





Abstract
In vivo animal models can offer valuable information on several aspects of asthma pathogenesis and treatment. The mouse is increasingly used in these models, mainly because this species allows for the application in vivo of a broad range of immunological tools, including gene deletion technology. Mice, therefore, seem particularly useful to further elucidate factors influencing the response to inhaled allergens. Examples include: the role of immunoregulatory mechanisms that protect against T‐helper cell type 2 cell development; the trafficking of T‐cells; and the contribution of the innate immunity. However, as for other animal species, murine models also have limitations. Mice do not spontaneously develop asthma and no model mimics the entire asthma phenotype. Instead, mice should be used to model specific traits of the human disease. The present task force report draws attention to specific aspects of lung structure and function that need to be borne in mind when developing such models and interpreting the results. In particular, efforts should be made to develop models that mimic the lung function changes characteristic of asthma as closely as possible. A large section of this report is therefore devoted to an overview of airway function and its measurement in mice.
⇓The vast efforts that have been put into asthma research in the past have clearly increased insight into the pathogenesis of the disease. At the same time, current gene technology identifies an ever increasing range of molecules that could be involved in the sensitisation process to an allergen and/or the development of a chronic inflammatory process in the mucosa of the lower airways. This evolution tends to confer an image of overwhelming complexity. The resulting challenge is to identify the exact functional role of the various molecules that may be involved. For obvious ethical reasons, studies in humans are restricted to a combination of morphological assessment and in vitro experimentation. Hence the continuing need for in vivo animal models, since they allow for a more functional assessment of a given molecule, relating it, amongst other features, to the dynamics of the inflammatory process or to alterations in airway behaviour.

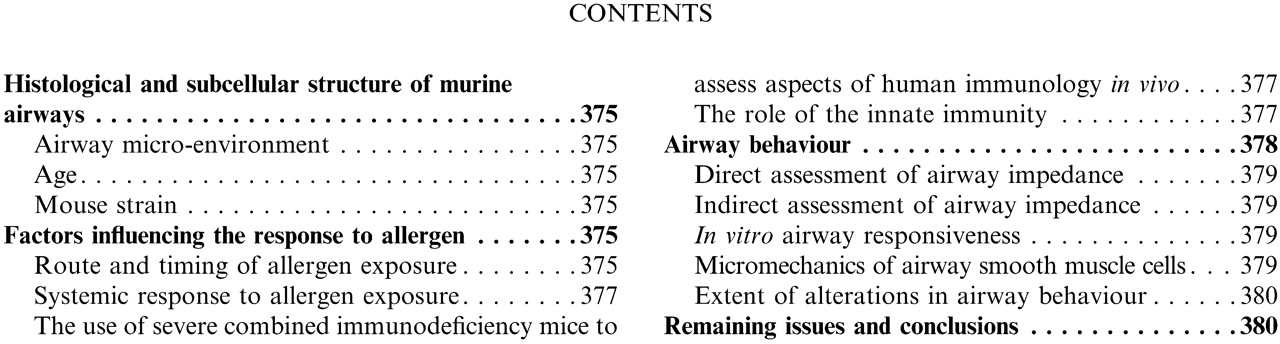
Epithelial-mesenchymal trophic unit. NEB: neuroendocrine body.

Interaction between CD8 cells and antigen presenting cells. Tc: Cytotoxic T cell; DC: dendritic cell; IL: interleukin; Th: T‐helper cell; IFN‐γ: interferon‐γ; IgE: immunoglobulin E.
The mouse is increasingly being used for the development of in vivo asthma models. An obvious advantage is that it is a nonendangered species that offers wide availability of genetically characterised inbred strains at relatively low cost. Above all, this species allows for the application in vivo of an extremely wide diversity of immunological tools, including gene deletion technology. The use of “knock-out” mice has obvious advantages when evaluating the functional role of a given cell or mediator in a complex situation, such as allergen-induced airway inflammation and its relationship with altered airway behaviour. Cell-derived products, such as cytokines, exert their effect in a paracrine or autocrine manner. This implies some uncertainty as to whether exogenously administered antagonists, such as monoclonal antibodies, can reach sufficiently high concentrations for a long enough time at the intercellular level to achieve full antagonism. Genetic manipulation can circumvent this problem. In addition, as possibilities for conditional deletion increase, the opportunity arises to time the switching off of the gene of interest. This will allow evaluation of the role of gene products in adult life that are essential for the early stage of murine development and the constitutional deletion of which can therefore prove fatal. Conditional deletion also allows evaluation of the functional role of a gene product, specifically during the process of primary antigen sensitisation or, conversely, during secondary antigen exposure to memory cells. However, as for other animal species, murine models also have limitations. Mice are not human and do not spontaneously develop asthma. Therefore, apart from the difficulties in transposing murine to human data, it has to be borne in mind that mice are probably more suitable for modelling a trait associated with asthma, rather than for modelling the entire asthma phenotype.
