





Abstract
Introduction TBX4 mutation causes small patella syndrome (SPS) and/or pulmonary arterial hypertension (PAH). The characteristics and outcomes of PAH associated with TBX4 mutations are largely unknown.
Methods We report the clinical, functional, radiologic, histologic and haemodynamic characteristics and outcomes of heritable PAH patients carrying a TBX4 mutation from the French pulmonary hypertension (PH) network.
Results 20 patients were identified in 17 families. They were characterised by a median age at diagnosis of 29 years (0–76 years) and a female to male ratio of three. Most of the patients (70%) were in New York Heart Association (NYHA) functional class III or IV with a severe haemodynamic impairment (median pulmonary vascular resistance (PVR) of 13.6 (6.2–41.8) Wood units). Skeletal signs of SPS were present in 80% of cases. Half of the patients had mild restrictive or obstructive limitation and diffusing capacity of the lung for carbon monoxide (DLCO) was decreased in all patients. High-resolution computed tomography (HRCT) showed bronchial abnormalities, peri-bronchial cysts, mosaic distribution and mediastinal lymphadenopathies. PAH therapy was associated with significant clinical improvement. At follow-up (median 76 months), two patients had died and two had undergone lung transplantation. One-year, three-year and five-year event-free survival rates were 100%, 94% and 83%, respectively. Histologic examination of explanted lungs revealed alveolar growth abnormalities, major pulmonary vascular remodelling similar to that observed in idiopathic pulmonary arterial hypertension (IPAH) and accumulation of cholesterol crystals within the lung parenchyma.
Conclusion PAH due to TBX4 mutations may occur with or without skeletal abnormalities across a broad age range from birth to late adulthood. PAH is usually severe and associated with bronchial and parenchymal abnormalities.
Abstract
PAH due to TBX4 mutations may occur with or without skeletal abnormalities across a broad age range from birth to late adulthood. PAH is usually severe and associated with parenchymal abnormalities, alveolar and pulmonary vascular remodelling, and low DLCO. http://bit.ly/38jM37U
Introduction
Pulmonary arterial hypertension (PAH) is a severe, progressive and uncommon disease affecting the small pulmonary arteries. Pulmonary vascular remodelling is mainly characterised by abnormal proliferation of endothelial cells, smooth muscle cells, myofibroblasts and pericytes, leading to increased pulmonary arterial pressure (PAP) and right-ventricular failure [1, 2]. Several medical conditions are related to the development of PAH, such as connective tissue diseases, congenital heart diseases, portal hypertension and HIV infection [3]. In the absence of an associated condition, exposure to drugs and toxins causing PAH, or a familial history of PAH, the disease is classified as idiopathic [3]. The main cause of heritable PAH is mutations in the bone morphogenetic protein receptor type 2 gene (BMPR2), which was first described in the year 2000. Since then, novel mutations have been identified in different genes of interest (TBX4, ATP13A3, GDF2, SOX17, AQP1, ACVRL1, SMAD9, ENG, KCNK3, CAV1, GDF2, BMP10) [4].
In 2013, array comparative hybridisation and sequencing of a population of children presenting with PAH associated with mental retardation and/or dysmorphic features led to the identification of a new gene of interest, T-box transcription factor 4 (TBX4) [5]. The TBX4 gene is a member of the T-box gene family encoding transcription factors that play varied and important roles throughout development, especially in the development and branching of the lungs and in lower limb formation [6–9]. Mutations in this gene cause small patella syndrome (SPS), also called coxo-podo-patellar syndrome, an autosomal dominant skeletal dysplasia affecting the lower limbs that is characterised by hypoplasia or aplasia of the patella, ossification defects of the ischia and inferior pubic rami, as well as anomalies of the feet such as a large gap between the first and second toes [10].
To date, TBX4 mutation is reported to be one of the main genetic causes of PAH in children and is mainly reported in childhood-onset PAH [11–14]. Because of its pivotal role in embryogenesis, especially in the lungs and lower limbs, heritable PAH patients carrying TBX4 mutations may present with a distinct phenotype. To test this hypothesis, we have studied the clinical, functional, radiologic, histologic and haemodynamic characteristics, as well as long-term outcomes of PAH patients carrying a TBX4 mutation, from the French pulmonary hypertension (PH) network.
