





Abstract
Cerebral hypoxia may promote cerebral damage in patients with obstructive sleep apnoea (OSA). We investigated whether OSA patients experience nocturnal cerebral hypoxia that is prevented by continuous positive airway pressure (CPAP).
OSA patients using CPAP underwent sleep studies including pulse oximetry (arterial oxygen saturation (SpO2)) and near-infrared spectroscopy to monitor cerebral tissue oxygenation (CTO) at baseline and after 2 weeks on either subtherapeutic or therapeutic CPAP according to randomised allocation. Changes in oxygenation at end of the 2-week intervention were compared between groups.
Among 21 patients (mean apnoea/hypopnoea index 50.3 events·h−1), OSA recurred in all nine patients using subtherapeutic CPAP and in none of the patients using therapeutic CPAP: mean (95% CI) between-group differences in changes of oxygen desaturation index from baseline to 2 weeks +40.7 (31.1–50.4) events·h−1 for SpO2 and +37.0 (25.3–48.7) events·h−1 for CTO (both p<0.001). Mean nocturnal SpO2 and CTO decreased more in patients using subtherapeutic versus therapeutic CPAP: −2.4 (−3.4–−1.1)% and −3.8 (−7.4–−0.1)%, respectively; both p<0.03. Severe CTO drops ≥13% associated with cerebral dysfunction in previous studies occurred in four out of nine patients using subtherapeutic CPAP, but in none out of 12 patients using therapeutic CPAP (p=0.01).
In patients with OSA, CPAP withdrawal resulted in nocturnal cerebral deoxygenation, suggesting a role of cerebral hypoxia in predisposing untreated OSA patients to cerebral damage.
Abstract
Untreated obstructive sleep apnoea predisposes to severe cerebral hypoxia during sleep http://ow.ly/mZ1030jAxZZ
Introduction
Obstructive sleep apnoea (OSA) is a highly prevalent sleep-related breathing disorder associated with adverse vascular outcomes [1, 2]. OSA has been associated with neurocognitive impairment [3] and increased risk of stroke and other manifestations of ischaemic cerebrovascular disease in epidemiological studies [4–7]. Proposed underlying mechanisms explaining the association between OSA and cerebral damage include cerebral hypoxia related to repetitive arterial oxygen desaturations, augmented sympathetic activity, endothelial dysfunction and impaired cerebrovascular autoregulation in response to blood pressure surges and intermittent hypoxaemia. A causal relationship between OSA and hypertension [8], as well as between OSA and peripheral vascular dysfunction [9] has been shown. However, less is known on cerebrovascular function and oxygenation in OSA patients. In a previous study using near-infrared spectroscopy (NIRS) to monitor cerebral oxygenation during sleep in OSA patients discontinuing their continuous positive airway pressure (CPAP) therapy for a few nights we observed considerable intermittent and sustained nocturnal cerebral deoxygenation, in particular at altitude [10]. The cerebral tissue oxygen saturation during sleep reached levels similarly low to those associated with cerebral dysfunction in patients undergoing unilateral carotid artery clamping during neurosurgery in previous studies [11]. The current trial was designed to test the hypothesis that OSA induces cerebral hypoxia that might expose patients with OSA to an increased risk of cerebral ischaemia that can be prevented by CPAP therapy. The study was performed concurrently in participants of an investigation on the effects of OSA on coronary perfusion during CPAP withdrawal [12].
Methods
Trial design
This study was conducted as a part of a randomised controlled trial assessing the effect of CPAP-withdrawal on myocardial perfusion in patients with OSA using long-term CPAP therapy (NCT01797653) [12]. Participants of that study were asked to undergo nocturnal monitoring of cerebral oxygenation in addition to cardiorespiratory sleep studies while being treated with therapeutic or subtherapeutic CPAP. The study was approved by the local ethics committee (KEK-ZH-Nr. 2012–0511) and written informed consent was obtained from all participants.
