, such as tumour necrosis factor (TNF)-α antibodies and receptor antagonists in arthritis [16–18], and anti-immunoglobulin (Ig)E antibodies in asthma [19]. Moreover, antibody trials on COPD are also underway that target IL-1β (canakinumab) [20]. Currently a number of anti-GM-CSF and anti-GM-CSFRα [4] approaches are in early clinical development for rheumatoid arthritis. To date, only a single study has been conducted investigating the role of GM-CSF in a CS-exposure model via intranasal delivery [10]. In the present study, we continued this work by evaluating a neutralising antibody to GM-CSF (22E9) and a GM-CSFRα (CAM-3003) antibody in the subchronic CS model dosed via the systemic compartment to determine the effects of inhibiting this pathway, via a more clinically relevant route of antibody administration. Moreover, we also explored the role of GM-CSF pathway blockade on downstream cytokines, and its effects on dendritic cell (DC) subsets in the lung and on CD8+ T-cell activation.
MATERIALS AND METHODS
Animals
Female BALB/c mice (6–8 weeks of age) were purchased from Charles River Laboratories (Montreal, QC, Canada). IL-1 receptor knockout and wild-type (WT) control C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained under specific pathogen-free conditions, on a 12-h light–dark cycle, with food and water provided ad libitum. The Animal Research Ethics Board of McMaster University (Hamilton, ON, Canada) approved all experiments.
FDCP-1 cell proliferation assay
FDCP-1 cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Burlington, ON, Canada) containing 10% fetal bovine serum (FBS; JRH Biosciences, Lenexa, KS, USA) and 5 ng·mL−1 murine GM-CSF (R&D Systems, Minneapolis, MN, USA). Prior to the assay, cells were cultured in the absence of GM-CSF for 30 h at 37°C in 5% CO2. Cells were resuspended at 1.5×105 cells·mL−1 in medium and 100 μL of this suspension was added to each well of a flat-bottomed, 96-well plate. Cells were incubated at 37°C in 5% CO2 with antibody in the presence of 1–2 pM murine GM-CSF for 16 h. 20 μL tritiated thymidine (5.0 mCi·mL−1) was added to each well and incubated for 4 h prior to harvesting on GF/C Unifilter™ plates (Perkin Elmer, Woodbridge, ON, Canada). Thymidine incorporation was determined using a scintillation counter (Packard Topcount; Packard Instrument Company, Meriden, CT, USA). Data were analysed using Prism software (GraphPad, San Diego, CA, USA).
CS-exposure protocol
Mice were exposed to CS from 12 2R4F reference cigarettes with filters removed (Tobacco and Health Research Institute, Lexington, KY, USA) for ∼50 min, twice daily, for 4 days using a whole body smoke exposure system (SIU-48; Promech Lab AB, Vintrie, Sweden) as described previously [21]. CS exposure followed an initial acclimatisation period whereby on day 1, mice were placed into the exposure box for 20 min, on day 2, for 30 min and on day 3, for 50 min. Control animals (sham), were exposed to room air only.
i.p. administration of antibodies
BALB/c mice were injected i.p. with 400 μg anti-GM-CSFR (CAM-3003; mouse IgG1) or anti-GM-CSF ligand (MP122E9 clone; rat IgG2a; R&D Systems) antibody 12 h prior to each of the first daily CS exposures. Mice were sacrificed 12–18 h after the last CS exposure.
Bronchoalveolar lavage and differential cell counting
Bronchoalveolar lavage (BAL) fluid was collected as follows. Briefly, lungs were dissected, cannulated and instilled with 0.25 mL ice-cold 1× PBS, followed by 0.2 mL 1× PBS. Total cell numbers were counted using a haemocytometer. Cytospins were prepared for differential cell counts and stained with Hema 3 (Biochemical Sciences Inc., Swedesboro, NJ, USA). 500 cells were counted per cytospin to identify mononuclear cells, neutrophils and eosinophils.
ELISA and meso-scale discovery analysis
ELISA kits for GM-CSF were purchased from R&D Systems and used according to manufacturer’s protocol. Cytokine detection of interferon (IFN)-γ, IL-1β, IL-4, keratinocyte-derived cytokine (KC) and IL-12 was performed using the multi-array murine pro-inflammatory and T-helper cell (Th) type 1/Th2 cytokine panel detection system (MesoScaleDevices, Gaithersburg, MD, USA).
mRNA expression Fluidigm analysis
RNA was extracted from a single lobe using the Qiagen RNeasy Fibrous Tissue kit according to manufacturer’s protocol (Qiagen, Hilden, Germany). RNA was quantified and normalised, and integrity assessed using the Agilent RNA 6000 Nano Kit (Agilent, Santa Clara, CA, USA). cDNA generation was carried out with the Super Script III kit (Life Technologies, Carlsbard, CA, USA). Relative transcript expression was assessed using the Fluidigm Biomark Dynamic Array (Fluidigm, Amsterdam, the Netherlands) loaded with probes for transcripts of interest.