Methods
Patient selection
We conducted a retrospective population-based study of PAH patients carrying a TBX4 mutation from the registry of the French PH network (French PH Referral Center, Hôpital Bicêtre, Université Paris Sud, Le Kremlin-Bicêtre and its 25 associated centres across France). This registry was set up in agreement with French bioethics laws (Commission Nationale de l'Informatique et des Libertés no. 842063) [15]. Genetic counselling was offered to all patients with idiopathic, familial, or drug-induced PAH [16]. All patients provided written informed consent prior to genetic analysis.
Genetic analysis
PAH predisposing genes, including the TBX4 gene, were screened by next-generation sequencing (NGS)-based gene panel analysis as described previously [17, 18]. Briefly, a custom gene panel including established PAH and pulmonary veno-occlusive disease (PVOD) genes (BMPR2, TBX4, EIF2AK4, CAV1, KCNK3, SMAD9, ACVRL1, ENG, BMP9) was used for NGS-targeted capture. Libraries were prepared according to the KAPA Library preparation protocol for Illumina platforms (KAPA Biosystems, Cape Town, South Africa), captured using the SeqCap EZ Choice Library (Roche Sequencing, Pleasanton, CA, USA) following the manufacturer's protocol and sequenced using the MiSeq system (Illumina, San Diego, CA, USA). Data were analysed by a bioinformatic pipeline developed by GenoDiag Inc (Paris, France), allowing the identification of variants and copy number variations (CNVs). Each base needed to be covered by at least 40 reads to be validated. All variants of interest were validated by Sanger sequencing. CNVs were validated by multiplex ligation-dependent probe amplification (MLPA) when available or by quantitative PCR. TBX4 genetic testing has been performed since 2014.
Clinical, functional, haemodynamic and radiologic characteristics
We reviewed clinical data (age, medical history and physical examination), dyspnoea assessed by modified New York Heart Association (NYHA) functional class, non-encouraged 6-min walk distance (6-MWD) and pulmonary functional tests (PFTs) including diffusing capacity of the lung for carbon monoxide (DLCO) corrected for haemoglobin (Hb).
Haemodynamic measurements on right-heart catheterisation (RHC) included mean PAP, pulmonary arterial wedge pressure (PAWP), right-atrial pressure and mixed venous oxygen saturation (SvO2). Cardiac output (CO) was measured by the standard thermodilution technique. Cardiac index (CI) was calculated as CO/body surface area. Pulmonary vascular resistance (PVR) was calculated as (mean PAP–PAWP)/CO. Precapillary PH was defined as mean PAP ≥25 mmHg, PAWP ≤15 mmHg and PVR >3 Wood units [1].
High-resolution computed tomography (HRCT) of the chest was independently analysed by an expert radiologist (M. Remy-Jardin) who was blinded to clinical and haemodynamic data.
Follow-up and clinical outcomes
Medical therapies approved for PAH (prostacyclin derivatives, endothelin receptor antagonists (ERA) and phosphodiesterase type-5 inhibitors (PDE5i)) were given to the patients according to the clinical judgement and discretion of the individual treating physicians. Clinical, functional and haemodynamic follow-up data were collected in the French PH registry. Time to death or lung transplantation was also recorded.
Statistical analysis
The analyses were carried out using the SPSS Base 20.0 statistical software package (SPSS Inc., Chicago, IL, USA). The variables were expressed as median (min–max) for quantitative variables and as n (%) for categorical variables. We used the Wilcoxon signed rank test to compare the NYHA functional class before and after treatment and the Wilcoxon matched-pair sign rank test to compare quantitative data before and after treatment. Overall survival was calculated from the time of the diagnosis of PAH and was estimated by the Kaplan–Meier method. Correlations between data were studied with Spearman's rank correlation coefficient.
Results
Patient population
Since 2014, 448 index patients (excluding related cases) were screened for PAH predisposing genes including TBX4. We identified 20 PAH patients (4.5%) in 17 unrelated families carrying heterozygous mutations in the TBX4 gene, bringing the frequency of TBX4 mutations in France to 6% and 3% in childhood-onset PAH and adult-onset PAH, respectively. Fourteen mutations were point mutations, two were CNVs and one was a variant located in the promoter region and classified as a variant of unknown significance (VUS) (table 1). However, the patient carrying this VUS presented with typical SPS foot abnormalities, leading to the genetic diagnosis of TBX4-linked PAH. The mutations of eleven patients have been reported previously [12, 17–19].
