





Abstract
Interstitial lung fibroblast activation coupled with extracellular matrix production is a pathological signature of idiopathic pulmonary fibrosis (IPF), and is governed by transforming growth factor (TGF)-β/Smad signalling. We sought to define the role of heat shock protein (HSP)90 in profibrotic responses in IPF and to determine the therapeutic effects of HSP90 inhibition in a murine model of pulmonary fibrosis.
We investigated the effects of HSP90 inhibition in vitro by applying 17-AAG (17-allylamino-17-demethoxygeldanamycin) to lung fibroblasts and A549 cells and in vivo by administering 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin) to mice with bleomycin-induced pulmonary fibrosis.
HSP90 expression was increased in (myo)fibroblasts from fibrotic human and mouse lungs compared with controls. 17-AAG inhibited TGF-β1-induced extracellular matrix production and transdifferentiation of lung fibroblasts and epithelial–mesenchymal transition of A549 cells. The antifibrotic effects were associated with TGF-β receptor disruption and inhibition of Smad2/3 activation. Co-immunoprecipitation revealed that HSP90β interacted with TGF-β receptor II and stabilised TGF-β receptors. Furthermore, 17-DMAG improved lung function and decreased fibrosis and matrix metalloproteinase activity in the lungs of bleomycin-challenged mice.
In conclusion, this is the first study to demonstrate that HSP90 inhibition blocks pulmonary fibroblast activation and ameliorates bleomycin-induced pulmonary fibrosis in mice.
Abstract
HSP90 inhibition might be a promising therapy option for PF by blocking exaggerated TGF-β/Smad signalling http://ow.ly/2iTe304ukgL
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and usually fatal lung disease of unknown aetiology, with high morbidity and mortality [1]. Histopathologically, IPF is defined by the usual interstitial pneumonia pattern, which is characterised by spatial and temporal heterogeneity, microscopic honeycombing and hyperplastic alveolar epithelium overlaying fibroblast foci. To date, there are only limited pharmacological options for IPF treatment [2].
In IPF, pulmonary fibrogenesis is believed to develop in response to recurrent alveolar epithelial micro-injuries irrespective of the undefined causes. Myofibroblasts are the key effectors of fibrogenesis, and are characterised by α-smooth muscle actin (SMA) expression and excessive production of extracellular matrix (ECM; mainly collagens) [1, 3]. Active interstitial myofibroblasts are suggested to result from transdifferentiation of resident fibroblasts, epithelial–mesenchymal transition (EMT) of alveolar epithelial cells and differentiation of circulating progenitor cells [4–6].
Overwhelming evidence supports a key role for transforming growth factor (TGF)-β in fibrotic diseases including IPF [7]. Through TGF-β receptors (TGF-βRs), TGF-β activates the canonical TGF-β-Smad signalling pathway and drives resident fibroblast transdifferentiation and EMT [8].
Evidence has been accumulating that heat shock protein (HSP)90 plays a large part in fibrogenesis [9]. For example, studies have shown that the TGF-βR signalling pathway is affected by HSP90 activity in renal fibrosis [10], dermal fibrosis [11] and cancer cell lines [12, 13], but little is known about the significance of HSP90 in IPF. HSP90 is ubiquitously expressed and serves as a molecular chaperone in multiple cellular responses, assisting in the stabilisation or degradation of proteins [14]. HSP90 is often overexpressed in malignant cells and is believed to be essential for the malignant phenotype [14, 15]; consequently, HSP90 inhibitors have been widely investigated as anticancer agents [15] and a proteomic study of IPF lungs revealed a profound upregulation of HSPs including HSP90 [16].
To test the hypothesis that HSP90 contributes to the development of IPF, we studied the effects of selective small molecule inhibition of HSP90 in interstitial lung fibroblasts (ILFBs), in the human alveolar type-II-like cell line A549, and in mice with bleomycin-induced pulmonary fibrosis.
Methods
Detailed methods are described in the online supplementary material.
Human samples
ILFBs were isolated by explant culture of lung tissue as described previously [17]. The study protocol for human tissue donation was approved by the ethics committee of the Justus-Liebig-University School of Medicine (31/93, 29/01 and 111/08: European IPF Registry), and written informed consent was obtained from each individual patient or the patient's next of kin.
