





Abstract
Airway remodelling is a complex process that involves all of the component tissues of the airway from the epithelium to the adventitia. Each of the changes has the potential to alter airway physiology so as to promote airway narrowing, hyperresponsiveness and inflammation. Structural changes, such as epithelial metaplasia, airway fibrosis and airway smooth muscle hyperplasia, have been successfully modelled in animals. These models are being extensively characterised and are providing valuable insights into mechanisms that are likely to be quite relevant to human asthma. Remodelling is induced by factors synthesised and secreted both by inflammatory cells and by structural cells, the latter frequently under the influence of the former. While information concerning the genesis of inflammation is abundant, the precise factors responsible for cellular hyperplasia, hypertrophy and altered matrix deposition are far from resolved. Elucidation of these factors will no doubt lead to novel therapies designed to prevent or reverse these changes.
- Airway epithelium
- airway hyperresponsiveness
- airway remodelling
- airway smooth muscle
- angiogenesis
- animal model
SERIES “AIRWAY REMODELLING: FROM BASIC SCIENCE TO CLINICAL PRACTICE”
Edited by L-P. Boulet and P.J. Sterk
Number 1 in this Series
Remodelling generally refers to the structural changes in the airways associated with asthmatic airway inflammation. These structural changes, which involve virtually every tissue, are accompanied by a myriad of changes in cellular phenotypes at the molecular level that, although not obvious on microscopy, are potentially of equal importance. The present review focuses on the often extensive structural alterations that constitute remodelling of asthmatic airways (summarised in fig. 1⇓). The review is not comprehensive but emphasises data from animal models that is supported by both human studies and data from cell culture systems.
In asthma, the epithelium is more fragile, as indicated by shedding and increased turnover of cells. Epithelial cells differentiate frequently into mucus-secreting goblet cells and mucus glands increase in number and size. Myofibroblasts, which have a mixed contractile and collagen synthesising phenotype, are likely to participate in subepithelial deposition of collagen and other matrix proteins that cause the classical thickening of the lamina reticularis 1. Similarly, altered matrix protein deposition contributes to remodelling of the submucosa and adventitia. Bronchial blood vessels increase in number and size 2, and bronchial smooth muscle increases in mass 3, 4. In addition, the inner border of the smooth muscle bundles is closer to the epithelium, suggesting that new muscle is preferentially added to the luminal side of the muscle 5, 6. In vitro evidence suggests that the phenotype of smooth muscle cells, which can be modulated by extracellular matrix (ECM), may regulate the ability of these cells to proliferate 7. This may also be a factor contributing to airway smooth muscle (ASM) remodelling in asthma. Finally, the peribronchial area is thickened and there is evidence of disrupted or damaged alveolar attachments and abnormal elastin content in peripheral airways from severe asthmatics 8.
The importance of remodelling seems beyond doubt, since asthma is a disease that is for the most part acquired over a protracted period of time. The gradual development of disease can be explained, at least in part, by the development of airway remodelling, whether at the tissue or molecular level. In some instances, asthma may come on quite quickly following an airway insult, perhaps best illustrated by irritant-induced asthma. Certain characteristic changes, such as growth of ASM, seem likely to contribute to airways hyperresponsiveness (AHR), but the consequences of other changes, such as subepithelial fibrosis, are not so intuitively obvious. Even the link between growth of ASM and AHR is deduced largely on the basis of modelling studies. One of the major challenges facing the field is to devise strategies that link structure and function directly. For example, it is difficult, if not impossible, to selectively modify ASM mass in animal models of allergic or nonallergic AHR in order to unequivocally define this relationship. Nevertheless, animal models provide an experimental system open to manipulation with which to investigate the link between decreased airway function and structural changes to the airways. In particular, murine models, in which gene expression can be manipulated at the genomic level, have increased the scope of investigation considerably.
THE PHYSIOLOGICAL CONSEQUENCES OF REMODELLING FOR AIRWAY CALIBRE AND FOR AIRWAY HYPERRESPONSIVENESS
The consequences of some of the structural changes in remodelled airways are consistent with a decrease in airway function. Goblet cell metaplasia and glandular enlargement can explain the excess mucus associated with asthma. However, mucus is only one of the components of the “mucous plugs” in the airways, others being extravasated protein and inflammatory cells 9. The obstruction of the airways by this protein-rich fluid is compounded by the disruption of surfactant function 10, which also contributes to the development of airway closure. These effects, which may be limited to the very small airways in human subjects, are likely to be more widespread in small animals, such as the mouse, used to model asthma 11. Indeed, in the mouse, allergen-induced AHR can be explained by airway closure and a modest increase in epithelial thickness 12, without the need to invoke more complex mechanisms. Interestingly, the published changes in AHR following allergen challenge in the mouse exceed the magnitude of the changes reported in human subjects or in animals larger than the mouse, suggesting that airway instability may be of particular importance in the mouse.