TBX4 mutations and signs of small patella syndrome (SPS) identified in pulmonary arterial hypertension (PAH) patients
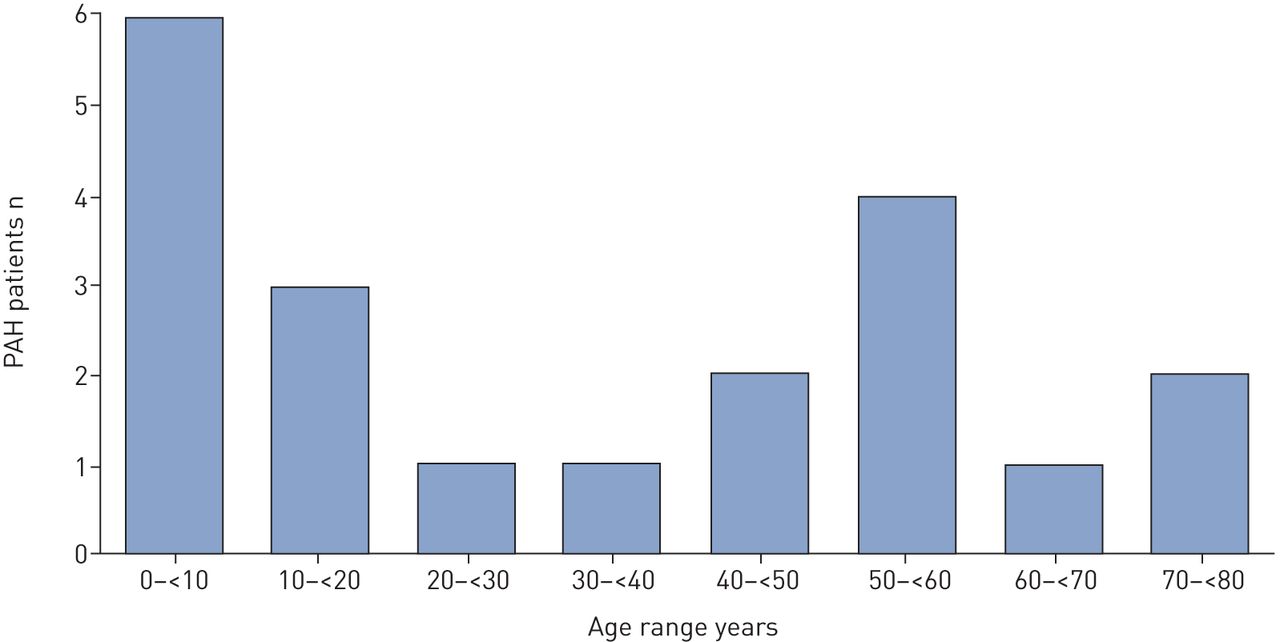
In this cohort, nine cases (45%) presented with childhood-onset PAH (<18 years old), 16 patients were sporadic cases and four were from the same family (figure 1). Median age at PAH diagnosis was 29 years (0–76 years), 6 years (0–17 years) in the childhood-onset group and 56 years (24–76 years) in the adult-onset group. Repartition of age at diagnosis is presented in figure 2. A female predominance was observed (female–male ratio of three), especially in the childhood-onset group, in which all patients were female.

Family tree of the familial case, whose four members presented with pulmonary arterial hypertension (PAH) and skeletal anomalies corresponding with incomplete small patella syndrome (SPS) and carried a mutation of the TBX4 gene.

Age at pulmonary arterial hypertension (PAH) diagnosis.
Two patients (10%) presented with atrial septal defects and one had patent ductus arteriosus. Among them, one child developed PAH 9 months after percutaneous closure of the atrial septal defect. Four patients had a history of mild asthma.
Clinical and functional characteristics
At diagnosis, two patients (10%) were in NYHA functional class II, 11 (55%) were in class III and three (15%) were in class IV. NYHA functional class was not evaluable in four children (20%) because of their age. At diagnosis, the 6-MWD was 388 m (224–613 m) in the whole cohort, 392 m (235–585 m) in adults and 290 m (224–613 m) in children. Forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC), FEV1/FVC ratio and total lung capacity (TLC) are presented in table 2. Fourteen patients had complete PFTs, showing normal values in seven patients (50%), mild obstructive limitation in four patients (28.6%) and mild restrictive limitation in three patients (21.4%). Moreover, DLCO was decreased in all patients with a median of 57.5% (43–69%) (table 2). This decrease was not associated with tobacco exposure since only two patients were smokers (14 pack-years and 10 pack-years, respectively).