As a result, when designing an in vivo mouse model, it is important to clearly decide which aspects of lung structure and/or function associated with asthma need to be addressed. Development of a representative model will therefore have to take into account knowledge of murine biology, as well as the characteristics of human asthma that need to be modelled. The present report attempts to highlight a few of these features.
Histological and subcellullar structure of murine airways
A trait associated with human asthma that currently incites intense scientific interest is airway remodelling. The pathogenesis and functional consequences remain to be fully established. When trying to model this trait in mice, it is important to bear in mind the structure of murine airways. The components of the tracheobronchial wall that are critical in determining the response to inhaled substances are organised as the epithelial-mesenchymal trophic unit. The airway wall consists of an epithelial, interstitial, nervous and immunological component (fig. 1⇑) 1–3. The basic assumption is that all of these compartments have an active interaction with each other. Perturbation of one compartment creates an imbalance in all parts of the airway wall, or rather, a metabolic response in one compartment will produce alterations in the other compartments. The pattern of epithelial repair after acute injury involves a number of essential elements. Injured cells are lost by exfoliation into the airway lumen. The remaining epithelial population at the site of injury undergoes a marked increase in proliferative rate, reaching a peak 2–4 days after injury. Complete re-establishment of cell density and organisation appears to rely on the epithelial populations in airway generations adjacent to the injury site, in most cases proximal to it in the airway tree. This includes prolonged proliferation past the proliferative upsurge that isassociated with the site of injury, the movement of cellsfromnontarget airways toward the site of injury, and reorganisation of phenotypes during the differentiation phase. Accompanying the re-establishment of cell density is the redifferentiation of pre-injury target cell phenotypic characteristics. Re-establishment of pre-injury steady state in the epithelium generally involves an extensive post-injury period (14–21 days). How interactive the epithelium is with the mesenchymal cells and the subepithelial matrix is unclear. However, fibroblasts proliferate at the site of injury in association with increased proliferation in the epithelium. Also, fibroblasts have been identified as a major source of the growth factors and other regulatory molecules known to enhance various aspects of epithelial cell function associated with repair.
It is important to note that several factors can influence the airway response to stressors. These include the airway micro-environment, age of animals at the time of exposure to injurants and mouse strain.
Airway micro-environment
First, a normal response to injury requires an intact tubular structure, including all components of the airway wall. The absence of an open tubular structure due to the collapse or rupture of airway wall integrity results in a transformation of the epithelial population. Secondly, the injury response varies markedly with the position of the target within the tracheobronchial airway tree. This is true for a wide variety of chemicals, including oxidant gases and bioactivated hydrocarbons. Regardless of the chemical or the phenotype of the target cell population, the sensitivity varies substantially between proximal and distal airways. This is at least partly related to position-dependent differences in the activity of enzymes recognised as participating in pathways that detoxify, neutralise or conjugate reactive compounds within cells, such as glutathione S‐transferase 4, 5.
Age
The age of the animals at the time of exposure to injurants plays a tremendous role in the pattern of injury and repair. Infants are much more susceptible to injury by bioactivated lung toxicants than adults of the same species. In addition, injury to young animals results in cessation of the cycle of repair in the proliferation and squamation phases, surviving cuboidal cells do not differentiate and squamated survivors do not recover to a cuboidal epithelial pattern. This arrangement of the airway epithelium persists for significant periods of time into adulthood.
Mouse strain
Comparison of acute injury responses and subsequent repair has established that the patterns are unique not only for the species, but even the strains under consideration, and that cells occupying identical positions in different species respond differently.
Factors influencing the response to allergen
Route and timing of allergen exposure
Immunoregulatory mechanisms
In atopic individuals, inhalation of nonpathogenetic allergens induces a preferential T‐helper cell (Th)2 development, whereas in nonatopic individuals, a Th1 response predominates. The predisposition to develop a Th2 phenotype is genetically determined, whereas environmental factors can probably contribute to further consolidate the overall skewing of the Th2 response. One aspect of this process that remains to be fully elucidated is the immunological mechanisms that protect against preferential Th2 development, such as interleukin (IL)-10 and transforming growth factor‐β secreting regulatory T‐cells 6, 7. This is one area where in vivo models could prove of interest.