On purely geometric grounds, the increase in thickness of the airways on the luminal side of the ASM will enhance airway narrowing for any given degree of smooth muscle contraction 13. The added tissue becomes a space-occupying factor that reduces luminal area. The thickened airway also results in a greater outer wall or adventitial area. Since the alveolar attachments constitute a part of the load on the ASM, changes in the coupling or the surface area over which the connections are applied will affect the pressure or load per unit area against which the activated ASM contracts. An increase in ASM mass is an intuitively obvious cause of AHR, and modelling studies suggest that the observed change in mass can account entirely for AHR in asthma 14. Interestingly, an increase in ASM mass in large airways distinguishes fatal from nonfatal asthmatics, whereas there is an overlap in the relationship to ASM mass in small airways 3. Remodelling of the larger airways may therefore have more importance for the severity of disease than the peripheral airway changes that also occur. The capacity to produce substantial narrowing of large airways in a synchronous manner may be an important part of severe asthma attacks. Conversely, given the very large number of small airways, synchronous closure in sufficient numbers to cause life-threatening airway narrowing seems less probable. It is of interest that, in an early study of allergen-induced AHR and remodelling in the rat, the change in airway responsiveness to methacholine was correlated with changes in ASM mass, specifically in the large airways 15. Subsequent reports have also demonstrated a correlation between ASM remodelling and airway responsiveness, although a definite cause-and-effect relationship has not been established 16, 17.
An increase in matrix protein deposition, particularly collagen, could also have an important effect on airway responsiveness, not only because of its space-occupying nature but also because of its effects on the mechanical properties of the airway wall. There is now abundant evidence that the forces exerted by the parenchyma on the airway wall during tidal breathing are a potent bronchodilating influence 18. However, stiffening of the airway wall caused by remodelling is predicted to result in a reduction of the stress exerted on the smooth muscle, since the cyclical stretching of the airway tissues would be less in asthmatic subjects. Although a reduced distensibility of airway walls has been reported for human asthmatic airways, it is not clear whether comparable changes in the airway properties occur in small animal models. A contrary point of view has been posited concerning possible beneficial effects of subepithelial collagen deposition 19. It has been argued that buckling of the epithelium that occurs during bronchoconstriction may be resisted to a greater extent in airways in which collagen deposition has occurred. Overall, it is likely that different factors will be important in causing airway obstruction and AHR in differently sized airways and in different species.
Two recent studies in animal models have addressed the effects of treatment on the reversal of airway remodelling. Data from Cho et al. 20 demonstrated that corticosteroid administration combined with allergen avoidance was required to reduce airway fibrosis and increased smooth muscle mass, even though allergen avoidance alone promoted resolution of inflammation and reduced mucus gene expression. In another study, administration of the cysteinyl leukotriene 1 (CysLT1) receptor antagonist, montelukast, reduced allergen-induced increases in ASM mass and subepithelial collagen deposition. Corticosteroids alone were without effect and did not further reduce airway remodelling when given in combination with montelukast 21. These inconsistencies regarding corticosteroids are likely to reflect differences in animal protocols. Resolution of this issue will require further investigation.
A recent novel approach to treatment of asthma through the destruction of airway smooth muscle has been reported. The technique called bronchial thermoplasty is similar to current ablation procedures popular in cardiology for the destruction of aberrantly conducting myocardial tissues. Bronchial thermoplasty employs radiofrequency waves to heat the airway tissues via a catheter placed within the airway during bronchoscopy. Experiments on dogs have demonstrated an apparently selective destruction of ASM 22. To date, results in humans have been promising 23 and support the notion of ASM as an important cause of airway hyperresponsiveness in asthma.
Finally, an unresolved issue in asthma is the mechanism of permanent loss of lung function with apparently irreversible airflow limitation. It is often attributed to remodelling, although formal proof is lacking. It is possible that fibrosis of the airways with contraction of the matrix results in loss of airway calibre. Refractory airway narrowing attributable to irreversible smooth muscle contraction has not been excluded, although it seems less plausible. To date, there is a dearth of information addressing this issue.
INFLUENCE OF INFLAMMATION AND INFECTION ON REMODELLING
The precise relationship between the various triggers of asthma and remodelling is poorly understood, although allergen exposure of sensitised subjects is the stimulus that is most strongly implicated in remodelling. Data from experimental asthma in several species have confirmed the propensity for allergen challenge and the subsequent inflammatory response, to trigger airway remodelling 16, 24, 25. Allergic inflammation is a complex process involving activation of T-cells, principally through antigen presentation by dendritic cells. Upon activation, T-cells in asthma produce a T-helper cell (Th) type 2 profile of cytokines that are thought to orchestrate recruitment and activation of other inflammatory cells, including eosinophils and mast cells that also participate in inflammation and remodelling in asthma.