Animal model and experimental plan
Adult male 12-week-old C57BL/6 mice (20–25 g body weight) were obtained from Charles River Laboratories (Sulzfeld, Germany). Both the university animal care committee and the Federal Authorities for Animal Research of the Regierungspraesidium Giessen (Hessen, Germany) approved the study protocol (GI20/10, 59/2012). Mice were given bleomycin (3.5 units·kg−1 in saline; Sigma-Aldrich, Munich, Germany) or saline alone via orotracheal instillation. From day 7 to day 21, mice received 17-DMAG (17- dimethylaminoethylamino-17-demethoxygeldanamycin; LC Laboratories, Woburn, MA, USA) 10 mg·kg−1 or 25 mg·kg−1 in saline or saline alone via oral gavage every 2 days.
On day 21, mice were scanned using micro-computed tomography (SkyScan, Kontich, Belgium) followed immediately by fluorescence molecular tomography (FMT; VisEn Medical, Bedford, MA, USA) to assess matrix metalloproteinase (MMP) activity. In addition, physiological measurements were performed on day 21 using the FlexiVent system (SCIREQ, Montreal, Canada), bronchoalveolar lavage fluid (BALF) was collected and lungs were fixed for histology or snap-frozen after euthanisation.
BALF cell count
Cytospins from BALF were stained using May–Gruenwald–Giemsa and inflammatory cells were counted using Leica DM6000 B microscope (Wetzlar, Germany).
Histological evaluation
To examine fibrosis and collagen deposition, mouse lung sections were stained with haematoxylin and eosin (H&E) or sirius red, respectively.
Immunohistochemistry
Lung sections were stained using the ZytoChem-Plus AP Kit or the ZytoChem-Plus HRP-DAB Kit (Zytomed Systems, Berlin, Germany), according to a previously published protocol [16].
Co-immunoprecipitation
Dynabeads Protein G (Life Technologies, Darmstadt, Germany) were incubated with TGF-βRII antibody (Santa Cruz Biotechnology, Heidelberg, Germany), followed by incubation with protein lysates.
Immunofluorescence
Cells were fixed with ice-cold methanol, incubated with primary antibodies overnight at 4°C, and then incubated with DyLight 549-conjugated secondary antibodies (Zytomed Systems) for 1 h at room temperature followed by 4′,6-diamidino-2-phenylindole (DAPI) staining for nuclei.
Collagen assay
Collagen was collected from cells using 0.5 mol·L-1 acetic acid and was assessed using the Sircol collagen assay kit (Biocolor, Carrickfergus, UK) according to the manufacturer's instructions.
In vitro scratch assay
Human ILFBs were seeded on 48-well plates. After serum starvation for 24 h, scratches were created. The images were captured by Leica real-time microscope system at the start of the experiment and 12 h later.
Active TGF-β1 quantification
Active TGF-β1 in mouse plasma and BALF was quantified in duplicates using standard ELISA (R&D Systems, Wiesbaden-Nordenstadt, Germany) according to the manufacturer's instructions.
Statistical analysis
Data are expressed as mean±sem. Statistical analysis between multiple groups was performed using one-way ANOVA with Newman–Keuls correction or t-test between two groups [18]. GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) was used for all statistics. p<0.05 was considered significant.
Results
HSP90 is increased in (myo)fibroblasts from lungs of IPF patients and bleomycin-challenged mice
Immunohistochemistry revealed the presence of HSP90α and HSP90β in the pulmonary interstitia of donor and IPF lungs, but with an increased signal in myofibroblasts and fibroblast foci in the IPF lungs (indicated by α-SMA staining; figure 1a and online supplementary figure S1A). In addition, Western blots showed a significant upregulation of HSP90β in cultured ILFBs (figure 1b) from patients with IPF compared with donors. Strong expression of both HSP90α and β was observed in abnormal bronchiolar structures overlying fibroblast foci (figure 1a and online supplementary figure S1A), as well as in hyperplastic bronchioles of IPF lungs (online supplementary figure S1A), compared with basal expression of both HSP90s in bronchiolar epithelium (online supplementary figure S1B) of donor lungs. Additionally, hyperplastic type-II alveolar epithelial cells (AECII; indicated by arrowheads and prosurfactant-protein C staining) near areas of dense fibrosis indicated robust immunostaining of HSP90β in IPF lungs compared with donor lungs, whereas HSP90α is absent in AECII (online supplementary figure S1B). Vascular smooth muscle cells (VSMCs; indicated by hash symbols and α-SMA staining) and the smooth muscle present in the wall of bronchioles (indicated by asterisks and α-SMA staining) showed expression of HSP90α, but not of HSP90β. In line with immunohistochemistry results, Western blots showed a significant upregulation of both HSP90s in lung homogenate from IPF patients compared with donors (online supplementary figure S1C).

