





Abstract
Idiopathic pulmonary fibrosis (IPF) is thought to result from aberrant tissue repair processes in response to chronic or repetitive lung injury. The origin and nature of the injury, as well as its cellular and molecular targets, are likely heterogeneous, which complicates accurate pre-clinical modelling of the disease and makes therapeutic targeting a challenge. Efforts are underway to identify central pathways in fibrogenesis which may allow targeting of aberrant repair processes regardless of the initial injury stimulus. Dysregulated endothelial permeability and vascular leak have long been studied for their role in acute lung injury and repair. Evidence that these processes are of importance to the pathogenesis of fibrotic lung disease is growing. Endothelial permeability is increased in non-fibrosing lung diseases, but it resolves in a self-limited fashion in conditions such as bacterial pneumonia and acute respiratory distress syndrome. In progressive fibrosing diseases such as IPF, permeability appears to persist, however, and may also predict mortality. In this hypothesis-generating review, we summarise available data on the role of endothelial permeability in IPF and focus on the deleterious consequences of sustained endothelial hyperpermeability in response to and during pulmonary inflammation and fibrosis. We propose that persistent permeability and vascular leak in the lung have the potential to establish and amplify the pro-fibrotic environment. Therapeutic interventions aimed at recognising and “plugging” the leak may therefore be of significant benefit for preventing the transition from lung injury to fibrosis and should be areas for future research.
Abstract
Vascular hyperpermeability in the fibrotic lung actively contributes to disease progression https://bit.ly/39HDEep
Introduction
Over the past few decades, insights into the mechanisms of endothelial cell biology have highlighted the role of the vasculature as an active participant in tissue repair and fibrosis. In 1989, Brown et al. [1] were the first to review the consequences of vascular permeability in the context of lung injury and lung fibrosis. Although primarily focussed on abnormal angiogenesis during tumour stroma formation, the authors note that lung injury and fibrosis are characterised by a hyperpermeable vasculature and extravascular coagulation.
However, since these initial discoveries, the role of vascular permeability in the fibrotic lung has not received as much attention as the injury sustained by the alveolar epithelium or the activation of the myofibroblast. In this hypothesis-generating review, we summarise the available imaging evidence demonstrating abnormal permeability in the lungs of patients with idiopathic pulmonary fibrosis (IPF) and then highlight data suggesting that persistent endothelial cell activation and hyperpermeability are not simply consequences of pulmonary fibrosis, but central contributors to its progression. We explore the molecular pathways regulating barrier function as well as the downstream consequences of increased permeability, and their roles in the development of pulmonary fibrosis. Finally, we discuss the therapeutic implications of these data and raise unanswered questions in the field and future directions for research in this area. Establishing a mechanistic link between vascular leak and disease progression and severity may elucidate future therapeutic targets for patients with pulmonary fibrosis. Indeed, if vascular leak predicts disease progression, therapeutic strategies targeted at limiting vascular permeability may be effective in identifying patients at high risk for decompensation, limiting disease progression and may permit earlier resolution of tissue injury and less fibrogenesis.
IPF pathogenesis
The prototypic fibrotic lung disease, IPF, is characterised by irreversible scarring of the lung parenchyma and a progressive decline in lung function. No curative medical therapies exist, and prognosis is generally poor, with the median survival ranging between 3–5 years from the time of diagnosis [2]. Current research suggests that the underlying pathogenesis of IPF is aberrant wound healing and tissue repair in response to repetitive lung injury [3, 4]. Damage to the alveolar epithelium catalyses a cascade of cellular and molecular events that, when appropriate in duration and magnitude, restore normal lung structure and function. However, when one or several of these injury responses occur in a dysregulated or overexuberant manner, persistent extracellular matrix deposition can result in progressive fibrosis and architectural distortion of the lung tissue.
In the search for novel anti-fibrotic therapies, investigative attention has historically been paid to protecting the epithelium from injury and preventing excessive fibroblast activation. These areas of focus have led to exciting advances in the field of fibrosis, and targets of these processes are in various stages of clinical trial development. However, the role of the pulmonary endothelium, in particular loss of barrier integrity and subsequent increase in vascular permeability, has been an underdeveloped area of fibrosis research. In the current paradigm of IPF pathogenesis, fibrosis is the result of multiple pro-fibrotic processes [5], including injury to the epithelium, an influx of immune cells, endothelial injury and dysfunction, and increased alveolar-capillary permeability with leakage of plasma proteins into the airspaces (fig. 1). Injured epithelial and endothelial cells in combination with activated immune cells release mediators and signal to recruit and activate fibroblasts to areas of injury. The activated fibroblasts, or myofibroblasts, lay down collagen and contract the tissue to form the fibrotic scar. Within this framework, endothelial permeability may be key to promoting several profibrotic responses including intra-alveolar coagulation and provisional matrix establishment, contributing to an environment in which an injury stimulus can generate fibrotic scar.