Amplification of the inflammatory response, including the T-cell component to that response, may also occur by nonantigen-specific pathways. For example, neurokinins, eicosanoids or other mediators may induce expression of cytokines and other inflammatory mediators from a variety of cell types. In addition, structural cells of the airways are now thought to play an active role in the induction and maintenance of airway inflammatory responses in asthma. Abundant data demonstrate that cultured epithelial cells, fibroblasts and ASM cells produce cytokines, chemokines and other mediators that promote inflammatory responses 26–28. Fewer in vivo studies have addressed the extent to which structural cells participate in airway inflammation and remodelling. Data from human biopsies indicate that airway epithelial, endothelial and ASM cells produce eotaxin and several cytokines implicated in airway remodelling 6, 29–31. Mathew et al. 32 have also demonstrated that allergen-induced airway inflammation in experimental asthma is dependent upon signal transducer and activator of transcription (STAT)-6 signalling and chemokine production by resident lung cells. The present authors (E.D. Fixman and J.G. Martin) have demonstrated that T-cells can be localised to the ASM layer in experimental asthma in the Brown Norway (BN) rat 25, though only the mast cell has been found in ASM bundles, in addition to the lamina propria and epithelial layer in human asthmatics 33–35. The factors that regulate the recruitment and/or maintenance of T-cells and mast cells in these tissue compartments are poorly understood.
A role for other airway inflammatory cells, including macrophages and neutrophils, in airway remodelling has not been ruled out. Macrophages have been shown to regulate inflammation and AHR in animal models of asthma 36, 37, and have been implicated in airway remodelling in murine models of asthma induced by fungal exposure 38. Though large numbers of neutrophils are present in the airways of a subset of severe asthmatics 39, it is not clear if they contribute to airway remodelling. Circumstantial support for a contribution of neutrophils to airway remodelling is provided by data from heaves in horses. Upon exposure to mouldy hay, these horses develop Th2-biased airway inflammation 40, which is accompanied by airway remodelling, particularly increases in ASM 41. Though modest numbers of eosinophils are present, the airways of these animals contain large numbers of neutrophils 40.
Recent studies have begun to clarify the role of the different cellular components of the inflammatory response in airway remodelling. In asthmatics, eosinophil-derived transforming growth factor (TGF)-β may regulate activation of fibroblasts and matrix deposition 42, as well as deposition of procollagen III and the proteoglycans, tenasin and lumican, beneath the epithelial basement membrane 43. Similarly, eosinophils, and possibly eosinophil-derived TGF-β, have been implicated in increases in collagen content and ASM remodelling in experimental asthma 44, 45. Moreover, in addition to eosinophils, other inflammatory cells may also alter ASM behaviour. For example, activated T-cells can be localised in apparent contact with ASM in vivo in rats 25 and direct contact with activated CD4+ T-cells induces ASM cell proliferation in vitro 25, 46. While T-cell–ASM cell contact is required for induction of ASM cell proliferation in vitro, ASM remodelling in vivo may also be regulated directly via T-cell cytokines and/or indirectly by factors induced by T-cells, such as endothelins and/or CysLTs 17, 47, 48. The extent to which T-cells contact ASM in vivo in human asthmatics remains uncertain and, therefore, the magnitude of the biological effect of this interaction requires further exploration. In addition to T-cells, mast cells have also been localised to ASM fibres in humans 34, although the outcome of mast cell interactions with ASM is not well understood. Mast cells express interleukin (IL)-4 and -13 in this location 35, and these cytokines, as well as lipid mediators such as CysLTs, may regulate ASM responses that contribute to airway remodelling and asthma pathogenesis. The regulation of airway remodelling by individual mediators is presented below.
The role of other triggers of airway remodelling, such as bacterial or viral infections, is less well explored. Recent data on human asthmatic subjects suffering from severe, late-onset, nonatopic asthma have suggested that infection with Chlamydia pneumoniae might contribute to the development of fixed airway obstruction 49. Subjects with elevated IgG antibodies for C. pneumoniae showed a more rapid decline in lung function, as estimated from the postbronchodilator forced expiratory volume in one second/forced vital capacity, a possible reflection of permanent loss of lung function. In addition, the effects of Mycoplasma infection on collagen deposition, alone and in combination with allergen challenge, have been studied in a murine model. Infection per se does not alter collagen deposition in the airway wall but, in conjunction with allergen challenge, increases the airway collagen at 6 weeks after challenge. The increase in lung collagen content is accompanied by an increase in TGF-β1 in the airway wall 50. Further studies of this sort are required to model the interactions of usual triggers of asthma with infection.