Participants
Patients aged 20–75 years with moderate to severe OSA, effectively treated by CPAP were recruited. Patients were eligible if they were treated with CPAP for >1 year, showed a minimal adherence of 4 h per night, and had an oxygen desaturation index (ODI) of ≥20 events·h−1 both at the time of initial OSA diagnosis as well as during a current 4-day period off CPAP (to confirm persistence of at least moderate OSA). Patients with previous respiratory failure (awake arterial oxygen tension <9.0 kPa or arterial carbon dioxide tension (PaCO2) >6 kPa), unstable coronary or cerebral artery disease, severe arterial hyper- or hypotension, Cheyne–Stokes breathing or a history of a sleeping-related accident were excluded.
Intervention and assessments
Outcomes were assessed at baseline on therapeutic CPAP and after 2 weeks of treatment with either therapeutic or subtherapeutic CPAP according to randomisation. Therapeutic CPAP was provided with a REMstar autoCPAP device (Philips Respironics, Murrysville, PA, USA) operated in the previous mode and with the nose or full-face mask that the patient was used to. Subtherapeutic CPAP was provided with REMstar autoCPAP device with modified tubing incorporating a restrictor at the airflow outlet and additional leaks near the mask to prevent rebreathing: maximal mask pressure was <2 mbar.
Polygraphic in-laboratory cardiorespiratory sleep studies measuring airflow, respiratory inductance plethysmography, finger pulse oximetry, electrocardiogram and transcutaneous carbon dioxide tension (Microgas; Radiometer, Basel, Switzerland) were performed along with cerebral NIRS. Regional cerebral tissue oxygenation (CTO) and cerebral total haemoglobin concentration (tHb) were continuously recorded over the night using a NIRO 200NX device (Hamamatsu Photonics, Hamamatsu City, Japan). Optodes were placed bilaterally on the skin high on the forehead where bone thickness is minimal (FP1/2 location of the 10/20 system), as previously described [10]. NIRS data were sampled at 1 Hz along with other polygraphic data in a polysomnography device (Alice 5; Philips Respironics). Subjective sleepiness was assessed using the Epworth sleepiness scale (ESS) [13].
Main outcomes of interest
The co-primary outcomes of the study were changes in mean nocturnal CTO and in CTO desaturation index. A CTO desaturation was defined as a ≥3% dip lasting for ≥10 s in association with an apnoea/hypopnoea-related arterial oxygen desaturation (arterial oxygen saturation measured by pulse oximetry (SpO2)-channel) (see further detailed explanations on CTO analysis in the online supplementary material).
Other outcomes of interest
Other outcomes were apnoea/hypopnoea-related CTO dips ≥13%; such desaturations were associated in previous studies in neurosurgical patients with neurophysiological signs of severe cerebral ischaemia [11], the cumulative night-time spent with apnoea/hypopnoea-related CTO dips of various severities including ≥13% and the night-time spent with sustained CTO desaturations ≥13% from wakefulness baseline, the mean nocturnal arterial oxygen saturation (SpO2), the ODI (≥4% dips in the SpO2 signal) and mean nocturnal transcutaneous carbon dioxide tension (PCO2). In order to evaluate the effect of cerebral deoxygenation on cerebral blood volume, as an index of the cerebrovascular response to hypoxia, coefficients of cross-correlation between both CTO and SpO2 and NIRS-derived total cerebral haemoglobin concentration, a NIRS-derived surrogate for regional cerebral blood volume, were computed. In theory, a perfect cerebrovascular autoregulation would result in a coefficient of cross-correlation of −1 between the two variables; this is because drops in CTO would be compensated for by an increase in blood volume to maintain oxygen delivery; conversely, a coefficient of +1 would indicate absence of such compensation (online supplementary material).
Randomisation and blinding
Patients were randomised to either subtherapeutic or continuation of therapeutic CPAP by a computer software minimising for differences in ODI, body mass index and vascular disease, as reported previously [12]. Patients and outcome assessors remained blinded to the allocation.