In the majority of current models, mice are sensitised by the intraperitoneal injection of ovalbumin, often together with a Th2 skewing adjuvant, such as alum. Sensitisation in itself induces production of ovalbumin-specific immunoglobulin (Ig)E. Upon secondary exposure to aerosolised ovalbumin, sensitised animals then develop in vivo airway hyperresponsiveness and eosinophil recruitment into the airways. However, it is well known that if mice are first exposed to an aerosol of ovalbumin, no or very low serum levels of antigen-specific IgE are induced 8. In addition, it was recently shown that ovalbumin inhalation prior to intraperitoneal sensitisation also affects antigen-induced airway changes. Mice were exposed to eight aerosols of saline (control) or ovalbumin (0.2% 5 min·day−1). After 2 weeks, mice were sensitised without adjuvant to seven injections of ovalbumin (10 µg intraperitoneal) on alternate days and 3 weeks later, repeatedly challenged by inhalation of either a saline or ovalbumin (0.2%) aerosol. Ovalbumin inhalations, prior to sensitisation, resulted in an almost complete inhibition of ovalbumin-induced eosinophil recruitment and increase in airway responsiveness. Moreover, no upregulation of serum IgE or IgG1 was observed. Instead, in mice exposed to ovalbumin inhalation prior to sensitisation, high serum levels of IgG2a were detected. This change in the Ig profile point to a shift from a Th2 to aTh1 dominated response. This phenomenon somewhat resembles the protective response of a nonatopic individual to allergen inhalation. Further perusing the mechanisms involved might therefore be relevant for human asthma.
Similarly, it has been shown that sensitisation and exposure to ovalbumin in rats and mice can induce activation of immunomodulatory CD8 cells that enhance the balance of Th1 over Th2 CD4 T‐cells and inhibit IgE synthesis 9. The mechanisms underlying this phenomenon have now been further clarified 10. First, CD8 T‐cells do not inhibit IgE by direct effects on B‐cells, such as cytotoxity. The observation that CD8 cells had no effect on IgE production in interferon (IFN)‐γ-/- mice clearly establishes the requirement for endogenous IFN production. The source of IFN‐γ production has been identified as the CD4 Th1 cell. Indeed, adoptive transfer of ovalbumin-specific CD8 T‐cells from IFN‐γ-/- mice to naïve wild-type mice as effectively inhibits IgE synthesis as IFN‐γ competent CD8 T‐cells, whereas CD8 T‐cells cannot inhibit IgE synthesis in IFN‐γ-/- mice, unless they are reconstituted with wild-type CD4 T‐cells. Subsequent experiments demonstrated that, in addition, IL-12 is essential for CD8 T‐cells to inhibit IgE. The overall emerging picture is that antigen-presenting cells (APC) interact both with CD8 and CD4 T‐cells. Interaction between CD8 T‐cells and the APC induces production of IL-12, which, in turn, stimulates CD4 T‐cells to produce IFN‐γ, thus delivering the signal to the B‐cell to inhibit IgE synthesis (fig. 2⇑). What remains to be further elucidated is how the CD8 T‐cell induces the APC to produce IL-12.
Growth factors
Differences in the response to allergens, depending on the route of administration, can also be used to unravel the fractional importance of growth factors in the pathogenesis of allergic airway inflammation. Adenoviral-mediated transfection of the airway epithelium with granulocyte-macrophage colony-stimulating factor (GM-CSF) has been shown to substantially enhance allergen-induced responses 11. In these experiments, Balb/c mice were repeatedly exposed to aerosolised ovalbumin, without intraperitoneal sensitisation or the use of adjuvants. In control animals, exposure to ovalbumin induced little airway change. However, in GM-CSF transgenic animals, ovalbumin exposure resulted in a clear increase in major histocompatibility complex (MHC) class II expressing cells (peaking at day 7), followed by CD3+ cells (peaking at day 11) and eosinophils (peaking at day 14). By day 28, ovalbumin-induced changes had subsided. However, in GM-CSF transgenic animals, a long-term recall phenomenon to renewed ovalbumin exposure could be demonstrated, suggesting the development of memory T‐cells.
The exact site of activation and trafficking of T‐cells in asthma is another element in the pathogenesis that needs to be further clarified. The same protocol of co-exposure to GM-CSF and ovalbumin in tumour necrosis factor (TNF)‐β-/- mice that lack local lymph nodes but retain a functional spleen, induced a similar degree of airway inflammation when compared with wild-type mice. Splenectomy abolished allergen-induced airway changes in TNF‐β-/- mice but not in wild-type animals. These results indicate that in wild-type animals, T‐cell activation occurs in the draining lymph nodes, with the spleen acting as back-up.