Heat shock protein (HSP)90 is increased in (myo)fibroblasts from lungs of interstitial pulmonary fibrosis (IPF) patients and bleomycin-challenged mice. a) Representative immunohistochemical staining of HSP90α, HSP90β and α-smooth muscle actin (SMA) in serial lung sections obtained from five organ donors and five patients with IPF. Myofibroblasts are indicated by α-SMA staining (arrowheads). Red colouring indicates positive staining. Scale bar=100 μm. b) Representative Western blots of HSP90α and HSP90β in interstitial lung fibroblasts (ILFBs) from donors and patients with IPF, with densitometric quantification. β-actin serves as the loading control. Data are presented as mean±sem of n=3 per group. *: p<0.05 versus donors. c) Representative immunohistochemical staining of HSP90α, HSP90β, thyroid transcription factor (TTF)1 and α-SMA in serial lung sections obtained from three sham controls and three bleomycin-challenged mice (Bleo). Myofibroblasts and type II alveolar epithelial cells are shown by α-SMA (arrowheads) and TTF1 (arrows) staining, respectively. Red colouring indicates positive staining for HSP90α, HSP90β and α-SMA; brown colouring indicates positive staining for TTF1. Scale bar=50 μm. d) Representative Western blots of HSP90α and HSP90β in mouse ILFBs from sham controls and bleomycin-challenged mice (Bleo), with densitometric quantification. β-actin serves as the loading control. Data are presented as mean±sem of n=3 per group. *: p<0.05 versus sham.
Furthermore, serial sectional immunohistochemistry exhibited increased interstitial immunoreactivity of HSP90α and HSP90β in myofibroblasts (indicated by asterisks and α-SMA staining; figure 1c and online supplementary figure S2A) in the lungs of bleomycin-challenged mice compared with sham controls. In addition, a pronounced overexpression of HSP90β (but not HSP90α) was observed in AECII in bleomycin-challenged lungs (indicated by arrows and thyroid transcription factor (TTF)1 staining; online supplementary figure S2A) compared with sham controls. Furthermore, Western blots demonstrated a two-fold increase of both HSP90α and HSP90β in ILFBs (figure 1d) derived from bleomycin-challenged mice compared with sham controls; however, only HSP90β showed significant increase in lung homogenates from bleomycin-challenged mice (online supplementary figure S2B).
17-AAG inhibits TGF-β1-induced ILFB transdifferentiation, collagen production and migration
Having observed the upregulation of HSP90 in ILFBs from fibrotic lungs, we employed 17-AAG (17-allylamino-17-demethoxygeldanamycin), a highly selective HSP90 inhibitor with a median inhibitory concentration of 5 nM in a cell-free assay, to investigate the function of HSP90 in TGF-β1-induced fibrotic responses in human ILFBs. Doses from 50 nM to 2 μM were reported in most of the in vitro studies; nevertheless, we determined to use 50 nM 17-AAG based on pilot studies (online supplementary figure S3). Western blots revealed that collagen I, fibronectin and α-SMA were all increased in ILFBs by stimulation with TGF-β1, indicating fibroblast transdifferentiation and excessive matrix protein production. This response was stronger in IPF ILFBs than in donor ILFBs. The effects of TGF-β1 were blocked by co-administration of 17-AAG (figure 2a). Moreover, extracellular collagen deposition examined by Sircol collagen assay was elevated two- and four-fold by TGF-β1 in donor ILFBs and IPF ILFBs, respectively, and this increase was prevented by 17-AAG (figure 2b). Next, we sought to determine the effect of 17-AAG on human ILFB migration (using in vitro scratch assay). Numbers of migrated cells to the scratch after TGF-β1 exposure were significantly reduced by 17-AAG (50 nM) treatment for both donor and IPF ILFBs (figure 2c). In parallel with the scratch assay, bromodeoxyuridine assay indicated that 17-AAG (50 nM) had no effect on ILFB proliferation (online supplementary figure S4). Taken together, these results suggest that 17-AAG suppresses fibroblast transdifferentiation and motility.