Isolation of lung mononuclear cells
Lung mononuclear cells were isolated as described previously [21]. Briefly, lungs were perfused with 1× PBS, and cell suspensions were generated by disaggregation and incubation for 1 h at 37°C in 150 U·mL−1 collagenase III in hydroxyethyl piperazine ethane sulfonic acid-buffered saline solution (Invitrogen). Debris was removed by passing through a nylon mesh. Cells were resuspended in 1× PBS containing 0.3% bovine serum albumin (BSA) (Invitrogen) or in RPMI supplemented with 10% FBS (Sigma–Aldrich, Oakville, ON, Canada), 1% l-glutamine and 1% penicillin/streptomycin (Invitrogen).
Flow cytometric analysis
1×106 lung mononuclear cells were washed once with 1× PBS/0.3% BSA and stained with directly conjugated primary antibodies for 30 min at 4°C. 105 live events were acquired using an LSR II (BD Biosciences, Mississauga, ON, Canada) flow cytometer and analysed with FlowJo software (TreeStar Inc., Ashland, OR, USA). The following antibodies were used: fluorescein isothiocyanate-conjugated anti-CD11c, phycoerythrin (PE)-conjugated anti-CD11b, PE-Alexa Fluor 610-conjugated CD8, PE-cy5.5-conjugated anti-CD19, PE-cy7-conjugated anti-CD69, allophycocyanin (APC)-conjugated anti-major histocompatibility complex (MHC) class II, Alexa Fluor 700-conjugated anti-CD86, APC-cy7-conjugated anti-CD45, Pacific Blue-conjugated anti-CD3 and Pacific Orange-conjugated anti-Gr-1. These antibodies were purchased from BD Biosciences (San Jose, CA, USA) or eBioscience (San Diego, CA, USA) except for CD86 (BioLegend, San Diego, CA, USA). Quantum dot (Qdot)605-conjugated anti-CD4 and Qdot655-conjugated anti-B220 were purchased from Invitrogen (Carlsbad, CA, USA).
Statistical analysis
Data were analysed using Prism Software version 5 (Graphpad) and expressed as mean±sem. Statistical analysis was performed by testing for a normal distribution using the Kolmogorov–Smirnov test followed by an unpaired t-test, one-way ANOVA or a two-way ANOVA with a Bonferroni post-test, as stated in figures. Nonparametric tests were undertaken using the Kruskall–Wallis test with a Mann–Whitney post-test. Differences were considered statistically significant when p-values were <0.05.