Data analysis
The sample size estimation was based on the main outcome of the trial evaluating coronary perfusion. There was no basis from previous studies to perform an a priori sample size estimation in the current exploratory study of CTO. Data are summarised as median (quartiles) and mean±sd, depending on distribution. The primary analysis was performed as per protocol. The treatment effect was determined by computing mean differences and 95% confidence intervals of changes in outcomes between baseline and the follow-up in patients randomised to therapeutic and subtherapeutic CPAP. A two-sided p<0.05 in independent t-tests or Mann–Whitney U-tests was considered statistically significant. Analyses were adjusted for differences in baseline mean oxygen saturation and oxygen desaturation index (SpO2 and CTO) using multiple regression. Cross-correlation analyses between both CTO and SpO2 and tHb were performed to evaluate changes in cerebral blood volume in response to cerebral tissue and peripheral arterial deoxygenation, as described in the online supplementary material.
Results
Participants
The patient flow is shown in figure 1. 26 patients with moderate to severe OSA included in the myocardial perfusion study [12] took part in the current NIRS study (therapeutic CPAP n=16 and subtherapeutic CPAP n=10). Data from five patients (one in the subtherapeutic and four in the therapeutic CPAP group) could not be analysed because of poor NIRS signal quality. Data from 21 patients were available for analysis. Characteristics of patients in the two study arms were similar (table 1).
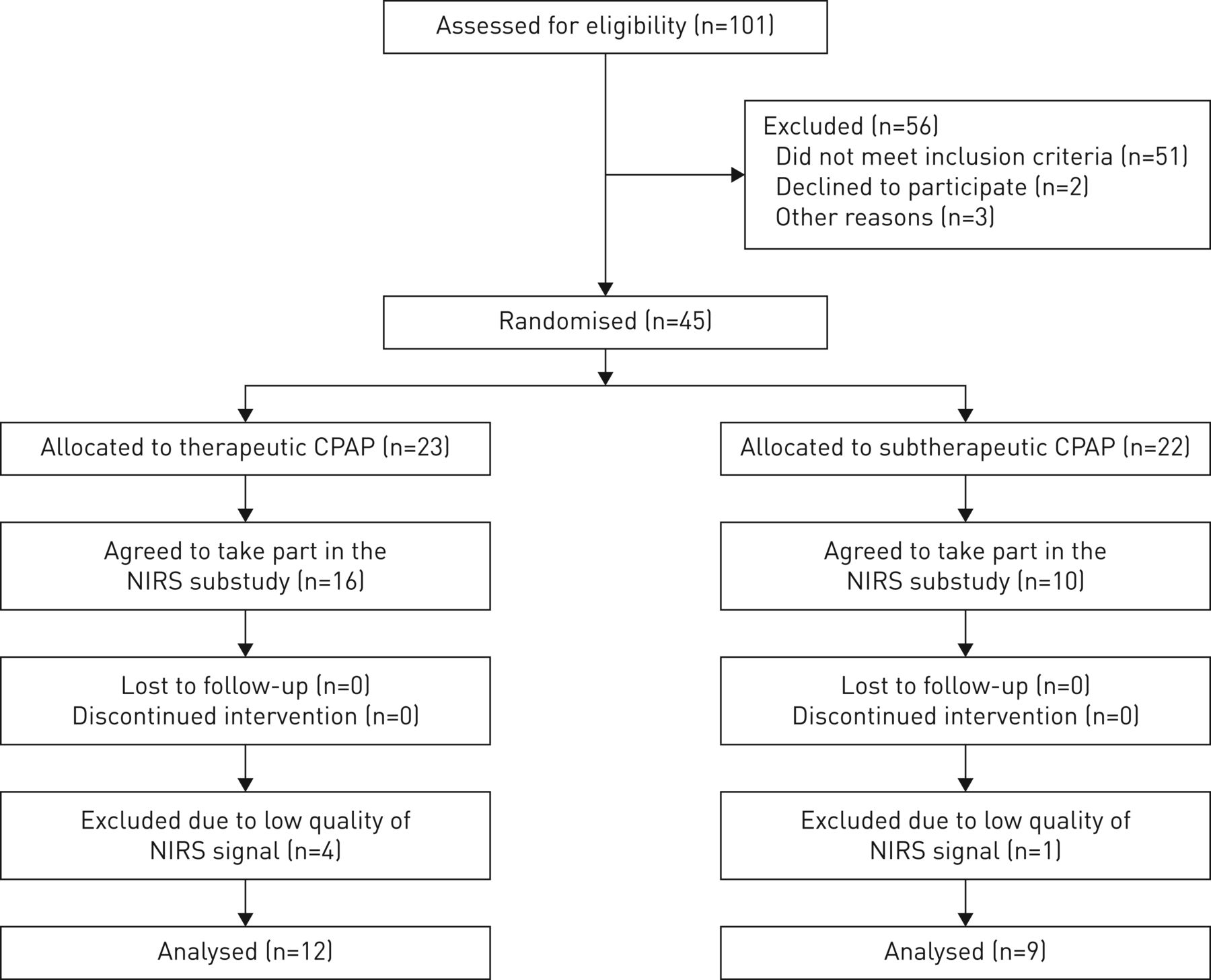
CONSORT patient flow diagram. CPAP: continuous positive airway pressure; NIRS: near-infrared spectroscopy.
Patient characteristics