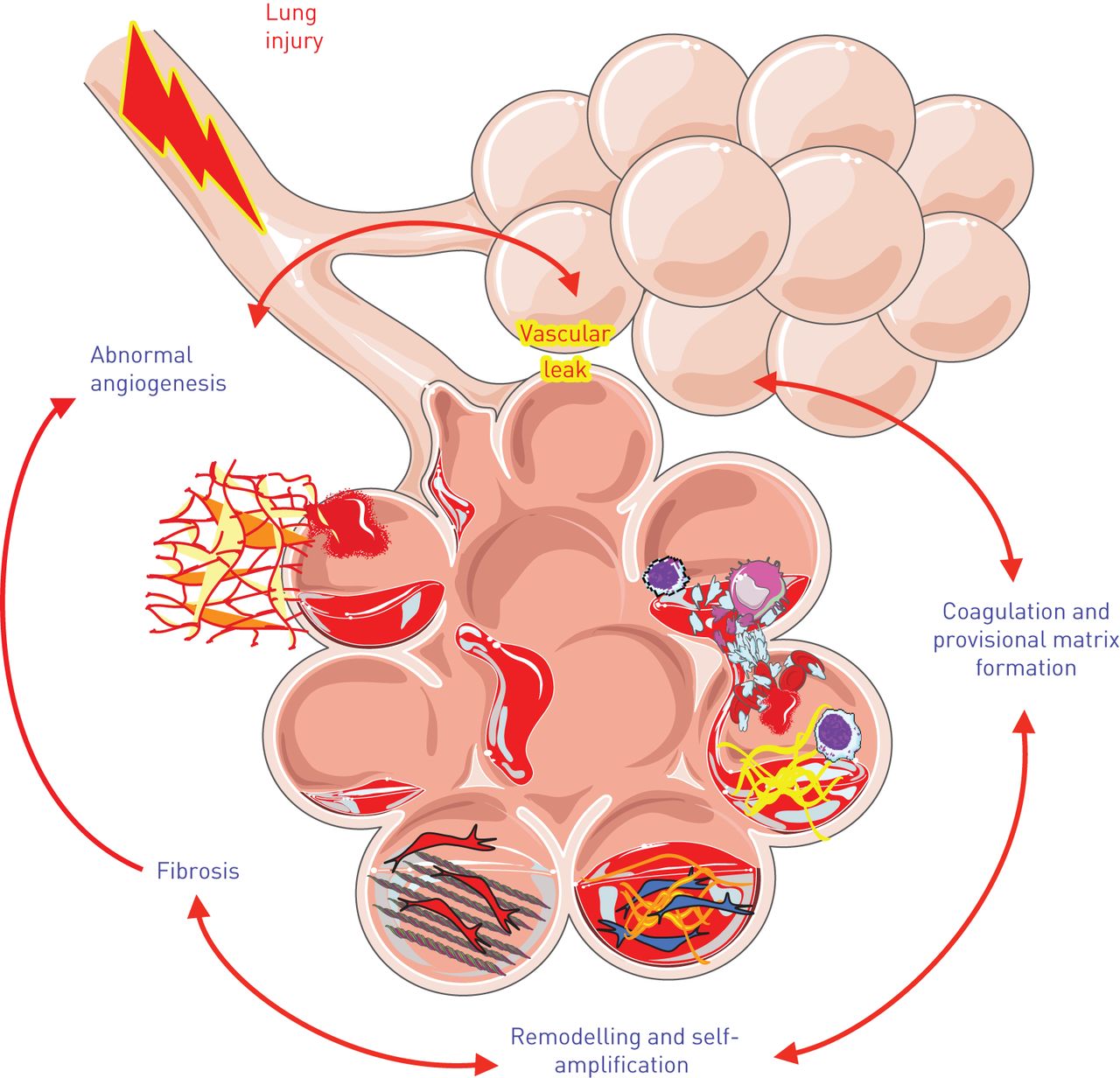
Integration of dysregulated vascular permeability into the paradigm of idiopathic pulmonary fibrosis (IPF) pathogenesis. Fibrosis is believed to develop as an aberrant response to repetitive tissue injury, which appears to primarily target alveolar epithelial cells (AECs) and the adjacent endothelium. Upon AEC injury, the adjacent endothelium becomes activated in response to inflammatory stimuli and increases its permeability as part of the normal repair response. With ongoing injury and increased vascular leak, abnormal angiogenesis is seen in addition to extravascular coagulation, provisional matrix deposition and vascular and airway remodelling as part of the fibrotic process.
The endothelial barrier in the lung
The alveolar–capillary interface consists of a microvascular bed that surrounds the alveolus and shares approximately 86 percent of its surface area [6]. Endothelial and epithelial cells with their fused basal lamina form a delicate layer, ensuring rapid and effective gas exchange while simultaneously providing a robust barrier against inhaled pathogens. Upon inflammatory stimulation, endothelial barrier function is dynamically altered to allow for transmigration of leukocytes and extravasation of pro-repair plasma proteins to the site of injury [7, 8]. This response usually results in tissue repair and restoration of homeostasis; however, in the case of ongoing injury, permeability may persist.
In homeostasis, endothelial cell barrier function is tightly regulated by cytoskeletal elements that allow for the formation of a confluent monolayer on the interior of the vascular lumen. On the cellular level, this is characterised by a thick cortical actin ring, which maintains a low level of intercellular junctional tension, thereby strengthening the endothelial barrier. To cross this barrier, molecules use either the transcellular or the paracellular route. Transcellular trafficking is accomplished via vesicular carriers (i.e. caveolae) or pore-like structures such as fenestrae and transendothelial channels. Tight junctions (TJ) and adherens junctions (AJ) maintain the connections between neighbouring cells and regulate the paracellular passage of plasma proteins and solutes. In addition to intercellular interactions, endothelial cells are tethered to the extra-cellular matrix (ECM) via interactions between cell surface integrins and their ECM ligands, which are organised in focal adhesion plaques [9].