RESULTS
CS-induced inflammation is associated with increased GM-CSF mRNA and protein expression
In order to investigate the impact of CS exposure on GM-CSF expression in the lungs, we exposed BALB/c mice to CS for 4 days. Control animals were exposed to room air only (sham). CS exposure led to a significant increase in total cell numbers in BAL, which predominantly reflected neutrophils (fig. 1a). CS significantly increased the levels of GM-CSF in the lung (fig. 1b).
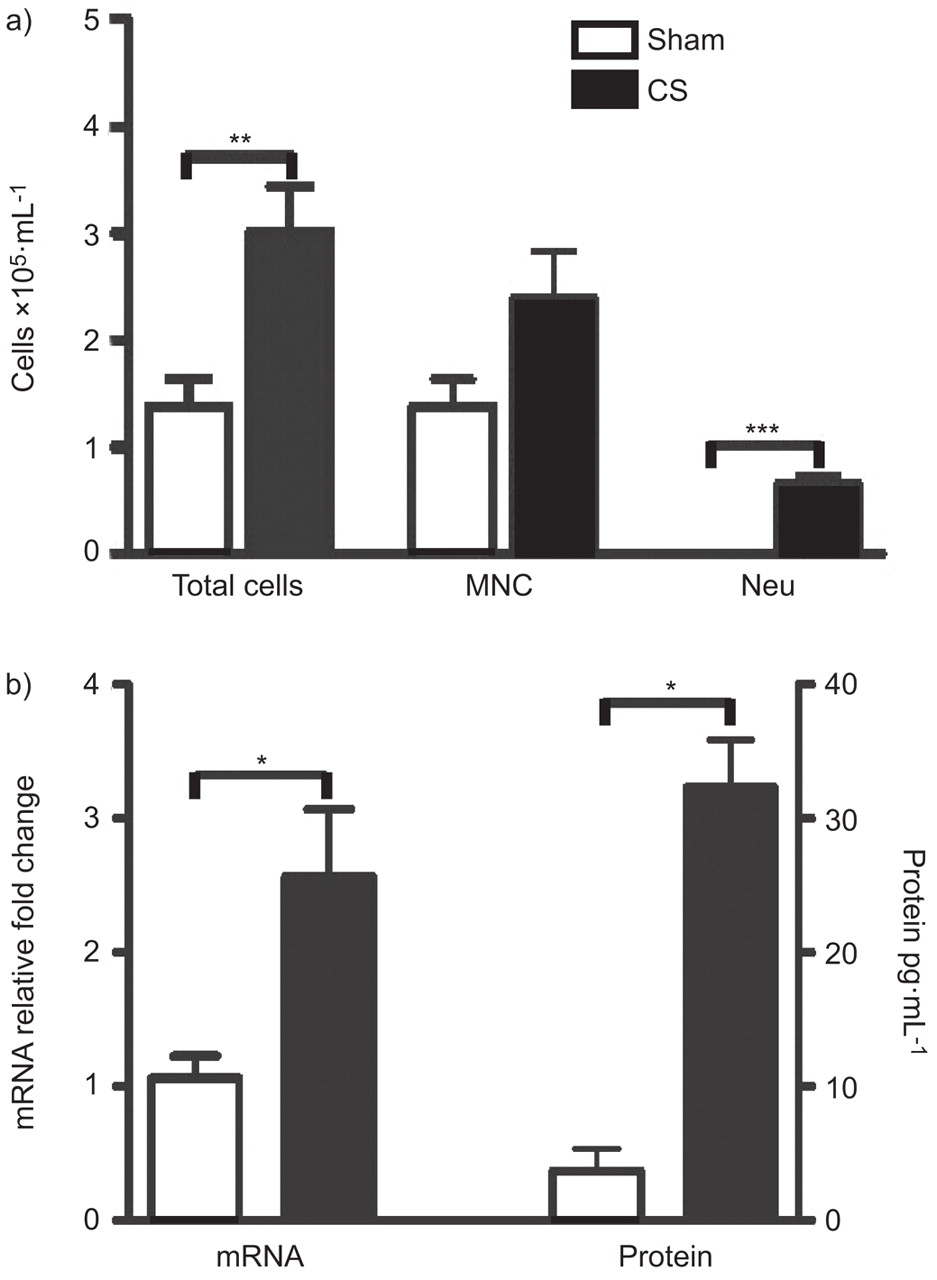
Bronchoalveolar lavage (BAL) cellular profile and granulocyte-macrophage colony-stimulating factor (GM-CSF) expression in cigarette smoke (CS)-exposed mice. BALB/c mice were sham- or CS-exposed for 4 days. a) BAL total cell number, mononuclear cells (MNC) and neutrophils (Neu). n = 5 per group. b) GM-CSF mRNA (sham: n = 3; CS: n = 8) and protein expression (n = 5 per group) in the lungs of sham- and CS-exposed mice. Statistical analysis was performed using the nonparametric Kruskall–Wallis test with a Mann–Whitney post-test. Bars represent means and whiskers represent standard errors of the mean. *: p<0.05; **: p<0.01; ***: p<0.001.
Mechanisms of GM-CSF expression
Although multiple pathways probably contribute to CS-induced inflammation, we next sought to investigate whether expression of GM-CSF following smoke exposure was IL-1 receptor (IL-1R)1-dependent. This experiment was based on a recent report by Doz et al. [22] demonstrating an important role of IL-1R1 in CS-induced inflammation. Consistent with the findings of Doz et al. [22], CS-induced neutrophilia was markedly attenuated in IL-1R-deficient mice (fig. 2a). Loss of IL-1R expression did not significantly affect the increase in mononuclear cells following CS exposure. We further examined GM-CSF mRNA expression in C57BL/6 (WT) and IL-1R-deficient mice that were either sham or CS exposed. GM-CSF mRNA expression was abrogated in the IL-1R-deficient mice (fig. 2b).