Effects of CPAP withdrawal on cerebral and peripheral arterial oxygen saturation and sleep apnoea
Physiological recordings in a patient during application of subtherapeutic CPAP are illustrated in figure 2. Apnoea/hypopnoea-related CTO dips were generally less pronounced than corresponding SpO2 dips and they were associated with a rise in tHb, the surrogate of regional cerebral blood volume, with a delay of ∼30 s. The outcomes assessed at baseline and at the end of the 2-week intervention period are summarised for the two groups in table 2 and are illustrated in figures 3 and 4.

Nocturnal recordings in a patient using subtherapeutic continuous positive airway pressure (CPAP). a) Arterial oxygen saturation measured by pules oximetry (SpO2), cerebral tissue oxygenation (CTO) and total haemoglobin (tHb, in arbitrary units) measured by near infra-red spectroscopy, heart rate (HR) and transcutaneous carbon dioxide tension (PtcCO2) over a period of 1 h; b) a 5-min zoomed-in portion of the same channels. SpO2 dips are larger than corresponding CTO dips. The increase in SpO2 dips over the period from ∼00:50 to 01:20 is associated with larger dips in CTO and a rise in transcutaneous PtcCO2 and tHb consistent with an inflow of blood into the brain region exposed to more severe hypoxia and hypercapnia during this period. In b), transient increases in tHb are seen in response to progressively larger SpO2 and CTO dips with a delay of ∼30 s. Over the same period, PtcCO2 is also increasing, but does not change with each event due to the slow response time of the measurement technique. Dashed horizontal lines in the CTO channel represent wakefulness baseline (a) and local baseline of CTO dips (b) and the corresponding 13% desaturation level associated with neurocognitive dysfunction in previous studies.