Clinical, functional, haemodynamic and biological data of pulmonary arterial hypertension (PAH) at diagnosis in patients carrying a TBX4 mutation (n=20)
Haemodynamic assessment at diagnosis
All patients underwent RHC at PAH diagnosis, showing severe pre-capillary PH with an elevated mean PAP of 59.5 mmHg (29–87 mmHg), a normal PAWP of 8 mmHg (4–14 mmHg), a decreased CI of 2.7 L·min−1·m−2 (1.48–7.60 L·min−1·m−2) and an increased PVR of 13.6 Wood units (6.2–41.8 Wood units) (table 2). No patient had acute vasodilator responses according to current international guidelines [1].
No significant differences in haemodynamic characteristics were observed between children and adults (data not shown). We found no significant correlations between PVR and DLCO (r=−0.24, p=0.47), FEV1 (r=0.12, p=0.71), FVC (r=−0.14, p=−0.63) or arterial oxygen tension (PaO2) (r=0.24, p=0.58).
Analysis of high-resolution computed tomography of the chest
Fifteen patients (75%) had analysable HRCT of the chest at diagnosis (table 3 and figure 3). Fourteen computed tomography (CT) images (93.3%) presented pulmonary abnormalities. Nine (60%) showed central and peripheral bronchial abnormalities, including thickened and irregular walls (n=7, 46.7%), bronchial and/or tracheal diverticula (n=5, 33.3%), mucoid impactions (n=3, 20%) and moniliform and cylindrical bronchiectasis (n=2, 13.3%). Four CT images (26.7%) showed the presence of peri-bronchial cysts communicating, in most cases, with bronchi. Hilar or mediastinal lymphadenopathies were observed in six patients (40%). Parenchymal abnormalities were mainly mosaic distribution (n=6, 40%), subpleural septal lines (n=4, 26.6%), micronodules (n=2, 13.3%) and ground glass opacities (n=2, 13.3%). Two CT images (13.3%) showed micronodules and sub-pleural neovascularity-like lesions. We found no significant differences in lung abnormalities, as highlighted by HRCT of the chest, between childhood-onset PAH and adult-onset PAH (data not shown).
Characteristics of high-resolution computed tomography (HRCT) of the chest in pulmonary arterial hypertension (PAH) patients carrying a TBX4 mutation (n=15)

High-resolution computed tomography (HRCT) of the chest of patients carrying a TBX4 mutation. Panels are as follows: a) 5-mm thick transverse computed tomography (CT) section of the upper lobes, viewed as a maximum intensity projection, in a 34-year old female patient showing bilateral micro-nodules suggestive of bronchiolitis; b) 1-mm thick transverse CT section of the upper lobes, viewed as a minimum intensity projection (same patient as a). The presence of a large, multi-loculated diverticula (arrow) directly connected to the posterior wall of the trachea (arrowhead) is noted, as is the additional presence of emphysema in both lungs; c) 2-mm thick coronal lung image obtained in the posterior part of the chest in a 24-year old male, showing dilated subpleural pulmonary vessels (arrowheads); d) 1-mm thick transverse CT section (lung image) obtained at the level of the tracheal bifurcation in a 17-year old female patient. The presence of a thin-walled cyst (arrow), adjacent to a peripheral bronchial division in the posterior segment of the right upper lobe, is noted; e) 3-mm thick transverse CT section (mediastinal image) obtained at the level of the right bronchus intermedius (same patient as d). Marked dilatation of the right and left bronchial arteries (arrows) is noted; f) coronal oblique reformation of the chest, viewed as a minimum intensity projection (magnified view of the left hilum), in a 56-year old male patient. The presence of bronchial wall irregularities and numerous adenolectases (arrows) in segmental and subsegmental bronchi of the left upper lobe is noted.
Lung histopathology
Lung samples were available in three patients (supplementary table S1). One sample was a surgical lung biopsy and two were lung explants (figure 4). All three showed major pulmonary arterial remodelling similar to that observed in idiopathic pulmonary arterial hypertension (IPAH), with plexiform lesions in two (66%) (supplementary table S1). Parenchymal abnormalities were found in all patients and accounted for patchy peribronchiolar and peripheral fibrosis, lymphoid nodules, peri-bronchial inflammation with cholesterol clefts and hypertrophy of smooth muscle. Alveolar septa were also interrupted and appeared rarefied, resulting in an appearance of “emphysematous dystrophia”. These changes are potentially related to small airway remodelling and altered lung development, as previously described in such patients [20].