Upon injurious stimulation, the endothelial monolayer is disrupted and paracellular gap formation is observed [10]. This response is mediated by dynamic actin reorganisation processes that allow for the transition from the quiescent to the activated cell phenotype. Actomyosin contraction and increased cytoskeletal tension lead to the formation of actin stress fibres that cause the disassembly of intercellular junctions, allowing the cell to contract. The subsequent creation of paracellular gaps disrupts the continuous monolayer and thereby increases its permeability. This is usually followed by tissue repair and restoration of homeostasis.
Abnormal permeability in patients with pulmonary fibrosis
Prior data suggest that alveolar–capillary permeability is abnormally increased in patients with pulmonary fibrosis and that the degree of increased permeability associated with outcomes [11–13]. Perturbations in alveolar–capillary permeability have previously been evaluated by measuring clearance of the inhaled radio-labelled molecule, technetium-99m diethylenetriamine pentaacetate (99mTc-DTPA). Clearance rates of 99mTc-DTPA have been demonstrated to be shorter in IPF and systemic sclerosis-associated interstitial lung disease (ILD) compared with subjects without known lung disease, suggesting increased disruption of the alveolar–capillary membrane in fibrotic ILD. In addition, faster clearance rates of 99mTc-DTPA (i.e. more extensive alveolar–capillary permeability) were found in subjects with higher risk of lung function decline [12] and greatest risk of death [1, 13]. Protein permeability index (PPI), a measure of alveolar–capillary permeability defined as the ratio of total protein in the bronchoalveolar lavage to total protein in plasma, has been demonstrated to be increased in IPF subjects compared with healthy controls and associated with poorer survival at 3 years [11].
In more recent studies, gadofosveset, an albumin-binding gadolinium contrast agent, was used in magnetic resonance imaging of the thorax to determine the degree of albumin extravasation as a surrogate for vascular leak in subjects with pulmonary fibrosis. This study demonstrated increased albumin extravasation in patients with pulmonary fibrosis compared with healthy control subjects [14]. Interestingly, albumin extravasation was increased in fibrosis patients in both radiographically normal and fibrotic lung regions. This finding suggests for the first time that radiographically quiet areas of the lung might be undergoing increases in permeability and that increased lung vascular permeability is an ongoing process in the IPF. The ongoing leak could thus set the stage for fibrosis, and imaging techniques that measure leak might allow us to identify patients with active disease in the pre-fibrotic stages. Additional research is needed to determine if measures of vascular leak can provide meaningful prognostic information, and whether abnormalities in endothelial permeability are a consequence of ongoing lung injury or an important contributor to fibrogenesis.
Vascular remodeling in the fibrotic lung
Pathologic examination of the endothelium in patients with pulmonary fibrosis demonstrates evidence of microvascular injury, necrosis and vascular remodelling including pulmonary systemic shunt formation [15]. As one example, microcirculatory changes in systemic sclerosis are some of the earliest pathologic abnormalities found and can precede the development of fibrosis by years [16]. Recent animal studies have demonstrated injury-induced phenotypic changes during the development of lung fibrosis in which endothelial cells lose some characteristic markers such as platelet endothelial cell adhesion molecule (PECAM)-1 and vascular endothelial cadherin (VE-cadherin), while gaining some mesenchymal markers such as α-smooth muscle actin (αSMA) and fibroblast-specific protein 1 (FSP-1) and recruiting macrophages to the site of injury [17–19]. Indeed, experimental models of pulmonary fibrosis have demonstrated a critical role for the haematopoietic-vascular niche where repetitive lung injury activates pulmonary capillary endothelial cells and perivascular macrophages, impairing repair and promoting fibrosis [20].
Some of the vascular remodelling in the fibrotic lung has been hypothesised to be the result of abnormal angiogenesis. Angiogenesis may play different pathogenic roles depending on the context and stage of the fibrosis [21]. Multiple studies have demonstrated temporal and spatial heterogeneity in capillary density in the fibrotic lung with increased capillary density in the more normal appearing areas of the lung and the border zones with a significant reduction in capillary density in the most fibrotic regions, in particular in the fibroblastic foci, and pulmonary systemic anastomoses in areas adjacent to the traction bronchiectasis and honeycomb changes seen in the peripheral, subpleural regions of the fibrotic lung [22–25]. The fibroblastic foci have been shown to have decreased expression of angiogenic factors such as vascular endothelial growth factor (VEGF), while they simultaneously have increased expression of angiostatic factors such as pigment derived epithelial factor (PEDF) [26]. There is some evidence that in acute exacerbations, there are increased and dilated alveolar capillaries, suggesting at least an association between disease activity and phenotype of the endothelium [27]. In pathological states, novel vasculature is generally characterised by weak and fragile vessels that are inherently leaky and often bleed [28]. The heterogeneity of these processes raises the important but unanswered question of whether the increase in vessel density in the non-fibrotic regions is cause or consequence of the fibrotic process and whether it is helpful or harmful to wound repair processes.
Similarly, lymphangiogenesis has been shown to be altered in IPF. Areas of fibrosis and particularly fibroblastic foci seem to be devoid of lymphatic vessels [27, 29]. In contrast, de novo lymphangiogenesis has been reported to occur in the alveolar spaces [30, 31], from which normally only 3–15% are associated with a lymphatic vessel [32]. These newly formed lymphatics are likely dysfunctional either because of fragmentation [33], or because of the presence of mural cells which obstruct the drainage of macromolecules [34]. Currently available data suggests that the abnormal microvascular changes, associated with hyperpermeability in the IPF lung, actively contribute to disease pathogenesis and could be of significant prognostic value, particularly for patients at risk of acute exacerbations.