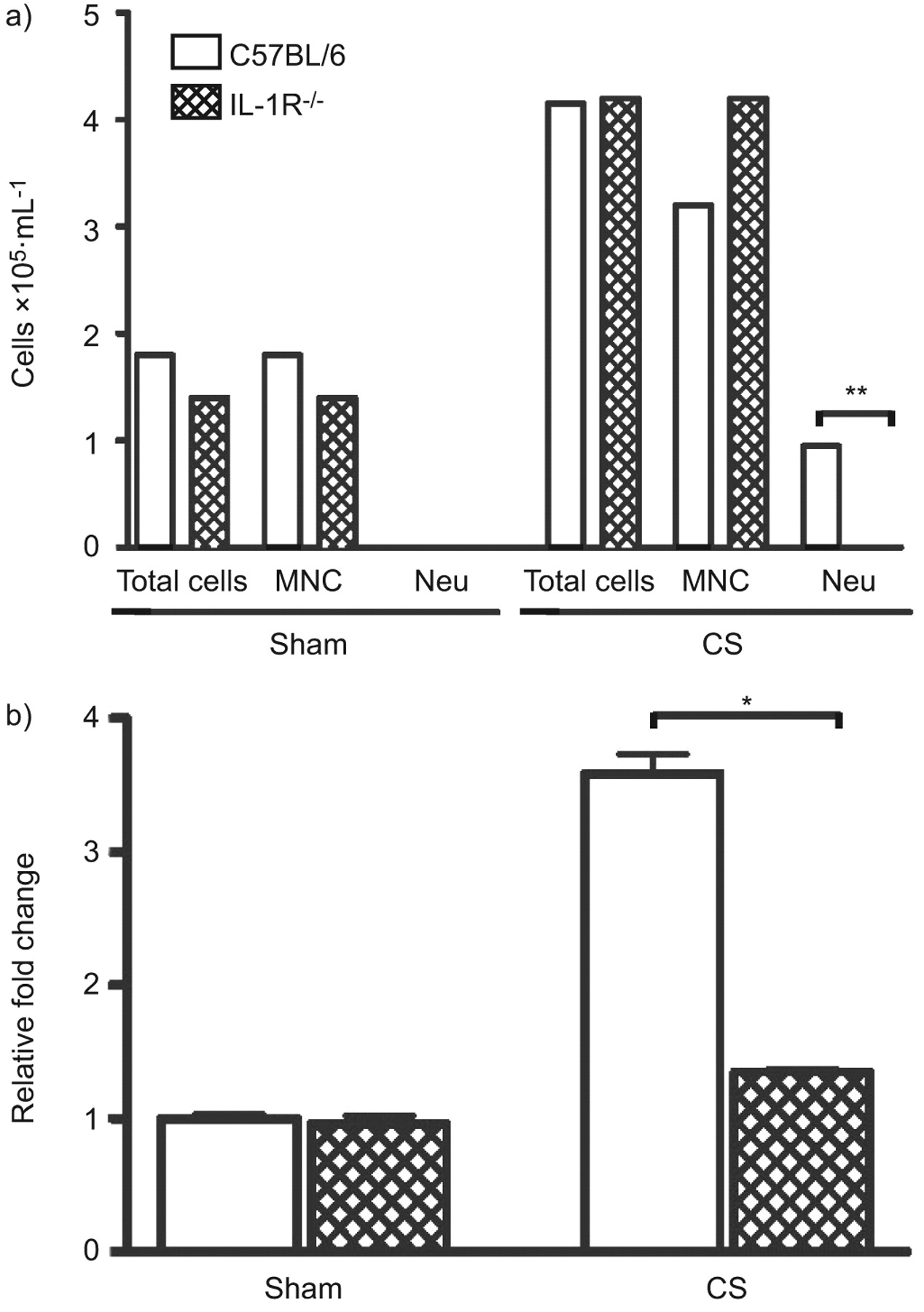
Bronchoalveolar lavage (BAL) cellular profile and granulocyte-macrophage colony-stimulating factor (GM-CSF) expression in cigarette smoke (CS)-exposed interleukin-1 receptor (IL-1R) knockout mice. a) Wild-type and IL-1R-/- mice were exposed to room air (sham) or CS for 4 days. BAL total cell number, mononuclear cells (MNC) and neutrophils (Neu) are shown. b) GM-CSF mRNA expression in sham- and CS-exposed wild type IL-1R-/- mice. Bars represent means and whiskers represent standard errors of the mean. n = 5 per group. Statistical analysis was performed using two-way ANOVA with a Bonferroni post-test for wild-type versus knockout. *: p<0.05; **: p<0.01.
Anti-GM-CSF ligand and receptor antibodies attenuate CS-induced lung inflammation
In order to test the importance of GM-CSF to CS-induced airway inflammation, we administered either anti-ligand (22E9) or receptor-blocking antibodies (CAM-3003) to sham- or CS-exposed mice. The activity of each antibody was confirmed in the FDCP-1 proliferation assay (fig. 3a). The median inhibitory concentration of 22E9 and CAM-3003 was 919 pM (95% CI 694–1,218 pM) and 130 pM (95% CI 99–171 pM), respectively. Due to the different potencies and isotypes of these antibodies, fully saturating doses of each antibody were used in these studies. Pharmacokinetic analysis of CAM-3003 was undertaken to confirm antibody exposure levels between sham- and CS-treated animals in both lung and blood compartments. No difference in antibody exposure was observed between sham- and CS-treated animals (fig. 3b). In vivo anti-GM-CSF and anti-GM-CSFR antibody administration resulted in a comparable and significantly attenuated CS-induced neutrophilic inflammation. Control mice receiving the isotype control antibody demonstrated no attentuation of CS-induced inflammation (fig. 3c and d). In order to determine changes in neutrophil numbers in the interstitium, flow cytometry of lung single-cell suspensions was undertaken. The percentage of Gr-1-high cells was significantly increased by CS exposure when compared with sham-treated animals (p<0.05). No significant difference was observed between CS-exposed isotype control-, anti-GM-CSF- or anti-GM-CSFR-treated mice (fig. 4).