17-AAG (17-allylamino-17-demethoxygeldanamycin) inhibits transforming growth factor (TGF)-β1-induced interstitial lung fibroblast (ILFB) transdifferentiation, collagen production and migration. a) Analysis of protein expression of collagen I, fibronectin and α-smooth muscle actin (SMA) by immunoblotting with densitometric quantification using β-actin as the loading control. The level of protein expression is presented as the fold change from the level in the unstimulated cells, which is presented as control. *: p<0.05; **: p<0.01 versus control; #: p<0.05 versus TGF-β1. b) Extracellular collagen production measured using Sircol soluble collagen assay. Collagen content is expressed as a percentage of collagen content in the control group. ***: p<0.001 versus control; ###: p<0.001 versus TGF-β1. c) TGF-β1-induced ILFB migration by in vitro scratch assay. Migrated cells were first visualised by phase-contrast and 4′,6-diamidino-2-phenylindole (blue) using the Leica real-time microscope. Representative images from one donor and one idiopathic pulmonary fibrosis (IPF) patient are shown, with red lines indicating the initial edges of the scratch. *: p<0.05; ***: p<0.001 versus control; #: p<0.05, ###: p<0.001 versus TGF-β1. ILFBs were stimulated using recombinant TGF-β1 (10 ng·mL−1) for 24 h (Western blots in (a)), 72 h (Sircol collagen assay in (b)) and 12 h (in vitro scratch assay in (c)), in the absence or presence of 17-AAG (50 nM). All panels represent results from cells of three different donors and three different patients with IPF, and all data are presented as the mean±sem of n=3 per group.
17-AAG blocks TGF-β/Smad signalling in ILFBs
Next, we questioned whether HSP90 regulates TGF-βR levels in human ILFBs. TGF-β1 led to increased levels of TGF-βRI and TGF-βRII in IPF ILFBs, both of which were reduced to control levels by co-administration of 17-AAG (figure 3a). However, donor ILFBs showed no decrease of both TGF-βRs when treated with 17-AAG (figure 3a). Nevertheless, TGF-β1-induced Smad2/3 activation indicated by Smad2 phosphorylation was blocked by 17-AAG in both donor and IPF ILFBs (figure 3a). Additionally, HSP90 expression in human ILFBs was not sensitive to TGF stimulation nor 17-AAG treatment (figure 3a).

17-AAG (17-allylamino-17-demethoxygeldanamycin) blocks transforming growth factor (TGF)-β/Smad signalling in interstitial lung fibroblasts (ILFBs) by diminishing TGF-β receptor (TGF-βR) levels via proteasome degradation, and heat shock protein (HSP)90 forms a protein complex with TGF-βRII. a) Western blot analysis of TGF-βRI, TGF-βRII, phosphor(p)-Smad2 (phospho-Ser465, phospho-Ser467), Smad2, HSP90α and HSP90β. β-actin serves as the loading control. The level of protein expression is shown as the fold change from the level in control group. All panels represent results from cells of three different donors and three different patients with idiopathic pulmonary fibrosis (IPF). Data are presented as mean±sem of n=3 per group. *: p<0.05; **: p<0.01; ***: p<0.001 versus control; #: p<0.05; ##: p<0.01; ###: p<0.001 versus TGF-β1. b)Representative Western blot analysis of TGF-βRI, TGF-βRII), p-Smad2 and Smad2. β-actin serves as the loading control. The experiment was repeated independently three times in one donor and one IPF patient. c) Western blots of HSP90β and TGF-βRII after protein lysate from donor and IPF ILFBs was immunoprecipitated (IP) with anti-TGF-βRII antibody or IgG antibody. The experiment was repeated independently three times; representative blots are shown. After 24 h of serum starvation, ILFBs were stimulated using recombinant TGF-β1 (10 ng·mL−1) for 24 h (Western blots and co-immunoprecipitation) in the absence or presence of 17-AAG. For MG-132, ILFBs were treated with 17-AAG (50 nM) in the absence or presence of MG-132 (30 μM) for 5 h, followed by 1 h stimulation with TGF-β1.