Other triggers, such as exercise or cold air hyperventilation challenge, are associated with the release of neurokinins and CysLTs 51, 52, both potential mitogens 53, 54. CysLTs have been implicated in airway remodelling in both murine and rat models of allergic asthma 17, 21, 48. However, whether repeated exercise challenge is a sufficient stimulus to cause airway remodelling or whether additional mediators present following allergic reactions are required, and not present after exercise challenge, is not clear. The high prevalence of AHR in athletes engaged in intense aerobic exercise suggests that repeated airway challenge resulting from exercise hyperpnoea may not be entirely benign 55. Whether this has anything to do with airway remodelling is unknown, but there is certainly evidence of airway remodelling of the asthmatic type present in elite cross-country skiers 56. Other intense airway challenges resulting from exposure to highly irritant gases, such as ozone, sulphur dioxide and chlorine, can cause substantial airway remodelling, including epithelial change and ASM growth in experimental asthma 57, 58. It is likely that oxidant injury is involved in all of the above mentioned stimuli, but the precise mechanisms await elucidation.
The notion of the epithelium as the tissue that drives many of the processes leading to asthma is not a new one 59. However, increasing understanding of the range of pro-inflammatory mediators and growth factors secreted by the epithelium has given the concept new momentum. Holgate et al. 60 have proposed an intimate relationship between epithelial damage and subepithelial remodelling processes. Moreover, they have proposed that remodelling in response to epithelial injury may precede significant Th2-related airway inflammation. Indeed, epithelial activation could occur through a variety of nonallergic mechanisms. The magnitude of the pro-inflammatory response of the epithelium, its production of factors promoting remodelling and its capacity to deal with insults such as oxidants, may affect the outcome 61.
TISSUE-SPECIFIC MECHANISMS DEMONSTRATED BY IN VITRO AND IN VIVO MODELS
Airway smooth muscle
Abundant data indicate that ASM mass increases in both human asthmatics as well as in animals with experimental asthma. In addition to an increase in the size and/or number of ASM cells, enhanced deposition of ECM in the interstitial space between muscle fibres may also contribute to the overall increase in ASM mass 62. Recent studies utilising bronchoscopic airway biopsies from asthmatic subjects have demonstrated an increase in muscle in the biopsies 5, 6. In addition, the inner border of muscle bundles is closer to the epithelium than normal, suggesting that new fibres have been added to the luminal aspect of the muscle. The number of nuclei seems to increase in proportion to the mass of muscle, suggesting that hyperplasia is responsible for the added muscle 63. There are also numerous reports of hyperplastic increase in ASM mass in animals. Using bromodeoxyuridine incorporation or proliferating cell nuclear antigen immunoreactivity, allergen-induced proliferation of ASM cells has been demonstrated 25, 64, 65. In the rat, as few as three allergen challenges over 2 weeks are sufficient to cause muscle to double in mass, suggesting that ASM has the ability to change in structure at a surprisingly rapid rate in response to external inflammatory stimuli 15, 25. In addition to rodents, an increase in ASM mass and evidence of hyperplasia have also been demonstrated in the heaves-affected horse 41 , suggesting that in this large animal with an asthma-like disease, the ASM is also plastic, undergoing quite dramatic structural changes in the presence of inflammation. To date, no studies have managed to capture active ASM proliferation in human asthmatic tissues (see 5, for example). However, the absence of proliferating cells does not prove that such a process does not exist. The adequate sampling of tissues and the relationship of the biopsy material to periods of acute asthma need to be considered.
Recently, information regarding the role of specific cytokines in remodelling of the ASM in experimental asthma has been obtained, primarily following repeated allergen challenge of mice lacking specific cytokines 66, 67. Interestingly, allergen-induced increases in ASM mass are either absent or attenuated in mice lacking IL-4, -5 or -13 66, 67. Abundant data support a role for TGF-β in ASM remodelling in asthma 43, 64. Studies carried out in transgenic mice overexpressing IL-11 in the airways have provided evidence that IL-11 may also participate in ASM remodelling 68. Though few studies addressing the role of other biochemical mediators in ASM remodelling have been performed, several studies have demonstrated that CystLTs are involved in allergen-driven ASM growth in the rat and mouse. Morphometric data indicate that hyperplasia of ASM in the ovalbumin-sensitised and -challenged rat can be prevented by CysLT1 receptor antagonists and 5-lipoxygenase inhibitors 17, 48. Administration of the CysLT1 receptor antagonist, montelukast, after repeated allergen challenge is sufficient to abolish the increase in ASM mass induced by repeated allergen challenge in mice 21. Endothelins have also been implicated in allergen-induced ASM hyperplasia in the rat 47. Nevertheless, endothelins and CysLTs are only weakly mitogenic in cell culture systems, suggesting that they may cooperate with other growth factors or cytokines to induce ASM growth responses 69, 70. IL-13 upregulates the CysLTR1 on cultured human ASM cells and augments mitogenesis induced by CysLTs in vitro, suggesting at least one possible mechanism via which Th2 cytokines may contribute to remodelling by a “priming” effect 71. In addition to an increase in ASM cell number, the increase in ASM mass in remodelled airways may result from hypertrophy of ASM cells 63, 72. A number of mediators, including TGF-β and endothelin-1, induce hypertrophy of ASM cells in culture 73–77. However, the extent to which ASM cell hypertrophy induced by these mediators participates in ASM remodelling in vivo is unclear.