Intracellular regulation of endothelial permeability
There are several signalling pathways that have been shown to regulate endothelial permeability, such as RhoA/Rho kinase (ROCK), sphingosine-1-phosphate/sphingosine-1-phosphate receptor 1 (S1P/S1PR1), VEGF and angiopoietin 1 and 2. In addition, cytokines and interleukins (IL) can increase vascular permeability. Many of these mediators have also been implicated in the development of lung fibrosis (fig. 2).

{kind=link}
{kind=link}
Cytoskeletal regulation of endothelial barrier function within an endothelial cell (EC). Under inflammatory conditions, mediators such as the bioactive lipid LPA, thrombin, vascular endothelial growth factor (VEGF), transforming growth factor (TGF)β, angiotensin 1 and 2, and multiple cytokines including interleukin (IL)-8 and IL-10 are increased. These ligands interact with EC surface receptors to induce changes in the cellular cytoskeleton. One of the effects of injury on the EC are increased contractile forces generated via the formation of actin stress fibres, predominate and lead to paracellular gap formation and loss of barrier integrity.
Rho kinases
The ROCKs are centrally involved in transitioning the endothelium to an activated phenotype. Many pro-inflammatory and pro-fibrotic mediators, including VEGF, tumour necrosis factor (TNF)-α, lysophosphatidic acid (LPA), transforming growth factor (TGF)-β1 and thrombin, all act by increasing ROCK-dependent actomyosin contractile tension in endothelial cells [35–38]. Two isoforms of ROCK, ROCK1 and ROCK2, have been identified. Despite their high sequence homology, ROCK1 and ROCK2 appear to have at least some isoform-specific effects, though the full details of their individual roles remain to be explored. Nonetheless, ROCK signalling has been implicated in the pathogenesis of fibrotic diseases in multiple organ systems [39–42]. Cellular contraction occurs directly via Rho-kinase dependent phosphorylation of myosin light chain (MLC), as well as indirectly via the phosphorylation of the targeting subunit (MYPT1) of the myosin light chain phosphatase (MLCP). Both lead to contraction and paracellular gap formation [43]. In addition to its cytoskeletal effects, Rho-kinase activity regulates pro-fibrotic transcriptional events, dependent on the myocardin-related transcription factor/serum response factor and HIPPO pathways [44, 45]. Non-selective pharmacologic ROCK inhibitors such as fasudil and Y27632 mitigate the increases in vascular permeability induced by thrombin and LPA in mouse models of lung injury [46–48] and attenuate the fibrotic response to bleomycin [40, 49, 50]. Although knockout of ROCK1 or ROCK2 is lethal in mouse models, haploinsufficiency of either ROCK1 or ROCK2 or both isoforms has been shown to be protective from bleomycin-induced pulmonary fibrosis through several mechanisms, one of which is a significant reduction in vascular permeability [40].
Sphingosine 1-phosphate
S1P is a potent barrier enhancing bioactive lipid which acts as an intracellular messenger or as an extracellular ligand for five cell surface receptors (S1PR1–5) [51, 52]. Vascular endothelial cells primarily express S1PR1, S1PR2 and S1PR3. S1P binding to the G-protein coupled S1PR1 receptor maintains vascular integrity by activating the small GTPase, Rac. In contrast to thrombin's effects on Rho-ROCK mediated cellular contraction, Rac activation promotes peripheral MLC phosphorylation, adherens junction assembly and cortactin translocation [53–55]. Produced by platelets, erythrocytes, haematopoietic and vascular endothelial cells, the biosynthesis and degradation of S1P is tightly regulated to maintain a stable plasma/tissue concentration in the range required for physiological function (∼0.5–1 µM) [56]. Infusion of S1P reduces lung micro-vascular leakage, leukocyte infiltration and cytokine release in in vivo models of acute and chronic lung injury [57–60]. Conversely, mice that do not make plasma S1P due to deletion of the enzymes that make S1P, sphingosine kinase (SPHK)1 and SPHK2, have impaired endothelial barrier function, increased pulmonary vascular leak and enhanced susceptibility to inflammation and fibrotic stimulation [61]. Similarly, numerous studies have shown that pharmacological inhibition of the endothelial S1PR1 also causes vascular permeability [62–64]. Disruption of endothelial barrier integrity by S1PR1 antagonism has also been shown to exacerbate an otherwise mild lung injury and, in doing so, shift the wound healing response from one which results in normal repair, to one which results in pathological lung fibrosis [63]. Taken together, these findings suggest that the S1P–S1PR1 axis plays a critical role in maintaining baseline vascular barrier integrity. Interestingly, circulating S1P levels are elevated in IPF patients [65]. Although the implications of this increase are not known, it is interesting to consider whether this increase could represent an endogenous attempt to restore barrier function. S1P signalling through another receptor S1PR2 appears to have opposing effects on the endothelium. S1P/S1P2 interactions signal through Rho/ROCK and induce increases in paracellular gap formation and increased vascular permeability [66]. In a mouse model, S1PR2 deficiency was protective against bleomycin-induced pulmonary fibrosis [67].