More interestingly, 17-AAG-induced reduction of TGF-βRs and Smad2 phosphorylation was restored by MG-132, a proteasome inhibitor (figure 3b). Furthermore, endogenous co-immunoprecipitation showed that HSP90β (figure 3c), but not HSP90α (online supplementary figure S5) co-immunoprecipitated with TGF-βRII, indicating that HSP90β and TGF-βRII are part of the same protein complex. More importantly, the complex formation of TGF-βRII and HSP90β was enhanced by TGF-β1 and disrupted by 17-AAG in IPF fibroblasts. Taken together, the result suggests that HSP90 regulates TGF-β/Smad signalling by stabilising TGF-βRs via forming a complex with TGF-βRII and 17-AAG disrupts the stability of TGF-βRs and promotes their proteasome degradation.
17-AAG prevents TGF-β1-induced EMT of A549 cells
In addition, we directed our study toward the effect of 17-AAG on TGF-β1-induced EMT of the A549 cell line (human AECIIs). Control A549 cells exhibited a cubic cell shape in cluster formation (epithelial phenotype); administration of TGF-β1 caused the cells to develop an individualised, elongated spindle-shape, whereas cells exposed to TGF-β1 and concomitant 17-AAG retained the epithelial phenotype (figure 4a). Additionally, immunofluorescence staining (figure 4a) and Western blotting (figure 4b) demonstrated that TGF-β1 exposure led to elevation of the mesenchymal marker α-SMA and fibronectin, and reduction of the epithelial marker E-cadherin in A549 cells, indicating an ongoing EMT event. The shift of markers induced by TGF-β1 was prevented by co-administration of 17-AAG, concomitant with decreased TGF-βRII expression and Smad2 phosphorylation (figure 4b). HSP90 expression in A549 cells was not sensitive to TGF stimulation nor to 17-AAG treatment (figure 4b).

17-AAG (17-allylamino-17-demethoxygeldanamycin) prevents transforming growth factor (TGF)-β1-induced epithelial–mesenchymal transition of A549 cells. a) Representative images of A549 cell morphology examined using i) phase contrast microscopy under 20× magnification and representative immunofluorescence staining of ii) E-cadherin, iii) fibronectin and iv) α-smooth muscle actin (SMA) with 4′,6-diamidino-2-phenylindole (blue) counter-staining for nuclei. Scale bars=20 μm. b) Representative Western blots of E-cadherin, fibronectin, α-SMA, TGF-β receptor II (TGF-βRII), phosphor(p)-Smad2 (phospho-Ser465, phospho-Ser467), Smad2, heat shock protein (HSP)90α and HSP90β, with β-actin as the loading control. A549 cells were stimulated using TGF-β1 (5 ng·mL−1) for 72 h in the absence or presence of 17-AAG (50 nM) or with 17-AAG alone after 24 h of serum starvation. Data are presented as mean±sem of n=3 per group. The experiment was repeated independently three times.
17-DMAG improves physiological parameters and suppresses inflammation in mice with bleomycin-induced pulmonary fibrosis in vivo
Based on the fact that HSP90 inhibition with 17-AAG blocks the fibrotic response to TGF-β in vitro, we performed in vivo studies with 17-DMAG (received from day 7 to day 21), a water-soluble HSP90 inhibitor, in mice with bleomycin-induced pulmonary fibrosis. Bleomycin causes significant loss of body weight (up to 20%) 1 week after instillation. The body weight gain of 17-DMAG-treated mice was effectively improved compared with bleomycin-challenged mice (figure 5a). Moreover, lung function assessment demonstrated a significant impairment of lung compliance in bleomycin-challenged mice compared with sham controls (0.025±0.006 mL·cm−1 H2O versus 0.035±0.005 mL·cm−1 H2O), which was dose-dependently ameliorated by 17-DMAG (0.028±0.007 mL·cm−1 H2O in the 10 mg·kg−1 group and 0.032±0.005 mL·cm−1 H2O in the 25 mg·kg−1 group) (figure 5b).