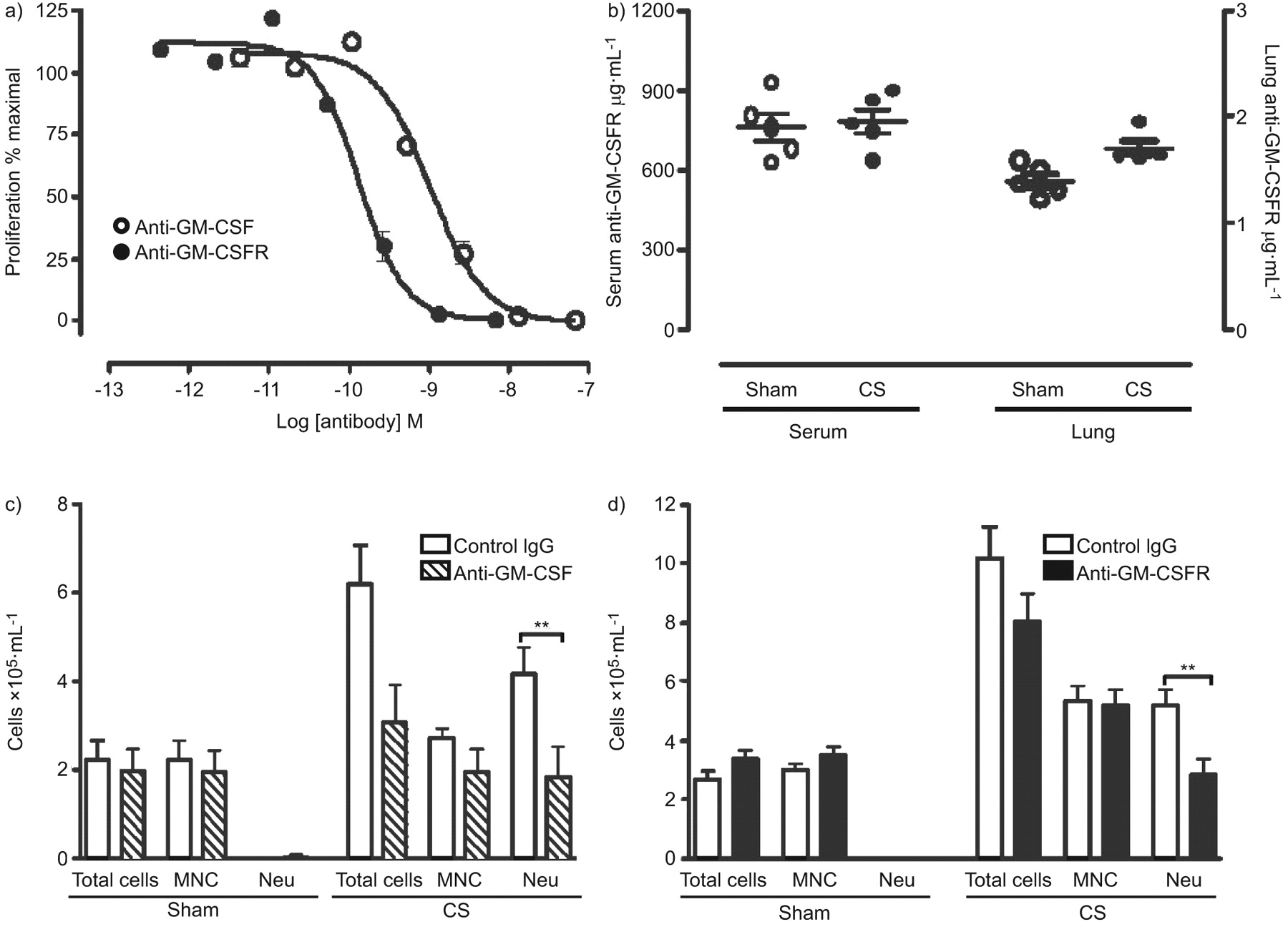
Granulocyte-macrophage colony-stimulating factor (GM-CSF) neutralising activity and the bronchoalveolar lavage cellular profile in cigarette smoke (CS)-exposed mice injected with anti-GM-CSF ligand or anti-GM-CSF receptor (anti-GM-CSFR) antibodies. a) Potency comparison of anti-GM-CSF and anti-GM-CSFR in a mouse GM-CSF FDCP-1 proliferation assay. Cells were incubated with mouse GM-CSF and a dilution series of antibody for 16 h. Cell proliferation was quantified following a further 4 h incubation with tritiated thymidine. b) Terminal exposure of anti-GM-CSFR levels in serum and lung homogenates from sham- and CS-exposed mice. BALB/c mice were exposed to CS for 4 days. Mice were injected i.p. with either c) anti-GM-CSF ligand or d) anti-GM-CSFR, or isotype control antibodies 18 h prior to daily CS exposures. Data show total cell numbers, mononuclear cells (MNC) and neutrophils (Neu). Bars represent means and whiskers represent standard errors of the mean. n = 5 per group. Statistical analysis was performed using a two-way ANOVA with a Bonferroni post-test for sham versus CS groups. **: p<0.01.