Lung histopathology of lung explant from a pulmonary arterial hypertension (PAH) patient with a TBX4 mutation. The histological analysis of lung explants showed pulmonary vascular remodelling with plexiform lesions and organised thrombi of some elastic pulmonary arteries. Small pulmonary arteries appeared thickened with marked intimal fibrosis (a). Plexiform lesions were also found close to or within the lumen of small pulmonary arteries (b and c). Eventually, organised thrombi were also identified in some elastic-type pulmonary arteries (d). Parenchymal alterations were also observed with patchy peribronchiolar and subpleural fibroses were identified (e and f). The fibrosis incorporated lymphoid nodules, remodelled vessels and cholesterol cleft deposits (f) (star). Eventually, alveolar alterations with rarefaction of alveolar septa resulting in an emphysematous pattern were also noticed (g). Dilation and remodelling of bronchial vessels were additionally found (h). Scale bars=200 μm (b, c) and 500 μm (a, d–h).
Skeletal anomalies
Clinical data on skeletal abnormalities were available in 15 out of the 20 patients (75%) and 12 of these (80%) showed clinical abnormalities. Nine patients (75%) showed a unique skeletal abnormality, two (17%) presented with two abnormalities and one (8%) with three abnormalities. No skeletal abnormalities were identified in three patients out of 15 (20%), a 14-year old, a 62-year old and a 56-year old, respectively. One of these three patients had normal lower limbs and pelvic radiographs. Ten patients, including five children and five adults, presented with foot abnormalities characterised by a large gap between the first and the second toes and flat feet. Disorders of the knees such as congenital dislocation of the patella or knee dysplasia were found in five patients (33%), including two children and three adults. One patient showed a lack of ossification of the ischia on pelvic radiography (table 1).
All the mutated members of the familial case and two parents (figure 1, III2 and III3), obligate carriers of the familial TBX4 mutation, presented with features of SPS.
Response to pulmonary arterial hypertension therapy
Nineteen patients received PAH therapy. Thirteen patients were re-evaluated with RHC in the first year after initiation of treatment with a median delay of 6 months (3–11 months). At reassessment, four patients (31%) were treated with ERA monotherapy, two patients (15.2%) were treated with PDE5i monotherapy, five patients (38.6%) were treated with oral dual combination ERA and PDE5i, one patient (7.6%) was treated with dual combination ERA and prostacyclin derivative and one patient (7.6%) received triple combination therapy including an ERA, PDE5i and prostacyclin derivative (table 4). Among the patients who received PAH therapy, three (22.8%) were treated with calcium channel blockers (CCB), despite the absence of acute vasodilator response.
Clinical, functional and haemodynamic data at diagnosis and reassessment after initiation of pulmonary arterial hypertension (PAH) therapy (n=13)
At reassessment, a clinical improvement was observed, with improved NYHA functional class (p<0.001) and increased 6-MWD (p=0.01). However, there was no significant haemodynamic improvement in mean PAP, CI or PVR (table 4).
Outcomes
At the last follow-up, 12 patients (60%) were treated with triple combination therapy (ERA, PDE5i and prostacyclin derivative) and six (30%) received oral dual combination therapy (ERA and PDE5i). One patient was treated with PDE5i and CCB and one patient received only CCB during 5 years. After a median follow-up of 56 months (6–239 months), two patients (10%) had died (one 67-year old patient died from right-heart failure after 53 months of follow-up and the other 74-year old patient died from an unknown cause after 23 months of follow-up). Two patients underwent double lung transplantation after 7 years and 22 years of follow-up, respectively. The 1-year, 3-year and 5-year event-free survival rates were 100%, 94% and 83%, respectively (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Time to death or lung transplantation of pulmonary arterial hypertension (PAH) patients carrying a TBX4 mutation. Event-free survival was estimated by the Kaplan–Meier method.