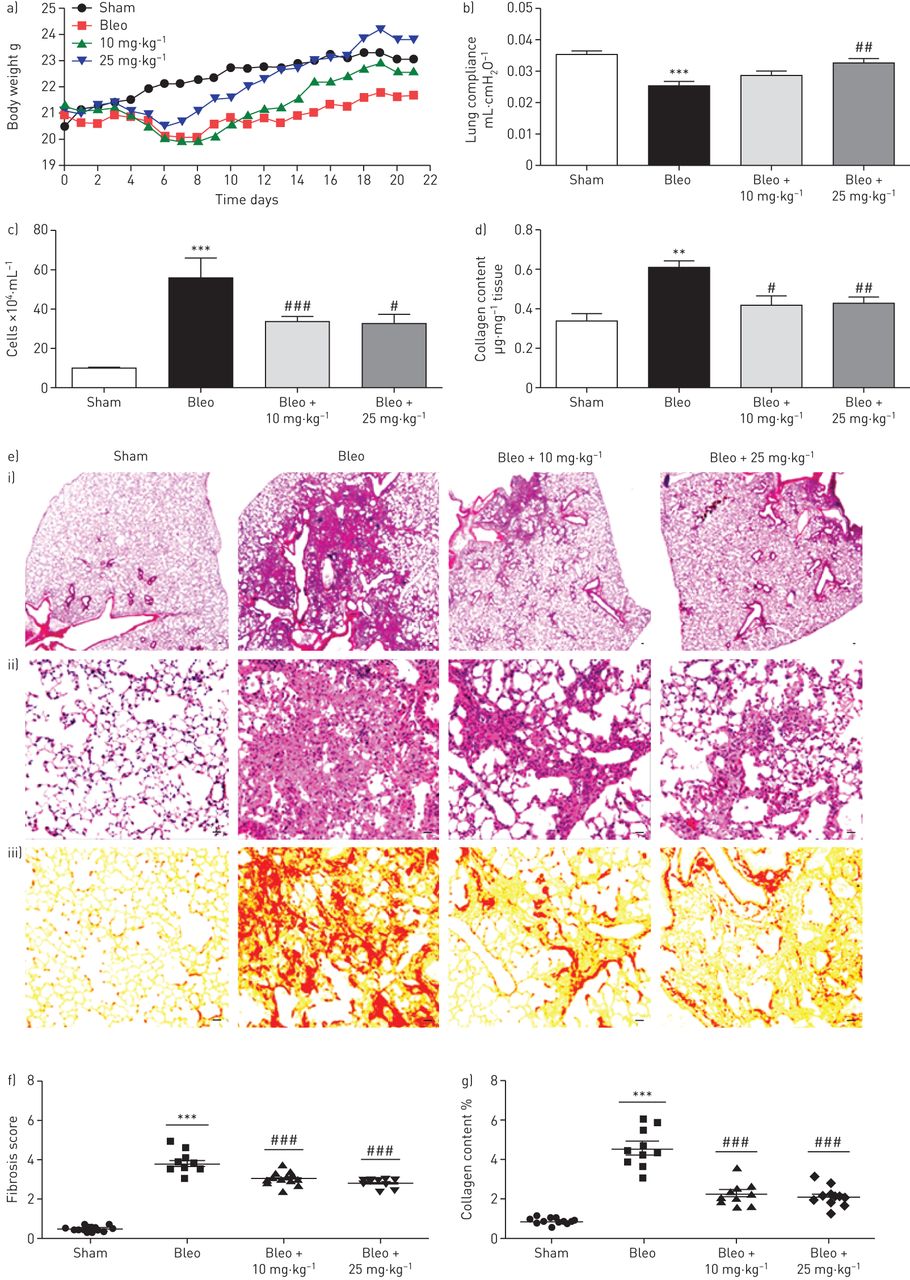
17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin) in vivo improves lung function and ameliorates pulmonary fibrosis in bleomycin-challenged mice. a) Body weight curve monitored throughout the whole experiment. b) Lung compliance measured at day 21 after instillation of bleomycin (Bleo) or saline (Sham), in the absence or presence of 17-DMAG treatment (10 mg·kg−1 or 25 mg·kg−1 every 2 days from day 7 to day 21). Data are presented as mean±sem of n=13–14 mice per group. c) Infiltrated inflammatory cells in bronchoalveolar lavage fluid (BALF) were counted. Data are presented as mean±sem of n=5–8 mice. d) Soluble collagen content measured using Sircol collagen assay. Data are presented as mean±sem of n=4–7 mice. e) Histological images of lung paraffin sections with i, ii) haematoxylin and eosin (H&E) staining for pulmonary fibrosis and iii) sirius red staining for collagen deposition. Scale bars=20 μm. f) Fibrosis score (based on H&E staining) expressed on a numerical scale ranging from 0 (healthy) to 6; and g) collagen content (based on sirius red staining) expressed as a percentage of the total area of the lung section. Data are presented as mean±sem of n=10 mice per group. **: p<0.01, ***: p<0.001 versus sham; #: p<0.05, ##: p<0.01, ###: p<0.001 versus bleo. All analyses were performed blind.
In addition, we analysed the BALF collected on day 21 and observed increased total number of inflammatory cells in bleomycin-challenged mice compared with sham mice (55.44±9.97×104 cells·mL−1 versus 8.96±0.509×104 cells·mL−1). 17-DMAG partially restored the inflammatory cell number in BALF (33.10±2.745×104 cells·mL−1 in the 10 mg·kg−1 group and 32.03±5.25×104 cells·mL−1 in the 25 mg·kg−1 group) (figure 5c), suggesting a favourable effect of HSP90 inhibition on sustained pulmonary inflammation in bleomycin-challenged mice.
17-DMAG decreases fibrosis scores and collagen deposition in bleomycin-challenged mice