Vascular endothelial growth factor
Originally known as vascular permeability factor, VEGF and its signalling pathways are aberrantly activated in the fibrotic microenvironment and contribute to persistent endothelial cell activation and hyperpermeability [68]. Specifically, VEGF-A signalling, through its tyrosine kinase receptors VEGFR1 and 2, is well studied in the context of wound healing and fibrosis [69–71]. VEGF induces vascular permeability through activation of Src, a tyrosine kinase. VEGF stimulation of its VEGFR receptor-2 activates Src, which in turn phosphorylates VE-cadherin, one of the key intercellular junction proteins. Phosphorylation of VE-cadherin increases vascular permeability [72]. In addition, VEGF also activates focal adhesion kinase (FAK), which phosphorylates b-catenin, leading to dissociation from VE-cadherin, which also weakens intercellular junctions, leading to increased vascular permeability [73]. Although several reports have indicated a protective role for VEGF-A–VEGFR2 signalling [74, 75], one target of the anti-fibrotic drug nintedanib recently approved by the US Food and Drug Administration (FDA) is VEGFR [76, 77]. Other targets include platelet-derived growth factor (PDGF) receptor and fibroblast growth factor receptor, and it is not clear which receptor is responsible for the anti-fibrotic properties of nintedinab.
Angiopoietin 1/2
Angiopoietin is another regulator of endothelial barrier function, though the effect may be isoform specific. Ang1 enhances barrier function through its receptor Tie2 while Ang2 acts as an antagonist for Tie2, increasing permeability [78]. A recent study demonstrated improved survival in a sepsis model by inhibiting Ang2 and activating Tie2 [79]. A study looking at Ang2 levels in patients with IPF found high Ang2 levels were negatively correlated with lung function, including diffusing capacity of the lung for carbon monoxide and change in % forced vital capacity (FVC) over 12 months. In addition, Ang2 was higher in patients undergoing acute exacerbations of IPF (AE-IPF), and higher Ang2 levels were associated with poor prognosis [80].
Cytokines and interleukins
Cytokines are known to regulate inflammation and permeability and have also been implicated in pulmonary fibrosis. TGF-β and IL-10 have been shown to be increased in the lungs of patients with IPF [81], while IL-2, IL-8, IL-10 and IL-12 have been shown to be increased in the sera of IPF patients [82]. Increased production of pro-inflammatory cytokines such as CXCL1 and IL-8 have been shown to be made by the bronchoalveolar lavage cells of patients with an acute exacerbation [83]. However, since much of the work on cytokines involved in vascular dysfunction and hyperpermeability has been connected to angiogenesis, the mechanistic link between their effects on the vasculature and the resultant fibrotic changes remains less well defined [84].
The anti-inflammatory, pro-fibrotic cytokine TGF-β contributes to vascular permeability in several ways. Perhaps most notably, it is a potent inducer of angiogenesis but can also induce apoptosis in activated endothelial cells [85]. Moreover, stimulation of TGF-β receptor 1 directly causes barrier dysfunction via a SMAD2, p38 and RhoA-dependent mechanism [38, 86].
Pro-inflammatory cytokines such as IL-1 and TNF may also play a role in the development of vascular dysfunction. Both isoforms of IL-1 (IL-1α and IL-1β) have been shown to induce endothelial cell permeability [87] and transient IL-1β overexpression increases lung vascular permeability, at least partially mediated via integrin αvβ6 and TGF-β-dependent mechanisms [88]. IL-1β levels are also increased in BAL fluid and correlate with poor outcome in IPF patients [89]. TNF disrupts the endothelial barrier in two major ways: it induces apoptosis in endothelial cells, via the autocatalytic activation of caspase-8 [89] and directly contributes to endothelial barrier dysfunction by downregulating the expression of VE-cadherin [90] and reorganising the cytoskeleton in a ROCK-dependent manner [91]. IL-1 and TNF may further enhance the inflammatory response by stimulating the production of other pro-inflammatory cytokines, such as IL-6 and IL-8 [92]. In vitro, IL-6 promotes endothelial permeability via the protein kinase C pathway [93] and genetic deletion of IL-6 attenuates alveolar capillary barrier dysfunction in a ventilator-induced lung injury model [94]. Similarly, IL-8 has also been implicated in vascular dysfunction and exerts its effects primarily via the recruitment of neutrophils to the site of injury. Neutralisation of IL-8 signalling blocks neutrophil infiltration and has been shown to be protective in various animal models of lung injury [95, 96].
In summary, cytokines may promote fibrogenesis by exacerbating vascular dysfunction and increasing vascular permeability. Whether through endothelial interaction, or by stimulating secondary pathways, aberrant or overexuberant cytokine signalling likely plays a role in the development of vascular leak and fibrosis in the lung.
Profibrotic consequences of chronic increased permeability
There are several consequences of chronically increased lung permeability that promote the development of lung fibrosis, including increased intra-alveolar coagulation, fibrin deposition, and the establishment of a provisional extracellular matrix, which matures into a stable, collagen rich matrix, the hallmark of fibrosis.
Intra-alveolar coagulation and fibrin deposition
With an increase in pulmonary endothelial permeability, the thin barrier that exists between plasma and tissue compartments during homeostasis is disrupted. Because of this loss of compartmentalisation, plasma proteins and immune cells leak into the airspaces. The plasma proteins include proteases such as thrombin, factor X1 and factor VII that activate the coagulation cascade, leading to intra-alveolar clotting, fibrin deposition and the formation of the provisional matrix. The provisional matrix inactivates surfactant, promoting alveolar collapse [97, 98]. Persistent alveolar collapse allows fibrotic remodelling to occur, which leads to induration and loss of lung function [99, 100]. There is evidence to suggest that fibrinolysis mediated by the activation of plasminogen to plasmin by urokinase-type plasminogen activator and tissue-type plasminogen activator, is suppressed in patients with pulmonary fibrosis, as evidenced by local increases in serpins, plasminogen-activating inhibitor-1 (PAI-1) and α2 antiplasmin [101], which could contribute to the persistence of fibrin in the airspaces.