Discussion
We describe a cohort of 20 PAH patients from a nationwide PH registry carrying a TBX4 mutation across a broad age range from birth to late adulthood. Our report describes the phenotype and outcome of a French mixed cohort of childhood-onset and adult-onset PAH associated with TBX4 mutation. PAH due to TBX4 gene mutations seems to occur with a bimodal distribution mode in our cohort, comprising two main peaks of occurrence: an early onset in childhood as described previously [5, 12, 14] and a later presentation in adulthood (figure 2). This observation is supported by the fact that TBX4 mutations are identified in 6% of paediatric index cases and in 3% of adult index cases. This consideration cannot be stated formally given the limited size of our population. However, a recent study of exome sequencing in a large cohort of 2572 adult and pediatric PAH cases finds the same type of presentational distribution in PAH patients carrying a TBX4 mutation [21]. A female predominance was observed, as described in other forms of heritable PAH [22, 23]. Clinical features of SPS were found in most patients but were frequently non-obvious. At diagnosis, patients presented with severe pre-capillary PH, usually associated with bronchial, parenchymal and vascular abnormalities, as demonstrated by HRCT of the chest and lung histology. After initiation of PAH therapy, a statistically significant clinical improvement was observed with no significant haemodynamic changes. Even if prognosis seemed to be better than in PAH associated with BMPR2 mutation it remained severe, with two deaths and two lung transplantations at follow-up.
A high penetrance of TBX4 mutation for skeletal abnormalities has been described [5]. In our cohort, 80% of patients presented with signs of SPS. However, signs were absent or non-obvious in some patients. Indeed, three out of 20 patients had no skeletal abnormalities, illustrating that PAH may be the only clinical manifestation of TBX4 mutation mimicking IPAH. Therefore, personal and familial signs of SPS must be systematically searched in every patient newly diagnosed with idiopathic or familial PAH and TBX4 mutations should be screened even in the absence of skeletal abnormalities. In the literature, this ascertainment was previously made by Zhu et al. [13], who identified 13 cases of PAH associated with a TBX4 mutation, including a single case of SPS and by Navas et al. [24], who identified four PAH patients carrying a TBX4 mutation with no skeletal abnormalities identified. By contrast, the penetrance of TBX4 mutation for PAH is incomplete, as previously reported with other PAH predisposing genes. In the present series, we have identified a small proportion of familial cases (four patients from one family and 16 sporadic cases) in comparison with what is usually observed in PAH associated with BMPR2 mutation [16]. In the reported family, all TBX4 mutation carriers presented with SPS but only four female members developed PAH, at an age of 1 year, 3 years, 17 years and 76 years, respectively, demonstrating that the manifestation of the disease is independent of the mutation type (figure 1).
The haemodynamic characteristics in our population are broadly similar to previous reports, showing severe pre-capillary PH at diagnosis without acute vasodilator response according to current guidelines. However, some patients presented with partial vasoreactivity in accordance with what has been reported in paediatric series [14]. Among the four patients who received CCB therapy only one presented stable disease, while the other three became worse requiring the use of PAH treatments. This haemodynamic profile was similar to the results observed in other heritable PAH populations, such as BMPR2 or ACVRL1 mutation carriers, for whom acute vasoreactivity was classically absent [22, 23].
We observed congenital heart diseases in 15% of our cases (two atrial septal defects and one patent ductus arteriosus): two in the nine childhood-onset PAH cases and one in the 11 adult-onset PAH cases. The paediatric report by Galambos et al. [14] describes 36.8% atrial septal defects, 21% patent ductus arteriosus, 5.3% ventricular septal defects and 5.3% patent foramen ovale. These differences may be explained at least in part by a higher incidence of CNV in the paediatric population. Indeed, the CNVs generally encompassed the whole TBX4 gene plus some other genes, including the TBX2 gene, which plays an important role in the embryologic development of the heart and is associated with cardiac septal defects [25, 26].