To understand the role of HSP90 in lung parenchyma remodelling, a comprehensive histological evaluation was performed. H&E staining revealed a significant distortion of bleomycin-challenged lungs, while 17-DMAG partially restored the lung architecture (figure 5e). The fibrosis score demonstrated a moderate degree of fibrosis in bleomycin-challenged mice compared with sham controls (3.83±0.56 versus 0.50±0.14) (figure 5e and f). 17-DMAG dose-dependently decreased fibrosis scores (3.06±0.38 in the 10 mg·kg−1 group and 2.85±0.25 in the 25 mg·kg−1 group). Furthermore, we examined collagen deposition in vivo using sirius red staining. Bleomycin-challenged mice exhibited excessive collagen deposition compared with sham controls (4.64±0.56% versus 0.94±0.19%), whereas 17-DMAG treatment narrowed the collagen areas (2.33±0.58% in the 10 mg·kg−1 group and 2.19±0.54% in the 25 mg·kg−1 group) (figure 5e and g). In addition, the data from morphometric measurement of collagen was confirmed by quantifying soluble collagen content in the lung homogenates (figure 5d).
17-DMAG reduces TGF-βRII expression in lungs and TGF-β1 level in plasma and BALF of bleomycin-challenged mice
Having demonstrated that TGF-βRII expression was abrogated by 17-AAG in vitro, we aimed to determine whether HSP90 inhibition results in reduction of pulmonary TGF-βRII expression in vivo, using comparative immunohistochemistry for TGF-βRII on lung tissue sections. We observed a significant overexpression and upregulation of TGF-βRII in hyperplastic AECII near areas of dense fibrotic remodelling (marked by arrows in figure 6b and asterisks in online supplementary figure S6B), as well as in α-SMA-positive myofibroblastic cells in areas of dense fibrotic remodelling (marked by arrowheads in figure 6a and asterisks in online supplementary figure S6a) in lungs of bleomycin-challenged mice, as compared to sham mice. Importantly, lungs from bleomycin-challenged mice treated with 17-DMAG presented neither significant immunostaining for TGF-βRII in hyperplastic AECII nor in the interstitium. Interestingly, TGF-βRII expression in VSMCs was generally present in lungs from all mice regardless of the 17-DMAG treatment (figure 6a and online supplementary figure S6A). Furthermore, Western blots of the mouse lung homogenates (figure 6c) confirmed that 17-DMAG treatment in vivo resulted in a significant reduction of TGF-βRII in fibrotic lungs induced by bleomycin.