Therapeutic intervention
The route and timing of allergen exposure used in murine models also need to be taken into account when evaluating potential novel forms of treatment. Therapeutic intervention that interferes either at the time of sensitisation or before the onset of airway inflammation might be less relevant to asthma than starting intervention only once airway inflammation has already been induced. As the relative importance and/or activation state of cells vary throughout the development of allergen-induced airway changes, so can the response to treatment. For example, it is well known that T‐lymphocytes, which are considered important for the initiation as well as the progression of allergic asthma, require different signals, depending on their activation status. Naïve T‐lymphocytes require at least two activation stimuli: a specific signal generated by ligation of the T‐cell receptor by the peptide-MHC class-II complex and a co-stimulatory signal delivered by CD28:B7.1/2 interaction. However, after sensitisation, memory T‐lymphocytes are present, which are less dependent on co-stimulatory signals for full activation 12, 13. During an ongoing response, recently activated effector T‐lymphocytes are present. Recently, it was shown that the inducible costimulator/B7RP‐1 co-stimulatory pathway predominates over the CD28:B7.1/2 pathway in regulating cytokine production from recently activated T‐cells. These differences in T‐lymphocyte phenotypes at different phases of the immune response may contribute to the effect of experimental treatment.
The effect of cytokines on T‐cells can also vary throughout their development. IL-12 and -18 induce preferential development of naïve T‐cells towards a Th1 phenotype. However, once polarised, Th2 cells lose expression of functional IL-12 or -18 receptors. This probably explains why a mixture of IL-12 and -18, when given to sensitised mice prior to ovalbumin inhalation, is far more effective at inhibiting allergen-induced hyperresponsiveness and bronchoalveolar lavage (BAL) eosinophilia than when given after repeated antigen inhalation, once the airway changes have already been established 14. Similarly, whereas IL‐4 is crucial for the development ofTh2 cells during primary antigen sensitisation, it is not sufficient to cause all allergen-induced airway changes. For example, it is increasingly clear that during secondary antigen exposure, IL-13 has a far larger influence on airway responsiveness 15, 16.
Systemic response to allergen exposure
It is increasingly recognised that asthma is a systemic disease that involves a profound cross communication between the airways and bone marrow 17. However, the degree of the contribution of bone marrow to the airway inflammatory process and the mechanisms by which the bone marrow is stimulated are still poorly understood. Mouse models seem suitable to further explore the signalling events. IL‐5 has been shown to be crucial for the induction of airway eosinophilia. For example, mice lacking IL‐5 fail to develop airway eosinophilia in response to allergen exposure. Furthermore, treatment of wild-type mice with a neutralising anti-IL‐5 antibody strongly reduces the eosinophilic inflammatory response induced by allergens. It has been reported that systemic administration of a neutralising IL‐5 antibody is far more effective at reducing airway inflammation than local administration into the airways. The inhibitory effects of systemic anti-IL‐5 were closely related to inhibition of allergen-induced bone marrow eosinophilia 18, 19. These conclusions are in line with studies showing that airway and bone marrow eosinophilia can be reconstituted in sensitised IL‐5-/- via systemic but not local overexpression of IL‐5. Overall, these data indicate that bone marrow is intricately involved in the induction of allergen-induced eosinophilic airway inflammation. Allergen inhalation has indeed been shown to cause an increase in IL‐5‐producing CD3+ cells in the bone marrow, resulting in an increased production of eosinophils. This doesnot exclude the possibility that, in addition, eosinophil precursors may migrate from the bone marrow to the lungs, locally generating terminally differentiating eosinophils.
The use of severe combined immunodeficiency mice to assess aspects of human immunology in vivo
One of the major strengths of in vivo animal models is that they allow for a functional evaluation of different pathogenetic pathways by using specific pharmacological or immunological interventions that are not (yet) available for human use. This approach is obviously hampered if differences exist between mouse and humans in the biological activity and/or structure of the mediator of interest. An example of a biological compound to which these limitations apply is T‐cell-derived cytokines, such as IL‐4. Because of the lack of cross-species reactivity, the biological activity of an antihuman IL‐4 approach cannot be properly evaluated in murine models. One possible approach to circumvent this problem is to use severe combined immunodeficiency (SCID) mice. Reconstitution of SCID mice with human peripheral blood mononuclear cells (Hu-PBMC) results in a human-mouse chimera with a functional human antigen-reactive system, enabling the in vivo study of human T‐cell biology. Exposure of Hu-PBMC SCID mice to an aerosolised allergen has been reported by different groups to induce airway hyperresponsiveness that would appear to be driven purely by human T‐cells 20, 21. These models allow further evaluation of the effect of specifically modulating human T‐cell biology on airway function.
The role of the innate immunity
The contribution of the innate immune system to the pathogenesis of asthma has long been largely neglected. However, more recent observations increasingly recognise the interaction between components of the innate and adaptive immune system. In recent years, in vivo murine models have been modelled to address several aspects of this interaction in relation to asthma pathogenesis.