One may hypothesise that pre-capillary PH in patients carrying TBX4 mutation could be a consequence of developmental disorders of the lungs and should be classified as PH due to chronic respiratory disease (group 3 of the PH classification) [27]. Analysis of HRCT of the chest demonstrated that 60% of patients presented with bronchial abnormalities, with central and peripheral thickening, irregularity of the bronchial walls and presence of tracheal or bronchial diverticula in one-third of patients. PFTs revealed an obstructive respiratory disease in 28.6% of patients and that 28.6% of patients showed decreased FEV1 at <80%. Furthermore, four patients had a history of mild asthma. Histological assessment of lung samples in three patients confirmed an abnormal distal lung development with emphysematous-like dystrophia appearing as disruption and rarefaction of the bronchiolar septation, abnormalities of bronchi with the presence of tracheal and bronchial diverticula and interstitial fibrosis. Furthermore, histologic examinations showed the presence of cholesterol cleft inclusions located in the perivascular connective tissue. This aspect is not a common feature of PAH but has been described previously in heterogeneous PH populations in which it is impossible to formally eliminate the presence of underlying TBX4 mutation or any developmental disorder [28–30]. These findings are reminiscent of those reported in the paediatric cohort of Galambos et al. [14]. Moreover, lung explants from our patients had a small size macroscopically, compatible to a certain degree with pulmonary hypoplasia, a lung development disorder classically defined by a ratio of lung weight to body weight of less than 0.012 after 28 weeks of gestation [20]. Finally, this hypothesis may still be supported by the low DLCO, which is not usually as pronounced in IPAH and heritable PAH [31]. These abnormalities may be related to pulmonary development disorders due to TBX4 mutations. However, some patients did not have CT abnormalities but developed PAH, arguing for the presence of isolated pulmonary vascular disease. Indeed, histologic assessment revealed pulmonary arterial remodelling as classically observed in PAH, including plexiform lesions (figures 3b and 3c) similar to those observed in IPAH or heritable PAH [2]. Moreover, we did not show significant correlations between PVR (reflecting the vascular anomalies) and FEV1, FVC or PaO2 (reflecting lung parenchymal involvement) or DLCO (reflecting the involvement of the alveolo-capillar barrier). This finding argues for a specific involvement of pulmonary arteries in this heritable form of PAH and against a mechanism merely due to parenchymal involvement.
Even if it might have started early, during lung development, pulmonary vascular involvement could progress slowly in TBX4 mutation carriers. This could explain the relative long-term stability with PAH treatments. Despite initially severe PAH, requiring a combination of PAH therapies and, in turn, the use of triple combination therapy in 60% of patients, clinical status improved despite the absence of significant haemodynamic improvement. The overall survival rates of our cohort seem to be better than those observed in other forms of IPAH and heritable PAH, but they must be interpreted with caution because of the retrospective nature of the study, the small size of the studied population and the variable duration in follow-up [32].
Most of the radiologic or histologic abnormalities observed in our patients could be related to the embryologic roles of the TBX4 gene. In utero, TBX4 is abundantly expressed in the pulmonary mesenchyme at critical stages of development, where the transcription factor activates genes that are essential for branching morphogenesis, including FGF10 and Nkx2.1. The association of TBX4 mutations with PAH further raises the possibility that TBX4 regulates genes that influence pulmonary vascular development and function [33]. Using a transgenic mouse line (Tbx4 lung enhancer), it was recently shown that angiogenic vessels originating from the extra-pulmonary site initially serve as a stem, and trigger lung mesenchymal stem cell differentiation and commitment to endothelial progenitor cells, which then coalesce into pre-existing vessels and become the major source for lung vasculature [34]. In alveolar capillary dysplasia, a disease mainly caused by FOXF1 mutations and characterised by pulmonary vein misalignment, capillary paucity and alveolar misdevelopment, decreased FOXF-1 and/or its downstream transcription factor TBX4 disrupt lung microvessel formation and homing to alveolar epithelium [35]. The involvement of FGF signalling in early lung development has been thoroughly investigated. FGF9 positively regulates the expression of Fgf10 via the upregulation of TBX4 in the lung mesenchyme. Epithelium-derived FGF9 and mesothelium-derived FGF9 induce proliferation in both the epithelial and mesenchymal compartments of the developing lung and are also involved in pulmonary vascular development [36]. It has been shown that TBX4 is highly specific in lung fibroblasts and that TBX4 is required for cell proliferation and collagen gel contraction capacity [37]. Transcriptome analysis has revealed that TBX4 could broadly regulate fibroblast-related pathways and partly contribute to super-enhancer-mediated transcriptional programmes. TBX4 knockdown and subsequent RNA-seq were performed in adult fibroblasts. Among the significantly regulated genes, KCNK6, CYP1B1, HMOX1 and ESM1 have been shown previously to be important in PAH pathogenesis [38]. Thus, TBX4 deficiency may compromise lung development and pulmonary vasculature in particular, through insufficient angiogenesis related in part to low FGF expression. In adults, TBX4 is mainly expressed in pulmonary fibroblasts, where it regulates genes and pathways involved in PAH.