Consistent with the concept of aberrant repair, the chronicity of alveolar fibrin deposition is thought to be a determinant of subsequent fibrotic remodelling [102]. The full nature of fibrin's contribution to IPF pathology remains unclear however, as gene deletion of the fibrin precursor fibrinogen is not protective in experimental lung fibrosis [103, 104]. Several groups have shown that the receptor-mediated actions of coagulation proteases such as thrombin play more of a fibrogenic role than the accumulation of fibrin [105–108]. Thrombin signalling through its protease activated receptors (PARs) contributes to fibrogenesis by inducing both endothelial gap formation in a Rho-ROCK dependent manner, as well as stimulating the expression and activation of several pro-fibrotic mediators including connective tissue growth factor (CTGF) [107, 109] and lung-injury associated chemokines CCL2 and CCL7 [110]. The absence of PAR1 signalling has been shown to be protective from bleomycin-induced pulmonary fibrosis in a mouse model [108]. Increased permeability induced by the S1PR1 functional antagonist FTY720 promotes the development of fibrosis in a manner dependent on thrombin signalling through its major receptor, protease-activated receptor-1 (PAR1) [106]. Intra-alveolar thrombin cleaves and activates PAR1 receptors on epithelial cells. PAR1, through a RhoA/ROCK dependent pathway, induces epithelial αvβ6 integrin expression, which bind and activate latent TGFβ from LAPs [106, 111].
Direct thrombin inhibition and PAR-1 deficiency are protective in several animal models of lung fibrosis and therefore provide a compelling rationale to target selective aspects of the coagulation system as part of a therapeutic approach in IPF. In addition, it is important to note that dysregulated fibrinolysis processes may equally contribute to pathogenesis. PAI-1 overexpressing mice for instance, or mice genetically deficient in urokinase and plasmin, display enhanced fibrosis in experimental models, whereas PAI-1 knockdown has been shown to be protective [112, 113]. Nonetheless, the clinical development and application of anticoagulants in lung fibrosis has been challenging. The complexity of haemostatic processes makes the beneficial and potential deleterious effects of anticoagulation difficult to balance.
In a small cohort of Japanese patients with IPF requiring hospital admission, anti-coagulation with low molecular weight heparin, followed by warfarin treatment upon hospital discharge had beneficial effects on mortality [114]. However, a subsequent phase III, randomised, placebo-controlled trial of warfarin for treatment of IPF was terminated early due to futility and increased mortality in the warfarin arm [115]. Although the causative factors that contributed to increased mortality are not fully understood, it has been suggested that nonselective depletion of vitamin K dependent factors, such as activated protein C, could be detrimental in IPF [116]. The activation of the anti-inflammatory protein C-axis has been reported to have protective effects on endothelial barrier functioning, as well as facilitating physiological repair processes [116–118].
Extracellular matrix formation
Endothelial permeability enables paracellular movement of diverse substances such as coagulation proteases, matrix metalloproteinases, fibrin degradation products, platelets, fibronectin, cytokines and immune cells from the vasculature into the airspaces, where they can accumulate in high concentrations. In the airspaces, extravasated fibrinogen rapidly clots to crosslinked fibrin, which in addition to its fluid trapping properties, provides a pro-angiogenic and pro-fibrotic stroma. In a process known as “organisation” or “remodelling,” a complex interplay of cells, cytokines and proteases replaces the fibrin/fibronectin-rich provisional matrix with mature connective tissue over time [119–121]. It is thought that the maturation process of the provisional matrix provides the template for subsequent fibrotic remodelling [122, 123].
The provisional matrix
This initial matrix primarily acts as a scaffold and is central to repair during normal wound healing. It allows binding of growth factors, promotes adhesion between cells and matrix, and supports proliferation, migration and growth of fibroblasts, endothelial cells and leukocytes [124]. However, in the fibrotic microenvironment, physical and post-translational modifications that occur during myofibroblast remodelling and persistent vascular leak instead promote tissue destruction and fibrosis. In addition to the coagulation proteases, such as thrombin, fibrin, and plasmin described above, other components of the provisional matrix implicated in aberrant injury repair are described below.
Matrix metalloproteinases
The plasmin-dependent activation of several matrix metalloproteinases (MMPs) plays an important role in normal ECM homeostasis, in particular its degradation [125]. Paradoxically, several MMPs including MMP1, MMP7, MMP8 and MMP9 are overabundant in the lungs of IPF patients [126] and have been implicated in disease activity and progression. These MMPs have other non-degradative effects beyond their role as ECM modulators. MMP-mediated proteolysis of latent forms of fibrogenic growth factors contribute to fibroblast proliferation and activation [127–129]. Mice deficient in several of these MMPs, including MMP7 and MMP8, are protected from bleomycin-induced pulmonary fibrosis [127]. Not all MMPs are profibrotic though, mice deficient in MMP19 have increased pulmonary fibrosis in response to bleomycin challenge [126]. MMP19 is upregulated in hyperplastic alveolar epithelial cells (AECs) which suggests an attempted repair response. By binding macrophage antigen-1, fibrin also activates macrophage-dependent upregulation of proinflammatory cytokines, adding another layer of pro-inflammatory stimulation that has historically been underappreciated [130].