Natural killer cells
There is much evidence to suggest that natural killer (NK) cells form an important link between the innate and adaptive immunity. Infectious agents or oxidant stress have been reported to stimulate not only macrophages and epithelial cells but also NK cells, to produce a range of cytokines including CSF‐1 and GM-CSF. One result of this, amongst others, is an upregulation of nuclear factor-κB expression, enhancing the acute inflammatory response, but also activation of fibroblasts to produce growth factors, thus contributing to the persistence of the inflammatory response. Macrophages and NK cells also express high levels of lyn kinases. Their activation has been shown to profoundly enhance both the degree and persistence of antigen-induced airway eosinophilia. Conversely, antagonising NK cells with anti-NK1.1 antibodies during the sensitisation period prevents the development of antigen-induced eosinophilia. The same treatment has no effect when given during secondary antigen exposure 22.
Endotoxin
A major role of the innate immune system is toprotect against infectious organisms. It is increasingly recognised that microbial components can have an important impact on the sensitisation to allergen, as well as on the clinical manifestations of asthma. Exposure to high levels of endotoxin early in life has been reported to protect against the persistence of a Th2 skewed immune system in genetically predisposed individuals 23, 24. The probable explanation for this observation is that, through activation of the Toll-like pattern recognition receptor, endotoxin stimulates dendritic cells to produce IL-12. The presence of this cytokine favours differentiation of naïve T‐cells towards a Th1 phenotype upon MHC class II-dependent antigen presentation by dendritic cell. This phenomenon is arguably one of the clearest examples of an interaction between the innate and adaptive immune system.
Once asthma has been developed, the effect of endotoxin exposure is different, as a relationship has been described between the severity of asthma and the endotoxin content of house dust samples. This phenomenon has been modelled in recent in vivo mouse models 25–27. From these models, it emerges that the lipopolysaccharide (LPS)-mediated effects are largely regulated by the endogenous production of TNF. Intranasal administration of LPS in C57Bl/6 mice induces an increase in TNF levels in BAL fluid, followed by recruitment of neutrophils. The TNF increase is accompanied by protein leakage in BAL fluid. The inflammatory changes are paralleled by an increase in enhanced pause (Penh), suggesting an enhanced resistance to airflow. It is important to note that the LPS-mediated effects are relatively resistant to the effect of steroids. Relatively low doses prevent the neutrophil recruitment, whereas high doses are required to also inhibit TNF release, protein leakage and increase in Penh.
Bacterial superantigen
Other microbial components that could profoundly affect the adaptive immune system are bacterial superantigens that are known to cross-link MHC molecules on APC to the T‐cell receptor on T‐cells. In nonsensitised animals, superantigen has been reported to induce a self-limiting airway neutrophilia. In contrast, in sensitised animals, superantigens largely enhance the antigen-induced inflammation, causing increased recruitment not only of neutrophils, but also eosinophils and lymphocytes, to the airways 28. Strikingly, the staphylococcal enterotoxin B‐induced enhancement of the allergen inflammation is very similar in Balb/c and C57Bl/6 mice, even if C57BL/6 mice develop less pronounced airway responsiveness to allergen challenge than similarly sensitised and exposed Balb/c mice.
Airway behaviour
Although in vivo murine models of allergic airway inflammation can provide data that are relevant to the pathophysiology of asthma, it should be remembered that the main feature of asthma is altered airway behaviour, resulting in an episodic difficulty of breathing. Therefore, representative in vivo models should mimic the main functional characteristics of the disease as closely as possible. These can be summarised as follows 29.
1) Spontaneous and variable airflow limitation.
2) Excessive airway narrowing. Asthmatics are not only more sensitive to bronchoconstrictor stimuli, but also more responsive. The latter characteristic includes a rise or disappearance of the maximal response plateau to bronchoconstrictor stimuli, as observed in nonasthmatics, and reflects potentially life-threatening excessive airway narrowing 30. This represents one of the major abnormalities of the disease. The main cause of this is altered airway mechanics, presumably due to changes in the cellular structure and extracellular matrix of the mucosa, lamina propria, the smooth muscle layer itself, and the adventitial area. This seems to highlight the fact that asthma is not caused by a single defect at the airway level.
3) Impaired modulation of airway narrowing by deep inspiration. One of the strongest modulators of airway narrowing in normal subjects is taking a deep inspiration. The impairment of this mechanism observed in asthma has been linked to several possible mechanisms 31, 32: reduced smooth muscle stretch by impaired airway parenchymal coupling secondary to peribronchial inflammation or loss of alveolar attachments; reduced stretch of airway smooth muscle, secondary to airways remodelling and stiffening of the airway wall; primary or secondary reduction in actin-myosin cross-bridge cycling rate in chronically shortened airway smooth muscle, leading to a stiff and contractile “latch” state; changes in airway smooth muscle plasticity after chronic shortening, due to changed arrangement of contractile filaments to the cytoskeleton; impaired inhibitory bronchoprotective mechanisms, such as endogenous nitric oxide.