Flow cytometric analysis of neutrophils (Gr-1-high) in sham and cigarette smoke (CS)-exposed mice injected with anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand or anti-GM-CSF receptor (GM-CSFR) antibodies. BALB/c mice were exposed to CS for 4 days. Mice were injected i.p. with either a) anti-GM-CSF or b) anti-GM-CSFR antibodies, or isotype control antibodies. Neutrophils (Gr-1-high, major histocompatibility complex class II-high) were examined in whole-lung, single-cell suspensions by flow cytometry. Statistical analysis was performed using two-way ANOVA with a Bonferroni post-test for sham versus smoke.
Anti-GM-CSF ligand and receptor antibodies attenuate CS-induced DC expansion and T-cell activation
It is widely accepted that GM-CSF promotes DC maturation, a critical step in antigen-driven T-cell responses (reviewed in [4]). Therefore, we investigated whether intervention with anti-GM-CSF or receptor antibodies attenuated DC expansion and T-cell activation following CS exposure. Anti-GM-CSF ligand antibody intervention abrogated CS-induced increases in myeloid DCs (mDCs) (CD11c-high MHC II-high) and plasmacytoid DCs (pDCs) (CD11c-high MHC II+ B220+) DCs (fig. 5a and b). Administration of an anti-GM-CSFR antibody also reduced the frequency of mDCs (fig. 5c and d), while no effect was observed on pDCs.

Flow cytometric analysis of dendritic cell (DC) subsets and activated T-cells in cigarette smoke (CS)-exposed mice injected with anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand or anti-GM-CSF receptor (GM-CSFR) antibodies. BALB/c mice were exposed to CS for 4 days. Mice were injected i.p. with either a, b, e) anti-GM-CSF ligand antibodies or c, d, f) anti-GM-CSFR antibodies, or isotype control antibodies. a, c) Myeloid DCs (CD11c-high, major histocompatibility complex (MHC) class II-high B220-) and b, d) plasmacytoid DCs (CD11c-high, MHC II-high B220+), or e, f) activated CD69+ CD4+ and CD8+ T-cell subsets were examined in whole-lung single-cell suspensions by flow cytometry. Statistical analysis was performed using a two-way ANOVA with a Bonferroni post-test for sham versus CS. Ig: immunoglobulin. *: p<0.05; **: p<0.01.
Anti-GM-CSF ligand antibody intervention abrogated CS-induced increases in activated CD69+ CD4+ and CD8+ T-cells compared with isotype control-treated animals. In contrast to ligand-blocking antibody, a reduction was only observed for CD69+ CD8+ T-cells, but not CD4+ T cells (fig. 5e and f), in mice treated with an anti-GM-CSFR antibody.
Anti-GM-CSFR antibody treatment reduces IL-12, matrix metalloproteinase 12 mRNA and protein expression in CS-exposed BALB/c mice
In order to study the mechanisms of CS-induced inflammation, we analysed the expression of lung cytokines and chemokines, as well as matrix metalloproteinases (MMPs), using Fluidigm analysis. CS exposure caused a significant increase in mRNA expression for: the cytokines IL-12p40, IL-1β, IL-33, IL-6, mucin 5AC, and TNF-α; the chemokines KC, monocyte chemotactic protein-1, C–C motif chemokine ligand (CCL)3, CCL4, CCL9, colony-stimulating factor-3 and CXC motif ligand (CXCL)2; and MMP9 and MMP12 (table 1). Anti-GM-CSFR antibody significantly attenuated the mRNA expression of IL-12p40 (fig. 6a) and MMP-12 (fig. 6b) relative to isotype control antibody treatment (fig. 6 and table 1). No significant difference was observed with TNF-α or CXCL2 relative to isotype control (fig. 6c and d).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Interleukin-12 p40, b) matrix metalloproteinase 12, c) tumour necrosis factor-α and d) CXC ligand 2 mRNA expression. BALB/c mice were exposed to room air (sham) or cigarette smoke (CS) for 4 days. Mice were injected i.p. with anti-granulocyte-macrophage colony-stimulating factor receptor (GM-CSFR) or isotype control antibodies. All values were normalised to three housekeeping genes and expressed as the relative fold change over sham-exposed mice. Bars represent means and whiskers represent standard errors of the mean. n = 5 per group. Statistical analysis was performed comparing CS-exposed control immunoglobulin (Ig)G versus anti-GM-CSFR antibody-treated mice using the Mann–Whitney U-test. ns: nonsignificant. **: p<0.001.