Platelets and fibrin degradation products
Platelets comprise the early wound clot and recent studies have reported increased platelet reactivity in IPF patients [131, 132]. Degranulated platelets also produce pro-fibrotic mediators such as PDGF and TGFβ [133]. This has led to the suggestion that the sequestration of activated, cytokine-producing platelets in the provisional matrix may contribute to pathogenesis of pulmonary fibrosis. Fibrin degradation products are bioactive as well and have been reported elevated in the serum of IPF patients [134]. Elevated serum D-dimer levels have been associated with an increased risk of acute exacerbation in patients with interstitial lung disease [135]. Although their pathological roles remain unclear, fibrin degradation products seem to represent ongoing endothelial activation and contribute to inflammation, given their potent angiogenic and macrophage chemoattractant activity [136].
Fibronectin
Fibronectin is a key component of the matrix. Plasma-derived fibronectin is initially deposited during fibrin polymerisation. It provides binding sites for receptors, growth factors and integrins. Described as the “gateway matrix,” fibronectin transitions the early provisional matrix to a more mature and permanent state [137]. Importantly, fibronectin is bioactive and serves as a depot for TGF-β latent complex, which is physically coupled to the fibrillary fibronectin matrix where it can be mechanically regulated (by simply stretching or contracting the matrix) [138]. Furthermore, the structural nature of fibrillary fibronectin is an important topographical cue for invading fibroblasts, which are known to align along the direction of fibres (in a process known as contact guidance) [139–141]. Fibronectin deposition contributes to the progressive stiffening of the maturing matrix. This is of importance in the context of the mechanically active myofibroblast, whose phenotype is described by its contractile function and its association with a stiffening microenvironment [142, 143]. Finally, fibronectin serves as the functional scaffold for nascent collagen deposition of fibroblasts and represents a critical step of transitioning the provisional matrix to permanent scar tissue [137].
Acute exacerbations of IPF
In addition to playing a role in the chronic injury seen in IPF, there is increased vascular permeability in the lungs of patients who experience an AE-IPF, which is known to lead to significantly increased morbidity and mortality, usually through an acceleration of the fibrotic process [144]. An AE-IPF is characterised radiologically by new, bilateral, ground-glass opacities and/or consolidation, superimposed on the underlying usual interstitial pneumonia pattern [144, 145]. Histopathological evaluations of AE-IPF lungs show diffuse alveolar damage (DAD), oedema and fibrin deposition in both the interstitial and alveolar spaces [146, 147] and serum biomarker profiles report increased markers for epithelial and endothelial injury and coagulation [148, 149]. Taken together, these data suggest a state of active tissue injury in the AE-IPF lung that is associated with vascular hyperpermeability and activated coagulation.
Intra-alveolar fibrin accumulation is a hallmark of lung injury and correlates with disease severity and outcomes in both patients with acute lung injury/acute respiratory distress syndrome and animal models of diffuse alveolar damage [150–153]. Animal models of lung fibrosis share some of the stages of the fibrotic response to lung injury that are seen in patients. For instance, bleomycin-induced lung injury is characterised by an early exudative phase, with increased tissue factor pathway activation and a reduced fibrinolytic capacity [154–157]. Likewise, in patients with fibrotic lung disease, increased vascular permeability, procoagulant activity and fibrin turnover are correlated with loss of lung compliance.
There have been few clinical trials targeting this population of patients with AE-IPF; however, some of the trials completed targeted intra-alveolar coagulation. Thrombomodulin, an endothelial cell membrane protein which binds to and inactivates thrombin, has been trialled in small non-randomised studies for AE-IPF [158, 159]. In one study, patients who presented with AE-IPF and elevated BAL D-dimer levels, were treated with recombinant soluble thrombomodulin and their 3-month mortality was half (30%) of the 3-month mortality of a historical control group of patients who did not receive thrombomodulin (65%) [159]. Although not a randomised controlled trial, these results suggest that microvascular dysfunction and subsequent intra-alveolar coagulation may be important to the development and prognosis of AE-IPF. In a second case–control study which also used historical controls, survival was significantly improved with 6 days of recombinant thrombomodulin therapy [158]. A pre-clinical study also demonstrated that recombinant thrombomodulin can inhibit bleomycin-induced pulmonary fibrosis and TGFβ1-driven exacerbation and progression of pulmonary fibrosis, in a manner dependent on its anti-coagulative effects [160]. However, a recently published randomised controlled trial demonstrated no difference in 90-day survival in patients treated with recombinant human soluble thrombomodulin, thrombomodulin alpha, versus placebo in AE-IPF [161].
Therapeutic implications
As these data suggest, there is evidence for increased pulmonary endothelial permeability and increased intra-alveolar coagulation in patients with IPF [162]. Either process thus provides a potential target for therapy. Permeability-targeted therapies are beginning to emerge in clinical trials (table 1). The ROCK2 isoform-specific inhibitor KD025 is undergoing phase 2 clinical trials in IPF patients (NCT02688647). This is encouraging because the pleiotropic effects of ROCK have raised concerns about potential adverse effects of non-selective inhibition, including systemic vasodilation and hypotension. Isoform-specific targeting could provide a method to limit these potential adverse effects. Nintedinab, as previously mentioned, inhibits multiple tyrosine kinases including VEGFR. Although the VEGF receptor is only one of many targets of nintedinab, one can speculate that the reduction in the rate of confirmed acute exacerbations of IPF seen in the phase III trials of nintedanib could be due to a reduction in vascular permeability through VEGFR inhibition [76]. A recent clinical trial of IPF patients with severely impaired gas exchange combined two drugs that target endothelial functions, nintedinab and sildenafil [165]. Although there was no significant difference in the primary endpoint in this study, there was a preservation of FVC in the treatment group, suggesting perhaps an anti-fibrotic effect due to modulation of the vasculature. Despite the negative results from the ACE-IPF trial using warfarin, some speculate that anticoagulant strategies targeting thrombin and/or its signalling pathways could be of clinical value for patients with lung fibrosis. As mentioned above, thrombin inhibition through thrombomodulin has been recently tried in small, non-randomised studies of acute exacerbations of IPF [159].