4) Closure of airways in the periphery of the lung 33.
5) The chronicity and persistence of these functional abnormalities over time 29, 31.
It should be remembered that current murine models do not match all of these functional characteristics of asthma. However, murine models of asthma can provide detailed information on the association between the (immuno)pathology and some of these various aspects of airway function in this disease. Development of murine models that model these issues as closely as possible requires insight in the structure/function relationship of murine airways.
It has been calculated that measurement of lung resistance (RL) together with compliance can offer an overall evaluation of lung function in small animals, including mice. Compliance is an index of functional stiffness of the lung. RL measures resistance to airflow and is determined by flow, airway size and lung tissue resistance (Rti). The relative contribution of each of these determinants to the overall RL in mice has been addressed in a limited number of studies 34. In mice, as in rats, the influence of the chest wall on airway mechanics is negligible. The chest wall in these species is far more compliant than it is in humans. As a result, mice have a lower functional residual capacity and compensate with a much higher breathing frequencies in order to reduce fluctuations in alveolar gas concentrations. The contribution of the various lung compartments to airway resistance (Raw) is less clear. The total RL consists of Raw+Rti. Rti largely depends on breathing frequency, being higher at low frequency and gradually decreasing with increasing breathing frequency 35. As in mice, this frequency is >120 min−1 and it can be anticipated that at baseline conditions, Rti contributes relatively little to the total RL.
Although it can be assumed that at baseline, RL largely reflects airflow resistance in the large airways, this does not necessarily mean that changes in RL as measured during the evaluation of airway responsiveness only reflect changes in the calibre of large airways 36, 37. First, in contrast to Rti, RL is dependent on the tidal volume, since RL decreases with increasing tidal volume as a reflection of parenchymal interdependence. Therefore, changes in lung volume throughout the construction of dose/response curves to a bronchoconstrictor agonist could in itself have an important effect on RL measurement. Secondly, bronchoconstrictor agonists also influence the repartition of RL. Increasing concentrations of methacholine affect both Raw and Rti. As a result, especially at increasing lung volumes (associated with a selective decrease in Raw), the relative contribution of Rti to RL increases. This is also dependent on the route of administration of methacholine. For the same increase in RL, the Rti component induced by aerosolised methacholine is far higher than after i.v. administration. i.v. administration largely causes contraction of airways of different size, whereas aerosol administration causes a contraction both in the airways and in the parenchyma, probably due to a far more inhomogenous constriction of peripheral airways. Therefore, methacholine-induced changes in RL do not allow definite conclusions that this is simply due to large airway constriction.
Additional elements that need to be taken into account when evaluating airway responsiveness in mice are that the basal degree of airway responsiveness, as well as the biological activity of different bronchoconstrictor agonists, can differ between various mouse strains.
As well as the question of which component of lung structure is reflected in a given lung function index, there is the question of what constitutes the correct technique to measure a given lung function index. Several techniques for measuring lung function in mice have been developed and, to date, none have been universally accepted. The difference between these various techniques makes interpretation difficult and between-laboratory comparisons are almost impossible. The available techniques for measuring lung function invivo can be classified according to whether airway function is assessed by a direct measurement of resistance (RL)/elastance (E) or by another measurement that is indirectly thought to be related to airway function 38–40.
Direct assessment of airway impedance
These techniques all have in common the measurement of airflow or lung volume change (ΔVL) and the driving pressure (Pdriving) responsible for achieving both the flow and lung volume change. The measured flow can be integrated with respect to time to calculate volume changes, whereas the measured ΔVL can be differentiated with respect to time to calculate flow. The resulting volume, flow and pressure signals are generally fitted to the equation of motion:  In other methods, E is calculated based on the change in ΔVL and Pdriving between end inspiration and end expiration (i.e. points of zero flow). Resistance is then calculated by relating flow to the nonelastic component of pressure during either inspiration or expiration (i.e. points of equal volume). Theoretically, when Pdriving is transpulmonary (requiring measurement of pleural pressure), then E and R are pulmonary (airway and lung tissue); when the driving pressure is transthoracic, then E and R are of the entire respiratory system (pulmonary plus the chest wall). However, as already indicated, the contribution of the chest wall in mice is considered to be minimal.
In other methods, E is calculated based on the change in ΔVL and Pdriving between end inspiration and end expiration (i.e. points of zero flow). Resistance is then calculated by relating flow to the nonelastic component of pressure during either inspiration or expiration (i.e. points of equal volume). Theoretically, when Pdriving is transpulmonary (requiring measurement of pleural pressure), then E and R are pulmonary (airway and lung tissue); when the driving pressure is transthoracic, then E and R are of the entire respiratory system (pulmonary plus the chest wall). However, as already indicated, the contribution of the chest wall in mice is considered to be minimal.