Effect of continuous positive airway pressure (CPAP) withdrawal
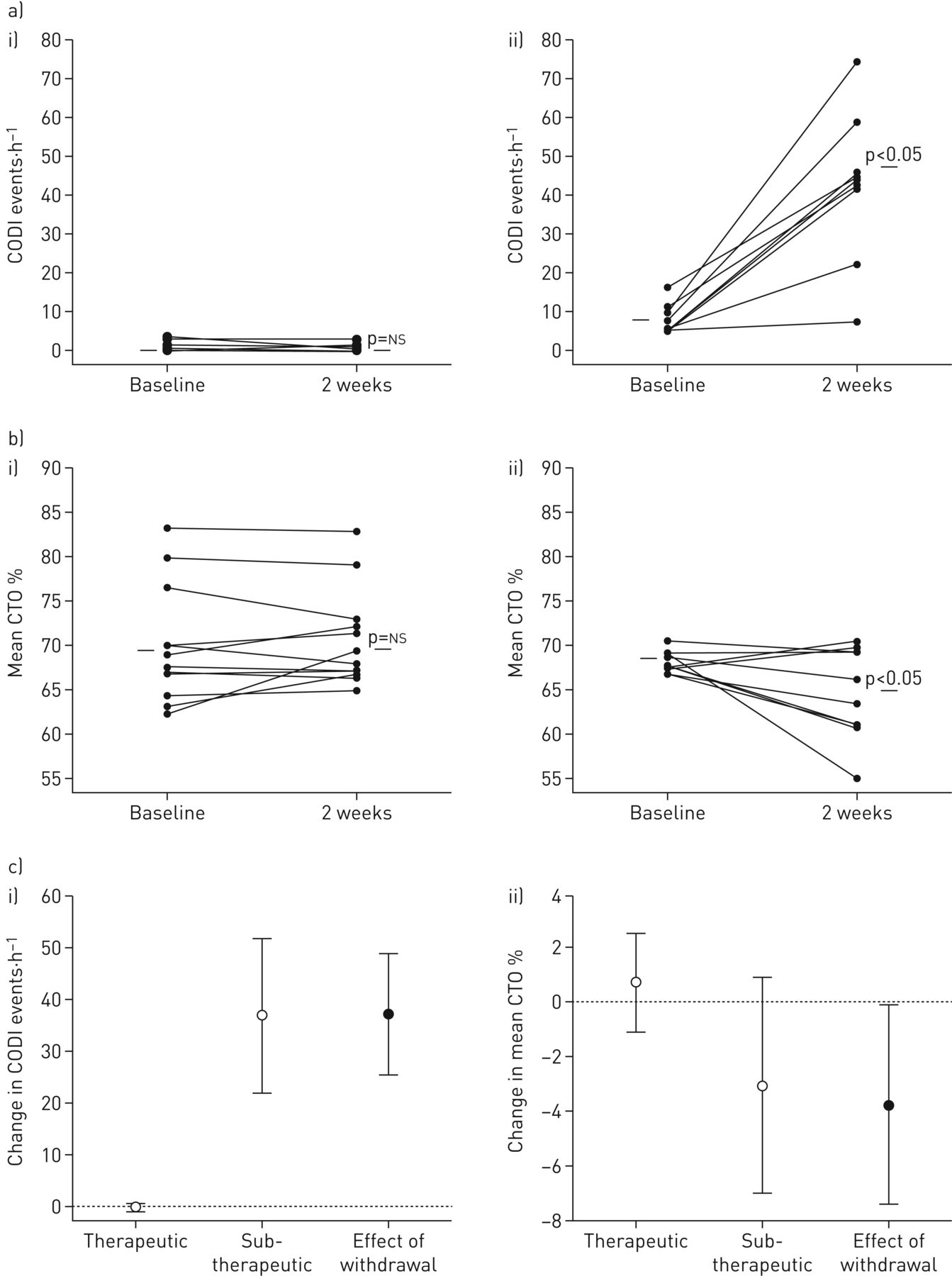
Continuous positive airway pressure (CPAP) withdrawal effect on cerebral oxygenation. Individual plots showing a) cerebral oxygen desaturation index (cODI) and b) mean nocturnal cerebral tissue oxygenation (CTO) in i) the therapeutic CPAP group and ii) the subtherapeutic CPAP group at baseline and at 2-week follow-up. c) Group change in i) cODI and ii) mean CTO.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Degree of cerebral hypoxaemia. Percentage of patients in each group reaching a certain degree of maximal falls in cerebral tissue oxygenation (CTO). A decrease in CTO of >13% was associated with cerebral dysfunction in neurosurgical patients in a previous study. *: p<0.05 (Chi-squared).