Clinical trials in pulmonary fibrosis that target vascular permeability or coagulation
Existing knowledge gaps and future directions
Although significant gains have occurred in the last decade with respect to knowledge about the connection between the pulmonary vasculature and fibrotic lung disease, there are gaps that remain in our understanding of the links between these processes. Primarily, it has not been demonstrated that vascular permeability is fundamental to fibrogenesis or whether it is a co-existent response to injury that exacerbates an independent fibrotic response. To support the hypothesis that it is a fundamental process, it would be necessary to inhibit endothelial permeability in the context of known fibrosis-inducing injury and demonstrate prevention of fibrosis. Other helpful evidence includes demonstration of correlation between the degree of permeability and outcomes in patients or animal models, and changes in permeability with current or new therapies, again correlated with outcomes. Beyond the question of necessity, parsing out which downstream consequences of increased permeability cause fibrosis would be helpful to develop targeted therapies. Whether the main mediators linking permeability and fibrosis are activated immune cells, cytokines or plasma proteins remains unanswered. Another important and clinically relevant question is whether inhalational injury to the epithelium, such as with viral infections or chemical exposures, directly injures the endothelium, or whether endothelial changes are secondary events. Similarly, the genetic and epigenetic mechanisms of how the vascular endothelium responds to injurious stimuli over time are not well understood. It is intriguing to speculate about the existence of genetic abnormalities that may predispose an individual to aberrant vascular responses to injury.
Going forward, new models and technologies may assist in delineating the link between vascular permeability and pulmonary fibrosis. Combining human, animal and ex vivo modelling systems with emerging methods such as single cell assay for transposase-accessible chromatin and RNA sequencing, may allow for additional insight into the genetic and molecular dynamics of lung fibrosis. Correspondingly, the implementation of bioinformatics pipelines to probe existing genome wide association study data sets for endothelial-specific variants could elucidate new mechanisms that contribute and/or predispose to abnormal wound repair. Animal models remain essential to answer many of these outstanding questions. However, in vitro multicellular systems offer advantages in that they use human cells and allow for the study of cell–cell interactions in a pathologically relevant microenvironment. Molecular imaging, as it continues to develop, may enable further quantification of permeability and other fibrosis-related processes in humans in vivo [169]. Given the data supporting a role for vascular permeability in promoting fibrosis, measuring the degree of permeability in IPF patients could potentially be a powerful tool for assessing disease activity and determining changes with anti-fibrotic therapies.
Ultimately, to support the hypothesis that vascular permeability is mechanistically tied to fibrogenesis, additional and improved models will be needed in which vascular permeability is either present or can be induced with or without lung injury to study the effects on other cell types and on tissue architecture and function. Using animal models and complex in vitro multicellular systems may eventually provide sufficient information to corroborate or reform our hypothesis.
Conclusions
In summary, evidence is accumulating that endothelial permeability plays a pathogenic role in the development of pulmonary fibrosis. Despite the recognition of increased alveolar capillary permeability in patients with pulmonary fibrosis and its association with outcomes, the important links between vascular permeability and subsequent extravascular coagulation and promotion of tissue fibrosis are still developing. As reviewed here, persistent vascular leak appears to contribute in a fundamental way to creating a pro-fibrotic intra-alveolar environment, facilitating the development of lung fibrosis when coupled with specific lung injuries and/or other aberrant wound healing responses to those injuries. Indeed, many of the intracellular pathways that regulate endothelial permeability during homeostasis and normal wound healing have also been shown to regulate lung fibrogenesis. Therefore, a therapeutic approach of limiting the extent of vascular leak and the downstream fibrotic responses it promotes, may be an effective strategy for treating pulmonary fibrosis. Consequently, detecting and resolving ongoing vascular leak may permit earlier resolution of tissue injury, or prevention of more permanent tissue damage.
While this review focusses on the mechanisms underlying increased endothelial permeability through paracellular gap formation, there are other ways to regulate vascular permeability. Morphological contributions, such as the integrity of the endothelial glycocalyx [170], the impact of fluid shear stress on endothelial cell alignment [171] and the pre-existence of paracellular gaps [172], all contribute. These will be important considerations for future research in this field.
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00100-2019.Shareable
Footnotes
Conflict of interest: C.K. Probst has nothing to disclose.
Conflict of interest: S.B. Montesi reports fees paid to institution and participating in research from United Therapeutics Corporation and Promedior, outside the submitted work.
Conflict of interest: B.D. Medoff has nothing to disclose.
Conflict of interest: B.S. Shea reports personal fees from Genentech and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: R.S. Knipe has nothing to disclose.
- Received July 3, 2019.
- Accepted March 26, 2020.
- Copyright ©ERS 2020
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- IPF pathogenesis
- The endothelial barrier in the lung
- Abnormal permeability in patients with pulmonary fibrosis
- Vascular remodeling in the fibrotic lung
- Intracellular regulation of endothelial permeability
- Profibrotic consequences of chronic increased permeability
- Acute exacerbations of IPF
- Therapeutic implications
- Existing knowledge gaps and future directions
- Conclusions
- Shareable PDF
- Footnotes
- References
- Figures & Data
- Info & Metrics