The main limitations of this study are first of all the retrospective data collection, with a potential underestimation of paediatric cases due to a lack of exhaustivity in genetic screening in early death. Moreover, the population is of limited size, even if it remains the largest adult PAH population of TBX4 mutation carriers to date. Notably, it is unlikely to gather a much larger cohort of patients, since the prevalence of TBX4 mutation currently ranges from 2.8% to 7.7% in childhood-onset PAH and from 1.5% to 2.4% in adult-onset PAH [12, 13, 17, 18]. Mutations in the TBX4 gene have probably also been underdiagnosed because, until recently, physicians were not systematically looking for skeletal abnormalities in PAH patients. Moreover, mutations in the TBX4 gene have not been tested for in all PAH cases due to its recent association with pulmonary vascular disease. It has been difficult to interpret the haemodynamic response to PAH therapy in this study because of incomplete haemodynamic follow-up in childhood PAH. Last, the pathologic assessments brought major information on pulmonary vascular and parenchymal involvements but this assessment was limited to a small number of patients, which makes it difficult to generalise the results to the whole population.
In conclusion, PAH due to TBX4 mutation is an autosomal dominant disease with incomplete penetrance for PAH and/or SPS, and generally occurs in sporadic cases. PAH in this population is severe and occurs across a broad age range from birth to late adulthood. Heritable PAH associated with TBX4 mutations often presents with bronchial and parenchymal radiological abnormalities and low DLCO.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02340-2019.SUPPLEMENT
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-02340-2019.Shareable
Acknowledgements
We thank all the physicians from the French Pulmonary Hypertension Network.
Footnotes
This article has an editorial commentary: https://doi.org/10.1183/13993003.00585-2020
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: P. Thoré has nothing to disclose.
Conflict of interest: B. Girerd has nothing to disclose.
Conflict of interest: X. Jaïs reports grants and personal fees from Actelion and MSD, and grants from Bayer, outside the submitted work.
Conflict of interest: L. Savale reports grants, personal fees and non-financial support from Actelion, grants and personal fees from MSD, and non-financial support from GSK, outside the submitted work.
Conflict of interest: M-R. Ghigna has nothing to disclose.
Conflict of interest: M. Eyries has nothing to disclose.
Conflict of interest: M. Levy has nothing to disclose.
Conflict of interest: C. Ovaert has nothing to disclose.
Conflict of interest: A. Servettaz has nothing to disclose.
Conflict of interest: A. Guillaumot has nothing to disclose.
Conflict of interest: C. Dauphin has nothing to disclose.
Conflict of interest: C. Chabanne has nothing to disclose.
Conflict of interest: E. Boiffard has nothing to disclose.
Conflict of interest: V. Cottin reports personal fees for advisory board work and non-financial (travel) support from Actelion, grants and personal fees for advisory board work, lectures and steering committee work, as well as non-financial (travel) support from Boehringer Ingelheim, personal fees for advisory board work and data monitoring committee work from Bayer/MSD and Galapagos, personal fees for advisory board work from Gilead and Novartis, personal fees for advisory board work, lectures and steering committee work, as well as non-financial (travel) support from Roche SAS, personal fees for lectures and non-financial (travel) support from Sanofi, personal fees for steering committee work and data monitoring committee work from Promedior, and personal fees for data monitoring committee work from Celgene and Galecto, outside the submitted work.
Conflict of interest: F.Perros has nothing to disclose.
Conflict of interest: G. Simonneau reports grants, personal fees and non-financial support from Actelion, Bayer, GSK and Merck, outside the submitted work.
Conflict of interest: O. Sitbon reports grants, personal fees and non-financial support from Actelion Pharmaceuticals, Bayer HealthCare and MSD, personal fees from Acceleron Pharmaceuticals, Ferrer, Gossamer Bio and United Therapeutics, and grants from GlaxoSmithKline, outside the submitted work.
Conflict of interest: F. Soubrier has nothing to disclose.
Conflict of interest: D. Bonnet reports personal fees for advisory board work and steering committee work from Actelion Pharmaceuticals, Eli Lilly and Novartis, outside the submitted work.
Conflict of interest: M. Remy-Jardin has nothing to disclose.
Conflict of interest: A. Chaouat has nothing to disclose.
Conflict of interest: M. Humbert reports personal fees from Acceleron, Actelion, MSD and United Therapeutics, and grants and personal fees from Bayer and GSK, outside the submitted work.
Conflict of interest: D. Montani reports grants and personal fees from Actelion and Bayer, and personal fees from GSK, Pfizer, MSD and Chiesi, outside the submitted work.
- Received December 10, 2019.
- Accepted February 5, 2020.
- Copyright ©ERS 2020
References