These principles have been applied in several models that, in general, perform measurements in tracheostomised and paralysed mice, using either a pneumotachograph, whole-body plethysmograph, flow interrupter method or forced oscillations.
Indirect assessment of airway impedance
Changes in the impedance of the mouse respiratory system will affect the Pdriving delivered by a ventilator to anaesthetised and tracheotomised animals. Therefore, impedance can be estimated by measuring the peak ventilator pressure or the overflow of a ventilator set to deliver via a T‐tube at a set volume and flow rate to the animals.
Another technique is the noninvasive whole-body plethysmograph. This technique is based on placing an unrestrained intact mouse in an essentially airtight chamber and measuring the pressure fluctuations within the chamber. Unlike direct measurement techniques, this pressure signal is not the pressure driving the respiratory system. The pressure increases occur as air is expanded due to the increase in temperature and the slight negativity of alveolar pressure during inspiration. Likewise, box pressure is decreased during expiration as expired air reduces in volume due to cooling and the slight compression of air within the thorax. Regardless of the mechanisms of the pressure fluctuation, a relationship termed Penh, based on the magnitude and timing of the pressure fluctuation, has been correlated with the magnitude of respiratory system resistance and dynamic compliance. It needs to be emphasised that Penh is not derived from first physical principles but based on empiric signal processing. Therefore, the absolute value of Penh has no physical meaning. In addition, a change in respiratory pattern in response to an intervention can influence Penh without a true change in airway calibre occurring. As a result, it has been recommended that, until the reliability of Penh as a surrogate for bronchoconstriction is further established, Penh measures should be confirmed with a parameter of airway obstruction which is based on physical principles 41.
In vitro airway responsiveness
An alternative approach to measuring lung function is to evaluate the in vitro responsiveness of murine airways. For the assessment of the underlying mechanisms of bronchial hyperresponsiveness and its pharmacological modulation in human airways, airway preparations are used that have been passively sensitised with IgE-rich serum, since lung resection material and therefore isolated airway preparation from patients with asthma are rare and difficult to obtain. So far, this model appears to closely mimic the characteristic features of bronchial asthma, such as the induction of specific responsiveness to allergens as well as nonspecific hyperresponsiveness to bronchospasmogenic stimuli, such as histamine and leukotriene C4 42, 43. It further allows prediction of drug effects, such as the effect of treatment with anti-IgE. A similar model in mice providing unlimited availability of tissue could obviously provide interesting additional data. Again, however, when developing such a model deemed to be representative of the human situation, a number of caveats need to be borne in mind. First, to study airway hyperresponsiveness in mice in vitro, airways from animals that have been already sensitised in vivo, either actively by repeated exposure to specific allergens or passively by the injection of specific IgE, are generally used. Whether in vitro reactivity of mouse airways sensitised in vivo resembles that of ex vivo sensitised human preparations has not been investigated to date. Secondly, there are obvious differences in the lung and the airway anatomy of humans and mice. The human tracheal diameter is ∼25 mm, while in mice it is <2 mm. Studies in mice generally use tracheal preparations. Human preparations of a similar size represent quite a different airway generation. If airway generations within the respiratory tract serve different functional tasks, comparability through size may not be sufficient. Finally, the airway response of mouse and human preparations to different agonists is not always comparable 44–46. For example, it has been shown that the cholinergic muscarinic subtype 3 receptor-mediated construction, as well as the adrenergic β2‐adrenoreceptor-mediated relaxation, is comparable in both species. However, airway responses to tachykinins are quite different between humans and mice. In human airways, neurokinin A‐induced bronchoconstriction is mediated through NK2 receptor activation, while substance P and other NK1-selective agonists have no effect. In mice, neither neuropeptide causes bronchoconstriction under basal conditions. However, carbachol-induced bronchoconstriction is relaxed by neurokinin A as well as substance P, thus illustrating the limitations of one model to predict responses in the other. The same is true for inflammatory mediators, such as histamine and leukotrienes, which are believed to play a central role in allergic bronchoconstriction in human airways. While both histamine and leukotriene C4 are potent constrictors of human airways, these mediators have almost no bronchoconstrictor effect in mice.