At baseline (on therapeutic CPAP), patients in both groups had a normal SpO2, apnoea/hypopnoea index (AHI) and ODI. At the end of the 2-week intervention, the number of cyclic dips in CTO ≥3% and in SpO2 was ≥4%, and the AHI had increased significantly more in the CPAP withdrawal group compared to patients continuing therapeutic CPAP. Moreover, the amplitude of apnoea/hypopnoea-related CTO dips ≥3% and the cumulative night-time spent in these dips were greater in the group using subtherapeutic CPAP (mean±sd amplitude 5.4±3.1%, cumulative night-time spent in CTO dips was ≥3% 186±117 min) compared to the group using therapeutic CPAP (3.5±0.8%; cumulative night-time spent in CTO dips ≥3% 3±16 min; p<0.001 for both comparisons) (figure 3). Major CTO dips ≥13% that were previously reported as the threshold for severe cerebral ischaemia, were observed in four out of nine patients treated with subtherapeutic CPAP (see example in figure 2), but in none of the patients on therapeutic CPAP (Chi-squared 6.6, p=0.01). The four patients with major apnoea/hypopnoea-related CTO dips ≥13% during subtherapeutic CPAP spent a median (interquartile range) duration of 16.6 (3.7–43.2) min in dips with such severe desaturation. Both the mean nocturnal CTO and SpO2 decreased significantly in the patients using subtherapeutic CPAP, while these variables remained unchanged in patients using therapeutic CPAP (table 2). The time spent with sustained CTO desaturations ≥13% below wakefulness baseline was significantly greater with subtherapeutic (mean±sd 34±65 min) compared to therapeutic CPAP (0±0 min; p<0.017).
CPAP withdrawal was associated with a significant increase in the peak negative cross-correlation coefficient between CTO and tHb at a mean±sd lag of 26±10 s (online supplementary table S1 and figures S1 and S2) while there was no significant change in the peak negative cross-correlation coefficient in patients using therapeutic CPAP. The lag time of the maximal cross-correlation coefficients did not change in either group. Correspondingly, the peak negative cross-correlation coefficient between SpO2 and tHb (at mean lag of 40±18 s) increased during the 2-week intervention in patients using subtherapeutic CPAP, but remained unchanged in patients using therapeutic CPAP (online supplementary table S2).
There was no change in the mean nocturnal transcutaneous PCO2. Subjective sleepiness assessed by the ESS significantly increased in response to CPAP-withdrawal when compared to continuing therapeutic CPAP (table 2).
Discussion
This randomised controlled trial in patients with moderate to severe OSA demonstrates that withdrawing therapeutic CPAP results in recurrence of nocturnal breathing disturbances causing major cyclical and persistent drops in CTO and SpO2 which is prevented by therapeutic CPAP. In several patients, the apnoea/hypopnoea-related cyclic drops in CTO during CPAP withdrawal were of a magnitude and duration reported to cause cerebral dysfunction in patients undergoing unilateral carotid artery clamping during neurosurgery [11]. Therefore, the current data support a potential role of untreated OSA in predisposing to neuronal damage with brain dysfunction and an increased risk of stroke.