The hyperplasia of smooth muscle in asthma may also be a consequence of recruitment of cells from the pool of subepithelial (myo)fibroblasts and circulating fibrocytes with the capacity to differentiate into cells indistinguishable from ASM cells 78. In this scenario, migration of mesenchymal cells from the circulation and the subepithelial space into the muscle bundles is required, and thus chemoattractants, such as platelet-derived growth factor and CysLTs, may also be important. Understanding mechanisms by which mesenchymal cells differentiate toward smooth muscle cells may also provide new targets for specific intervention in the remodelling process 79–81.
One interesting aspect of smooth muscle biology is the fact that smooth muscle cells in culture decrease expression of contractile proteins 82, 83. Moreover, smooth muscle cell phenotype may also regulate the ability of cells to proliferate in response to mitogen stimulation 84. Similarly, in vivo proliferation may be associated with “dedifferentiation” to a synthetic phenotype and loss of contractile protein expression 85. Whether the increase in ASM mass in asthmatic airways is associated with an altered phenotype is not clear. This could be a temporary phenomenon and the contractile phenotype might be restored following proliferation. However, ASM cells cultured from asthmatic biopsies retain the ability to proliferate more rapidly compared with those from nonasthmatic controls, providing evidence that changes in asthmatic ASM cell phenotype may be stable 86. Inflammation-induced alterations in ECM deposition may also regulate ASM cell phenotype and remodelling in asthma. ECM-dependent activation of integrins and other adhesion molecules regulates the phenotype of smooth muscle cells in vitro and during development, and thus it seems likely that similar regulation may occur in vivo in asthmatics 84, 87, 88, although the possible mechanisms by which changes in ECM regulate ASM (dys)function in asthma have not been explored.
Epithelium
It is not surprising that the epithelium is altered in asthma as it is subject to the initial insult of the triggers of asthma, for example, reactions to aeroallergens or respiratory viral infections. The relatively rapid turnover of the epithelium is another feature that may predispose to phenotypic changes in response to inflammation. Asthmatic airway epithelium may participate in airway remodelling in asthma through the production of mediators that induce remodelling of other airway tissues. For example, epithelial cell-specific production of growth factors, such as endothelins, is likely to induce fibrosis and ASM remodelling. Moreover, goblet cell differentiation is one of the more obvious features noted on light microscopy of asthmatic airways and mucus glands undergo hypertrophy and hyperplasia in more severe cases of asthma. Much of the in vivo work on mechanisms of mucus cell metaplasia has been carried out on murine models. In addition, in vitro data demonstrate that a variety of asthma-related cytokines induce expression of mucus genes, principally mucin (MUC)2 and MUC5AC, in cultured epithelial cells. The Th2 cytokines, IL-4, -5, -9 and -13, have all been implicated in MUC gene expression 89. IL-4 and -9 induce MUC2 and/or MUC5AC gene expression and mucus production in cultured human airway epithelial cells 90, 91. Similarly, transgenic mice expressing IL-4 or -9 in the lung exhibit increased expression of MUC5AC (and MUC2 for IL-9) as well as increased levels of mucus glycoconjugates in the epithelium 90–92. Moreover, IL-4 transgenic mice have hypertrophic, nonciliated cells in their conducting airways, due, in part, to the accumulation of mucus glycoprotein; lavage fluid from these mice also contains an increase in mucus-positive staining material. While IL-4 and/or -9 may directly regulate mucus production in some circumstances, evidence suggests that in vivo they do so indirectly via IL-13 93. Consistent with a role for IL-4 and/or -13 in mucus production, each of these cytokines induces activation of the Src homology 2 domain containing transcription factor STAT-6, and experiments using gene-deleted mice demonstrate that STAT-6 also regulates many features of acute asthma, including mucus overproduction. In fact, IL-13 transgenic mice expressing STAT-6 only in the epithelium exhibit IL-13-dependent mucus production 94. Finally, both IL-1β and tumour necrosis factor-α have recently been shown to induce MUC gene expression 95, though it is not clear if IL-13 also regulates these responses. In addition to these cytokines, neutrophil elastase regulates mucus production in vivo 96 and enhances MUC5AC mRNA stability in vitro, possibly in a reactive oxygen species-dependent manner 97. In addition, activation of receptors for epidermal growth factor or vascular endothelial growth factor (VEGF) has also been linked to MUC gene expression in asthma 98, 99.