Micromechanics of airway smooth muscle cells
Bronchial hyperresponsiveness is a cardinal feature of asthma. The mechanistic basis of this phenomenon remains tobe further understood. Inflammation-induced changes in the intrinsic contractility of airway smooth muscle is one of the elements thought to be involved. Techniques have been developed that allow assessment of the micromechanics of human airway smooth muscle cells in culture 47–50. This includes the magnetic twisting cytometry. Ligand-coated ferromagnetic beads are bound to a receptor of interest (e.g. integrin) and oscillatory mechanical torques are then applied to the beads by a sinusoidally varying external magnetic field. This applied field induces a torque that causes each bead to rotate towards alignment within the field, like a compass needle aligning with the Earth's magnetic field. Beads bind avidly to the cell and as the bead rotates it deforms the cytoskeleton. Bead rotation is impeded principally by the mechanical stresses that are developed within the cell body as it deforms. As the cell stiffens as a result of activation of contractile proteins, the amplitude of bead rotation decreases. Bead motions can be determined either by measuring changes of the remanent magnetic field due to bead rotations or, alternatively, by direct observation of bead displacements using light microscopy. This technology thus allows measurement of changes in cell stiffness and frictional mechanical properties in cells and has been shown to be extremely useful in elucidating gene function in human airway smooth muscle cells. Whether the same technique can be applied to smooth muscle from mice is currently under investigation.
Extent of alterations in airway behaviour
Bronchial hyperrresponsiveness in asthma is characterised by an important increase in the sensitivity to bronchoconstrictor agonists, when compared to normal subjects. Irrespective of the technique used to measure smooth muscle and/or lung function, representative murine models should try to induce a similar degree of hyperresponsiveness. In most of the currently developed models, this has not been achieved as, in general, only limited and short-lived hypersensitivity can be demonstrated. This may be due partly to the degree of morphological alterations induced in response to allergen exposure. In most murine models, only transient acute inflammatory changes are induced, which consist of an increase in vascular permeability and influx of inflammatory cells. These alterations are more pronounced in asthmatic airways. In addition, in human asthma, structural alterations, described as airway remodelling, are also present. These include an increase in goblet cells in the epithelium, mucous gland hyperplasia, subepithelial fibrosis with deposition of collagen and fibronectin, smooth muscle hypertrophy and/or hyperplasia and formation of new vessels. Mathematical models predict that airway remodelling profoundly influences bronchial responsiveness. In view of the importance attributed to remodelling, it seems highly relevant to develop in vivo models that accurately reflect this phenomenon. This would allow understanding of the functional consequences of these changes and the mechanisms underlying them and examination of ways of preventing or reversing these important alterations. Such studies are not easy or possible to perform in asthmatics because, at present, there are no adequate ways of assessing airway wall remodelling noninvasively.
Studies in transgenic mice have indicated that at least some of the features can be mimicked. For example, mice overexpressing IL‐6, -11, -13 and ‐5 show evidence of subepithelial fibrosis. Overexpression of IL-11 is associated with myocyte hyperplasia and overexpression of IL‐4, -13, ‐5 and ‐9 with mucus metaplasia 51–53. Clearly, aspects of airway wall remodelling can be mimicked in the mouse, but these studies are not a realistic model of asthma, since overexpression of these cytokines is an artificial situation. However, the possibility of turning cytokines on and off at will and expressing cytokines specifically to the airway epithelium usually by using an adenovirus vector have provided more valuable information on the capacity of certain cytokines to induce airway wall remodelling.
A more realistic approach is to use chronic allergen exposure to induce aspects of airway wall remodelling and, in turn, use these models to unravel pathophysiology. In these models of chronic exposure following sensitisation, persistent airway eosinophilia, with goblet cell hyperplasia and subepithelial fibrosis, is observed but there is little mention of the airway smooth muscle changes. Several models, displaying, to a variable extent, characteristics of remodelling, are currently undergoing further development 54, 55.
Remaining issues and conclusions
In vivo murine models can offer valuable information in several domains of asthma pathogenesis and treatment. Particular advantages of in vivo models are that they allow the study of the dynamics of certain phenomena and the relation of structure to function. At present, it is an illusion to think that a relevant overall asthma model exists in mice. Instead, the in vivo models should be used to model specific traits of the human disease. Several factors that need to be taken into account when developing such models and interpreting the findings have been touched upon. A number of other features, however, were not discussed. These include thechoice of allergen or bronchoconstrictor agonist when assessing airway responsiveness, whether mouse models can be used to model asthma traits, such as childhood asthma, the variability in baseline airway calibre and enhanced mucous production. Furthermore, it was felt that optimal use of the mouse as an asthma model requires additional insight into the structure and function of murine airways, including its neural control and the protective role of the airway mucosa against infections, as well as a further characterisation of the murine genome. Finally, it was clearly felt that further characterisation and development of in vivo mouse models will undoubtedly enhance the exchange and comparability of information emerging from different groups and increase the representativeness of the model for well-defined asthma traits.
- Received March 9, 2003.
- Accepted March 18, 2003.
- © ERS Journals Ltd










{kind=link}
{kind=link}
{kind=link}