Several epidemiological studies have shown a strong association between OSA and the incidence of stroke as well as other manifestations of ischaemic cerebrovascular disease [4–7]. Imaging studies have shown metabolic and structural changes in the brains of patients with OSA associated with cognitive dysfunction similar to that observed in patients with multi-infarct syndrome [14–19]. In a meta-analysis of prospective observational studies, the pooled relative risk of stroke in OSA, compared to the control group, was 2.0 (95% CI 1.4–2.9) [20]. Another meta-analysis including 8435 patients found a significant association between OSA and stroke risk with an odds ratio for incident stroke of 2.24 (95% CI 1.57–3.19) in OSA, which was even higher in males, and correlated with OSA severity [21]. Over 10 years, 14% of patients with severe OSA are predicted to experience a stroke [22]. Potential mechanisms explaining this association, besides the role of OSA in development of established risk factors for stroke such as hypertension and probably atrial fibrillation, are impaired cerebral perfusion by disturbed endothelial function [9] and cerebral autoregulation, autonomic dysregulation [23], repetitive shear stress by nocturnal blood pressure surges, blunted nocturnal dipping blood pressure pattern and increased intracranial pressure [24, 25] leading to a decreased cerebral perfusion pressure. Based on epidemiological observational studies, CPAP is suggested to improve the cerebro- and cardiovascular outcome in OSA patients [4, 26] and to reduce the risk of stroke. Robust evidence from randomised controlled interventional trials is missing. However, a matched analysis of CPAP adherent patients versus usual-care patients of the SAVE trial, a recent large randomised controlled trial on the effect of CPAP on cardiovascular events in OSA, has shown a significant reduction of cerebral events and stroke in the CPAP group (hazard ratio 0.52, 95% CI 0.30–0.90; p=0.02) [27].
The current study provides new evidence that OSA causes cerebral tissue hypoxia that is prevented by therapeutic CPAP. The mean nocturnal CTO measured during CPAP withdrawal of 65% in the current study is the same as that observed in our previous study in OSA patients discontinuing CPAP therapy for a few days studied at an altitude of 490 m before travelling to 2590 m [10]. The current data extend these earlier findings by providing detailed information on the magnitude and duration of cyclic and sustained CTO desaturations that occurred as a consequence of apnoeas/hypopnoeas over the course of entire nights. More than half of the patients in the current study revealed CTO desaturations >10% (figure 3), i.e. exceeded the decrease in mean nocturnal CTO of 8% associated with exposure to an altitude of 2590 m observed in our previous study [10]. The proportion (44%) of patients with very large CTO dips >13% during use of subtherapeutic CPAP was considerable. These patients spent a median time of 16.6 min with cyclic dips reaching this degree of cerebral hypoxaemia. Moreover, in the whole group of patients using subtherapeutic CPAP, the median time of the night spent with a CTO desaturation ≥13% below wakefulness baseline was 34 min. These findings are consistent with an exposure of the patients to cerebral hypoxia sufficiently severe to represent a risk of cerebral dysfunction and ischaemia [11]. For comparison, in a previous study, unilateral carotid artery clamping for only 2–3 min that caused CTO desaturations ≥13% in neurosurgical patients had induced cerebral dysfunction [11]. Other, uncontrolled observations in sleeping OSA patients during shorter periods of 1–2 h revealed a baseline CTO of 65% and desaturations up to 8% in OSA patients with a broad range of severity [28]. In one study, OSA patients had a lower mean nocturnal CTO (57%) than healthy controls (62%) and this was related in part to the older age of OSA patients; the extent of CTO desaturations was not reported [29].
Intact cerebral autoregulation is required to maintain a constant tissue perfusion during changes in blood pressure. Additional physiological mechanisms that control the cerebral perfusion contribute to the prevention of hypoxia and hypercapnia of the brain tissue [30, 31]. OSA was associated with impairment of cerebrovascular regulation in a population-based study [32]. Transcranial Doppler ultrasound and NIRS studies have suggested an ineffective autoregulation in OSA [33–37]. This finding led to the conclusion that OSA patients might be particularly susceptible to nocturnal cerebral ischaemia due to repetitive decreases in cerebral blood flow and hypoxaemia.