Extracellular matrix
Abundant data demonstrate that ECM changes occur in human asthmatics as well as in murine and rat models of allergic asthma 100. In the ovalbumin-sensitised and -challenged BN rat, the amount of collagen is increased in large airways in both the inner and outer layers of the airway wall. Fibronectin is also increased in airways of rats and mice repeatedly exposed to ovalbumin 100, 101 and corticosteroids administered at the time of ovalbumin exposure effectively inhibit these structural changes 101, 102. Similarly, proteoglycan and/or collagen deposition is increased in the airway walls of mice chronically exposed to ovalbumin 16, 21, 103. While an increase in ECM deposition is consistently seen in rodent models of allergic asthma, strain-related differences may also exist. For example, airway wall collagen deposition is increased in A/J but not C57BL/6 and C3H/HeJ mice following repeated allergen challenge 104.
There are multiple sources of evidence that Th2 cytokines mediate ECM deposition in airway remodelling. Murine models of asthma derived by upregulation of, or exposure to, specific cytokines, e.g. IL-5 or TGF-β, have shown evidence of increased collagen deposition and airway fibrosis 105. Similarly, allergen-induced collagen deposition is decreased in the airways of both IL-5 gene-deleted mice and eosinophil-deficient mice 45, 66 and ECM deposition is decreased in mice treated with an inhibitory IL-13 antibody at the time of allergen challenge 106 or with an inhibitory TGF-β antibody administered following induction of ovalbumin-induced inflammation 64. A role of eosinophil-derived TGF-β in ECM deposition in asthmatics is also supported by data from Flood-Page et al. 43, who demonstrated that treatment of asthmatics with humanised anti-IL-5 antibodies decreased not only ECM deposition, but also the number of TGF-β-expressing eosinophils and the concentration of TGF-β in the bronchoalveolar lavage fluid. A role for the profibrotic cytokines, IL-11 and -17, in airway remodelling has also been proposed 107. Finally, a variety of genes, whose products are likely to regulate airway fibrosis in asthma, were shown to be upregulated in mice 2 weeks after the final of a series of allergen challenges. These include TGF-β, plasminogen activator inhibitor-1, tissue inhibitor of metalloproteinase-1, connective tissue growth factor, α-smooth muscle actin, fibronectin and procollagen 1α2 108.
Interestingly, mice deficient in the transcription factor, T-bet, which regulates expression of the Th1 cytokine interferon (IFN)-γ and promotes Th1 type responses, spontaneously develop an asthmatic phenotype including AHR, as well as abundant deposition of collagen III below the basement membrane and an increase in α-actin-positive ASM cells and/or myofibroblasts. The lungs of these mice contain activated memory CD4+ T-cells that express high levels of Th2 cytokines (IL-4, -5 and -13), as well as TGF-β 109. It is not surprising that T-bet deficiency should have profound consequences for airway function, since IFN-γ is such an effective counter-regulatory molecule in the setting of allergic inflammation.
Angiogenesis
An increase in bronchial vessels in asthmatic subjects has been reported in numerous papers. As with the majority of the changes constituting remodelling, there is no definitive answer to the question of the causative mediators. Expression of VEGF, an obvious target of interest, is increased in asthma and co-localises with markers for macrophages, eosinophils and CD34+ cells, and mast cells 110, 111. In humans, the presence of VEGF-immunoreactive cells correlates with the number of blood vessels and to basement-membrane thickness 112. ASM cells in culture also release VEGF in response to treatment with the pro-inflammatory cytokines, TNF-α and IL-1β 113, or the Th2 cytokines, IL-4 and -13 114. In murine models of experimental asthma, VEGF levels are increased and VEGF receptor inhibitors reduce inflammation and AHR 99, 115. Transgenic mice in which VEGF is inducibly expressed in epithelial cells exhibit lung-specific increases in angiogenesis and oedema, as well as increased Th2-specific inflammation and mucus production 99. Prolonged VEGF expression in these mice results in abundant TGF-β expression accompanied by increased collagen deposition, as well as myocyte hyperplasia and smooth muscle remodelling. While mucus production is dependent upon IL-13 production in these mice, all other phenotypic changes are IL-13 independent. Interestingly, these mice are also more sensitive to allergen sensitisation than their wildtype counterparts and have exaggerated allergen-induced AHR and inflammatory responses.
SIGNALLING PATHWAYS THAT REGULATE CELL PROLIFERATION IN AIRWAY REMODELLING
As previously discussed, a variety of stimuli has been implicated in the proliferative responses that are likely to contribute to airway remodelling in asthma. These include agonists that activate receptor tyrosine kinases and G-protein-coupled receptors, as well as cytokines, reactive oxygen species, ECM components and mechanical forces, such as stretch and compression. Signalling pathways that regulate proliferative responses induced by many of these agonists in other cell types, including the mitogen-activating protein kinases (MAPK), extracellular signal-regulated kinases (ERK)1, ERK2 and p38MAPK, as well as Ras, phosphoinositide 3-kinases and Src also regulate in vitro proliferation of cells that contribute to airway remodelling in asthma 116–120. While there may be airway cell-specific signalling pathways that regulate proliferation, it is likely that extensive overlap with other systems exists. The propensity for cells to proliferate and contribute to airway remodelling is almost certainly regulated, at least in part, by inflammation-dependent phenotypic alterations of these airway cells 85, 121.