In order to confirm the mRNA expression data, we examined expression of cytokines IL-1β KC, IFN-γ, IL-2, IL-12 and IL-4. Increased expression of IL-1β, KC and IL-12 was observed in CS-exposed animals injected with isotype antibody control. The anti-GM-CSFR antibody significantly reduced expression of IL-12 in the BAL and lung homogenates, while IL-1β or KC levels remained unchanged. A similar observation was made for IL-1β levels in animals treated with the anti-ligand antibody (supplemental table 1). IFN-γ, IL-2 and IL-4 levels were all below the limit of detection in sham- and CS-treated groups (table 2).
DISCUSSION
Neutrophils and macrophages are regularly implicated in the pathogenesis of COPD. It has previously been shown that as the severity of the disease progresses, the proportion of inflammatory cells in the airways increases [2]. Therefore, it has been proposed that interfering with mechanisms that blunt the activity of these cells may provide clinical benefit. Consequently, GM-CSF has been implicated as a potential mediator in COPD.
Whilst GM-CSF is elevated in the lungs from patients with COPD or chronic bronchitis [8], interventions targeting this pathway in models of CS-induced lung inflammation have not been widely described [10]. Here we showed that, following 4 days of CS exposure, we observed a consistent increase in total cells in the BAL, the majority being neutrophils. Moreover, GM-CSF was elevated at both the transcript and protein levels in the lungs from these animals. Interestingly, whilst Vlahos et al. [10] demonstrated attenuation of CS-induced lung inflammation with antibodies to GM-CSF, they were unable to measure GM-CSF protein in the BAL. Likewise, they also demonstrated attenuation of neutrophils and macrophages; however, in our model of CS-induced inflammation, we observed an increase and subsequent attenuation of neutrophils only, using either antibody. It is of note that the type of CS-exposure system and route of antibody delivery were different and, thus, may account for the subtle differences observed between this study and that previously described [10].
In order to determine whether the GM-CSF pathway played a significant role in the recruitment of inflammatory cells into the BAL, we evaluated both an anti-GM-CSF neutralising antibody (22E9) and an anti-GM-CSFR α-chain antibody (CAM3003) in this model. To rule out the potential of antibody-dependent cellular cytotoxicity, the anti-GM-CSFR antibody was specifically expressed as a mouse IgG1 antibody. Consequently, no evidence of neutrophil depletion in peripheral blood was observed with this antibody (data not shown). Using either approach, we inhibited the recruitment of neutrophils in the BAL by dosing via the systemic compartment; however, we did not reduce neutrophil numbers in the tissue, as defined by flow cytometry. This observation in the BAL is consistent with the previous observation that intranasal delivery of the same GM-CSF antibody (22E9) was reported to blunt CS-induced neutrophil and macrophage recruitment in the BAL [10, 23]. Whilst we did not observe a significant increase in macrophages and lymphocytes upon CS exposure, we did observe a modest but significant change in the percentage of mature lung DCs that was attenuated with either antibody. In vitro, it has been shown that diesel exhaust particle-conditioned medium from epithelial cells can induce a GM-CSF-dependent DC maturation [24], suggesting that CS exposure may also contribute to DC maturation in a similar manner. This is supported by the observation that, in turn, both CD4+ and CD8+ T-cells increase CD69 expression following CS exposure as previously described [21], and that this was partially inhibited by blockade of the GM-CSF pathway. Interestingly only CD8+ T-cell activation was suppressed by both mechanisms, but the CD4+ CD69+ activated T-cells were not affected by treatment with anti-GM-CSFR. The reason for this is unclear and merits further investigation. Nevertheless, T-cells do not express the GM-CSFR α-chain; therefore, these data suggest that a reduction in the CS-induced T-cell activation maybe an indirect effect of GM-CSF stimulation of myeloid cells rather than a direct effect of GM-CSF on T-cells.
Recent studies [22] have shown that MyD88-, Toll-like receptor 4- and IL-1R-deficient mice all had reduced lung neutrophilia and cytokine levels in the lung following CS exposure. Therefore, we investigated GM-CSF expression in CS-exposed IL-1R-deficient mice. In this system, we confirmed that CS-induced neutrophilia was suppressed and that KC mRNA levels were significantly inhibited (data not shown). Based on our observations, it may be concluded that GM-CSF plays a central role in CS lung inflammation, potentially downstream of the IL-1R pathway. A similar observation has also been observed in a mouse model of IL-1β-driven monoarthitis [25].