In the current study we employed cross-correlation analysis to investigate the changes in tHb, the NIRS-derived surrogate of regional cerebral blood volume, following CTO and SpO2 desaturations based on an approach proposed in previous studies [34]. We found a negative peak in the cross-correlation coefficients between these variables at a lag in tHb of a few seconds, consistent with an influx of blood in response to hypoxia. The peak negative cross-correlation was larger during CPAP withdrawal than during therapeutic CPAP, suggesting that the greater degree of variation in CTO and SpO2 during OSA recurrence was met with larger changes in blood volume (online supplementary tables S1 and S2). Thus, some degree of response of the cerebral circulation to alterations in cerebral hypoxia seemed to be preserved in OSA patients chronically treated with CPAP even after short-term withdrawal. However, due to the delay and/or insufficient magnitude of this response, the brain was not protected from relevant desaturations, which was consistent with our previous observations [10]. Nevertheless, the desaturations in CTO were less pronounced than those of the arterial blood. Cross-correlation analyses between tHb and PaO2 would have provided additional comprehensive insights into the cerebral blood flow response to apnoea/hypopnoea-related changes in blood gases but were not feasible due to the slow response-time of transcutaneous capnography.
Despite a more pronounced fall in SpO2 than in CTO during apnoeic events, the treatment withdrawal effect on mean CTO during the whole night was more pronounced than on mean nocturnal SpO2 (−3.8% versus 2.3%; table 2), a finding that may be related to the different physiological basis of the two signals, i.e. CTO reflects an average of oxygen saturation in all vessels included in the NIRS sample volume (mainly capillaries) while SpO2 reflects oxygenation of the arterial blood.
A limitation of the current randomised controlled trial is the relatively low number of participants, not allowing any subgroup analysis to predict which patient characteristics were associated with the most pronounced cyclic and persistent cerebral deoxygenation. However, the randomised design and the application of a short-term CPAP-withdrawal in treated OSA patients allowed effectively investigating treatment effects. Nevertheless, the long-term consequences of OSA-induced cyclic dips and sustained nocturnal falls in cerebral oxygenation are not addressed in this study. Whether patients with OSA exhibit specific responses of cerebral perfusion to intermittent hypoxia, either pronounced due to endothelial dysfunction or attenuated due to hypoxic preconditioning cannot be answered by the current study which did not include a control group of healthy subjects. Interestingly, Rupp et al. [38] found relatively less pronounced CTO dips in response to intermittent 2-min cycles of hypoxia in healthy subjects applied over 45 min. The mean amplitude ratio of CTO versus SpO2 dips was 0.35, i.e. lower than the corresponding ratio of 0.48 in OSA patients using subtherapeutic CPAP in the current study.
Our non-invasive NIRS measurements recorded oxygenation of the frontal cortex only, while other regions that are also susceptible to hypoxia such as the hippocampus and the cerebellum were not studied. However, multichannel NIRS recordings revealed similar patterns of CTO changes in response to hypoxia and exercise in different regions of the prefrontal, premotor and motor cortex and magnetic resonance studies in patients with OSA identified structural changes in frontal and hippocampal territories, which were both improved with CPAP therapy [39, 40].
Conclusion
The current study shows that OSA results in intermittent and sustained nocturnal cerebral tissue deoxygenation to a degree reported to cause cerebral dysfunction. These findings suggest that patients with untreated OSA might be at increased risk of nocturnal cerebral damage.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
supplementary material ERJ-00032-2018_Supplement
Footnotes
Author contributions: Conception and design: K.E. Bloch, E.I. Schwarz, M. Furian, M. Kohker, J.R. Stradling. Data acquisition: E.I. Schwarz, C. Schlatzer. Analysis and interpretation of data: E.I. Schwarz, M. Furian, K.E. Bloch. Drafting the article: E.I. Schwarz, K.E. Bloch. Revising the article for important intellectual content and final approval: all authors.
Conflict of interest: J.R. Stradling reports personal fees (consultancy) from ResMed UK, outside the submitted work.
Support statement: This work was supported by Swiss National Science Foundation grants (32003B_124915 and 143365) and the Clinical Research Priority Program (CRPP) Sleep and Health of the University of Zurich. Funding information for this article has been deposited with the Crossref Funder Registry
This study is registered at clinicaltrials.gov, with registration number NCT01797653.
This article has supplementary material available from erj.ersjournals.com
- Received January 5, 2018.
- Accepted April 6, 2018.
- Copyright ©ERS 2018
References