Recent data from animal studies has implicated other signalling molecules in airway remodelling 64, 122, 123. The role of the nuclear factor (NF)-κB transcription factor in airway remodelling was assessed by generating gene-modified mice in which the gene encoding the inhibitor of κB (IκB) kinase β (IKKβ) was selectively deleted in airway epithelial cells 122. IKKβ regulates NF-κB activity and, in its absence, NF-κB cannot be activated. Nuclear translocation of the RelA subunit of NF-κB, induced by repeated ovalbumin challenge, is selectively lost in airway epithelial cells of IKKβ-ablated mice. Pathologically, compared with wildtype controls, these mice exhibit decreased ovalbumin-induced airway wall thickening and total lung collagen content. Moreover, ovalbumin-induced increases in mucus and airway eosinophils, and peribronchial CD4+ cells are also reduced. Decreased inflammation and remodelling in these mice are accompanied by lower levels of the chemokines eotaxin-1 and thymus- and activation-regulated chemokine, as well as TGF-β1. Together, these data suggest that epithelial cell-specific NF-κB participates indirectly in airway remodelling by regulating inflammatory responses that contribute to mucus production, fibrosis and airway wall thickening in experimental asthma.
A role for c-Jun N-terminal kinase (JNK), a member of the MAPK superfamily of serine/threonine kinases, in airway remodelling was assessed by treating mice with a JNK-specific inhibitor, SP600125, 2 h prior and 8 h following each of 15 ovalbumin challenges over the course of 3 weeks. Compared with control mice, SP600125-treated mice exhibited decreases in mucus levels and numbers of ASM cells 124. Overall inflammation and eosinophilia were inhibited, as was AHR, indicating a broad anti-inflammatory effect. In another study 123, allergic mice were treated with 32-deoxorapamycin (SAR943), an analogue of the anti-inflammatory macrolide, rapamycin. Rapamycin is a potent inhibitor of cell proliferation and targets the mammalian target of rapamycin (mTOR) protein and inhibits activation of p70S6K. In allergic mice treated with SAR943, inflammation and eosinophil numbers were diminished, as were mucus production and AHR 123. Markers of remodelling, bronchoalveolar lavage fluid fibronectin levels and airway epithelial cell proliferation were both inhibited in mice treated with SAR943. Finally, a role for Smad proteins, which regulate TGF-β-induced responses, in airway remodelling was recently proposed 64. Levels of phosphoSmad2, which promotes TGF-β-induced responses, are elevated in airways of mice repeatedly challenged with ovalbumin. Treatment of mice with TGF-β inhibitory antibodies diminished both airway remodelling and phosphoSmad2 levels, and caused an increase in levels of Smad7, an inhibitor of TGF-β-induced responses. These data suggest that targeting signalling molecules may be beneficial for the development of novel therapeutic strategies to inhibit airway remodelling in asthma. Nevertheless, ongoing efforts are required to ascertain whether there are dominant cytokine, growth factors and/or signalling molecules that control sufficiently key aspects of the remodelling features to render them viable as targets for intervention.
CONCLUDING REMARKS
The mechanisms by which remodelling occurs are gradually being elucidated (fig. 1⇓), although there are major gaps in present understanding of the cellular and molecular basis of many of the changes. What is clearly emerging is that remodelling is induced by factors synthesised and secreted both by inflammatory cells and by structural cells, the latter frequently under the influence of the former. However, the precise factors responsible for cellular hyperplasia, hypertrophy and altered matrix deposition are far from resolved. Elucidation of these factors will no doubt lead to novel therapies designed to prevent or reverse these changes, and have the greatest potential for curative pharmacological intervention.