In order to determine the mechanism of GM-CSF blockade, we investigated the change in expression levels of various proteins and mRNAs in the lung. Whilst CS exposure enhanced the expression of a range of cytokines, blocking the GM-CSF pathway did not appear to suppress cytokines typically associated with neutrophilic inflammation, such as IL-1β and KC. GM-CSF is known to activate neutrophils, promoting adhesion in pulmonary vascular endothelium [26], recruitment [27] and sensitisation to chemokines [28]. The lack of a direct effect on KC, IL-1β and TNF suggests that GM-CSF may operate downstream of these cytokines, potentially rendering neutrophils less responsive to the direct effect of these molecules, and may also account for there being no apparent difference between the percentage of neutrophils in lung homogenates from CS-exposed mice treated with either an anti-ligand antibody or anti-receptor antibody. It has also been demonstrated that GM-CSF promotes neutrophil survival by preventing apoptosis [29] and, thus, GM-CSF blockade may also have enhanced neutrophil apoptosis and clearance by macrophages. Further studies are warranted to better understand how neutrophil numbers in the BAL are attenuated and the activation status of resident neutrophils within the lung.
However, inhibition of the GM-CSF pathway during CS exposure did inhibit IL-12p40 and IL-12 production at the transcript and protein level, respectively. In vitro, GM-CSF has previously been shown to induce IL-12 from both DCs and macrophages [30]. In addition, mice deficient in GM-CSF exposed either to a viral [31] or bacterial [32] pathogen demonstrated a reduction in IL-12 production. Furthermore, overexpression of GM-CSF in the lungs of normal mice stimulates an increase in IL-12 production [33]. Similarly, we showed that IL-12p40 was upregulated in the lungs of CS-exposed mice and that these levels were attenuated with either antibody, indicating that IL-12p40 expression is downstream of CS-induced GM-CSF production. Chronic CS exposure in mice plus the addition of virus or viral mimetics (polyI:C) has been shown to promote an increase in IL-12p40 production [34]; however, this is the first time that this elevation has been shown in such an subchronic model. Nevertheless, relatively little is known about the expression of this molecule and its role in COPD [24] and, therefore, further studies are warranted.
In addition to an upregulation of IL-12p40, subchronic smoke exposure also stimulated an increase in MMP-12 mRNA, consistent with previous reports [35]. Tissue remodelling and proteolytic damage are hallmarks of COPD, and proteases, such as MMP9 and MMP12, have been shown to be elevated within the COPD lung [36, 37]. Furthermore, mice deficient in MMP-12 do not present with an emphysematous phenotype following chronic smoke exposure [38]. Both neutrophils and macrophages are known to be a rich source of MMP-12 [39] and their numbers correlate with disease severity in COPD [40]. In another model of CS induced lung inflammation, macrophages were described as being the main source of this protease [35]. In the present study, we have demonstrated that blocking GM-CSF signalling attenuated MMP-12 expression in lung tissue. These data are consistent with the observation that GM-CSF can induce MMP-12 production from human peripheral blood monocyte-derived macrophages [41, 42]. Interestingly, whilst we did observe a change in MMP-12 mRNA levels in the lung, as recently described [10], we did not observe a statistically significant change in either TNF-α mRNA or macrophage inflammatory protein 2α (fig. 6c and d) in this system, suggesting that the route of delivery may have subtly different effects on mRNA profiles within the lung. This difference was not due to antibody exposure, as pharmacokinetic analysis of CAM-3003 confirmed significant levels of antibody in both serum and lung homogenates (fig. 3b).
In conclusion, the present study reports that systemic delivery of anti-GM-CSF or anti-GM-CSFR antibodies attenuates CS-induced neutrophilia. Furthermore, GM-CSF appears to play a role in activating resident lung DCs and lymphocytes, providing a link between the initial innate response to subchronic CS exposure and supporting the adaptive response. Further studies are warranted to characterise this relationship and shed light on the potential role of this cytokine in COPD.
Acknowledgments
The authors gratefully acknowledge the expert technical support of J. Kasinska and S. Kianpour (both McMaster University, Hamilton, ON, Canada), J. Elvin (MedImmune Ltd, Cambridge, UK), P. Brohawn and A. Keller (both MedImmune LLC, Gaithersburg, MD, USA), and the Hybridoma team at MedImmune (Cambridge).
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
J.K. Nikota was supported by Ontario’s Early Research Award program (ERA). M.R. Stämpfli holds a Canadian Institutes for Health Research New Investigator award.
Statement of Interest
Statements of interest for N.H.E. Davis, E.S. Cohen, I.K. Anderson, A.J. Coyle, R. Kolbeck, A.A. Humbles, M.R. Stämpfli and M.A. Sleeman can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received May 14, 2010.
- Accepted February 2, 2011.
- ©ERS 2011
REFERENCES