{kind=link}
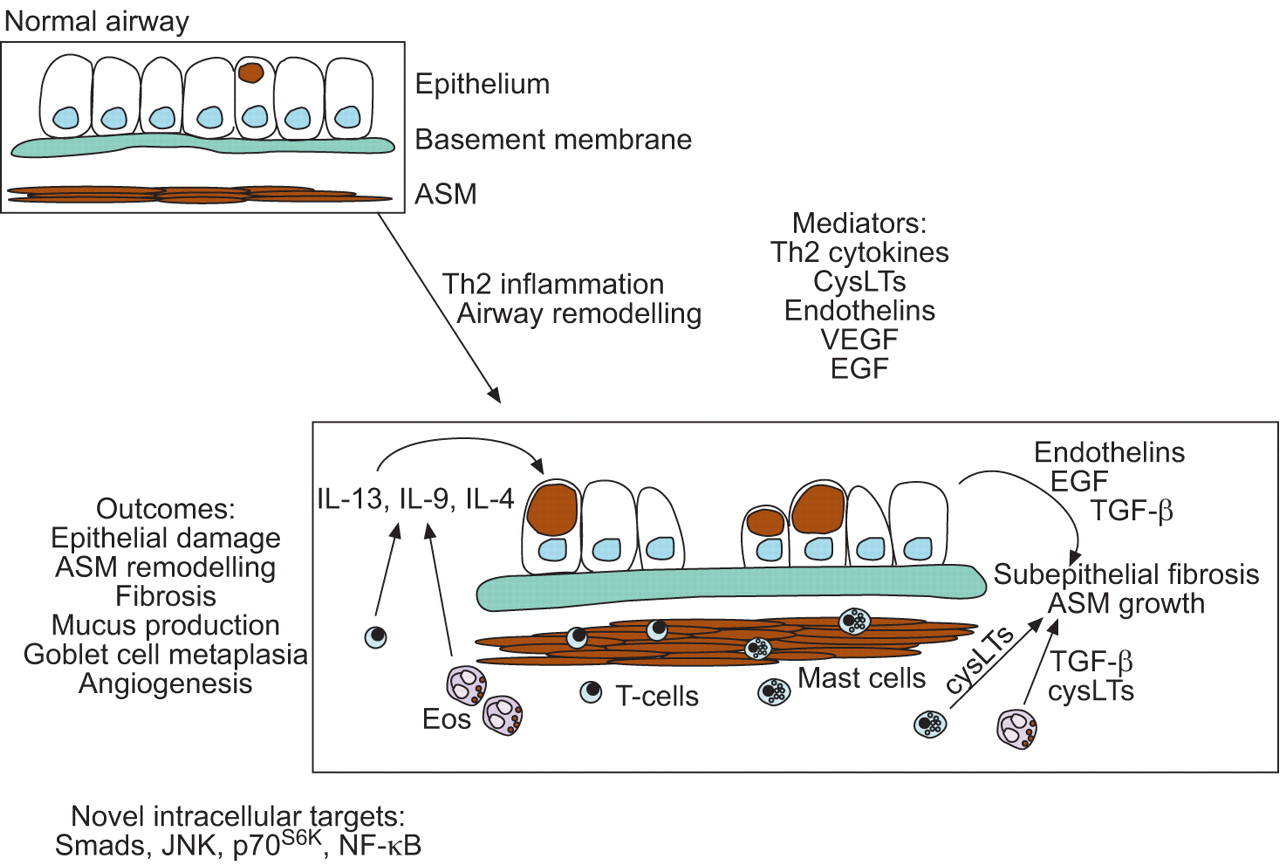
Mediators and outcomes of T-helper cell (Th)2-mediated inflammation and airway remodelling in asthma. Remodelling changes include damaged epithelium and an increase in production of mucus, as well as several mediators with the potential to induce airway remodelling, including endothelins, transforming growth factor (TGF)-β, and epidermal growth factor (EGF). Mediators that regulate airway remodelling, such as cysteinyl leukotrienes (CysLTs) and interleukin (IL)-13, are also produced by a variety of inflammatory cells present in asthmatic airways. In addition to epithelial changes, structural changes in the airways include an increase in collagen and proteoglycan deposition that contribute to fibrotic changes of the airways. Bronchial blood vessels increase in number and size and airway smooth muscle increases in mass. The inner border of smooth muscle bundles is also localised in closer proximity to the epithelium. Recent data from animal models have implicated transcription factors and signalling molecules in airway remodelling. These include Smads, c-Jun N-terminal kinase (JNK), p70S6K and nuclear factor (NF)-κB. ASM: airway smooth muscle; VEGF: vascular endothelial growth factor; Eos: eosinophil.
Footnotes
-
For editorial comments see page 231.
- Received April 18, 2006.
- Accepted November 13, 2006.
- © ERS Journals Ltd
References











Jump To
- Article
- Abstract
- THE PHYSIOLOGICAL CONSEQUENCES OF REMODELLING FOR AIRWAY CALIBRE AND FOR AIRWAY HYPERRESPONSIVENESS
- INFLUENCE OF INFLAMMATION AND INFECTION ON REMODELLING
- TISSUE-SPECIFIC MECHANISMS DEMONSTRATED BY IN VITRO AND IN VIVO MODELS
- SIGNALLING PATHWAYS THAT REGULATE CELL PROLIFERATION IN AIRWAY REMODELLING
- CONCLUDING REMARKS
- Footnotes
- References
- Figures & Data
- Info & Metrics