17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin) decreases transforming growth factor-β receptor II (TGF-βRII) expression in lung and reduces the level of TGF-β1 in plasma and bronchoalveolar lavage fluid (BALF) of bleomycin-challenged mice. a) Representative immunohistochemistry for α-smooth muscle actin (SMA) (marker for myofibroblasts and smooth muscle cells) and TGF-βRII in serial lung tissue sections of bleomycin (bleo)-challenged mice, 17-DMAG-treated bleomycin-challenged mice and sham-treated mice. In bleomycin-injured lungs, TGF-βRII was expressed in vascular smooth muscle cells (VSMCs), in α-SMA-positive myofibroblasts in areas of dense fibrosis (arrowheads), as well as in bronchial epithelial (BE) cells. Treatment of bleomycin-challenged mice with 25 mg·kg−1 17-DMAG from day 7 to day 21 resulted in marked reduction of TGF-βRII expression in the interstitium (arrows), as well as in BE cells, but not in VSMCs of lungs from treated mice. Finally, TGF-βRII was generally expressed in VSMCs and BE in lungs of sham-treated mice. b) Representative immunohistochemistry for proSP-C (marker for type II alveolar epithelial cells (AECII)) and TGF-βRII in serial lung tissue sections of bleomycin-challenged mice, 17-DMAG-treated bleomycin-challenged mice and sham mice. Hyperplastic AECII near areas of dense fibrotic tissue indicated robust overexpression of TGF-βRII (arrows) in comparison with normal AECII of healthy mice lungs (sham), which did not reveal significant expression of TGF-β RII (arrows). Of note, hyperplastic AECII in lungs of 17-DMAG-treated bleomycin-challenged mice (with marked reduction of fibrotic tissue) indicated no significant immunostaining for TGF-βRII (arrows). Strong immunostaining for TGF-βRII was also observed in the “fibrotic interstitium” of bleomycin-mice, whereas 17-DMAG-treated bleomycin-challenged mice revealed significant reduction of dense fibrotic tissue. Scale bars=50 μm. c) Representative Western blot analysis of TGF-βRII in mouse lung homogenates from sham and bleomycin-challenged mice with/without 17-DMAG treatment. GAPDH serves as the loading control. Data are presented as mean±sem of n=3 mice. Active TGF-β1 level was measured using ELISA in BALF d) and plasma e) obtained from sham- and bleomycin-challenged mice with and without 17-DMAG treatment. Data are presented as mean±sem of n=5–7 mice per group. *: p<0.05, **: p<0.01 versus sham; #: p<0.05 versus bleo; ##: p<0.001 versus bleo.
As the ligand of TGF-βRs, active TGF-β1 level in BALF and plasma was elevated in bleomycin-challenged mice compared with sham mice (213.60±54.08 pg·mL−1 versus 28.02±20.17 pg·mL−1 in BALF and 14.84±3.407 ng·mL−1 versus 6.99±2.06 ng·mL−1 in plasma); however, reversed TGF-β1 level was only observed in the lower dose group (79.36±2.73 pg·mL−1 in the 10 mg·kg−1 group versus 243.6±42.5 pg·mL−1 in the 25 mg·kg−1 group in BALF and 6.06±0.928 ng·mL−1 in the 10 mg·kg−1 group versus 14.03±1.542ng·mL−1 in the 25 mg·kg−1 group in plasma, figure 6d and e).
17-DMAG suppresses MMP activity in bleomycin-challenged lungs
MMP proteolytic activity is reported to reflect the severity of pulmonary fibrosis, and patients with IPF show elevated MMP levels in BALF [19, 20]. To monitor MMP activity in vivo, we introduced a MMP-responsive probe into mice and measured the signal using FMT. MMP activity was increased in lungs from bleomycin-challenged mice compared with sham controls (4.67±1.22 pmol versus 0.54±0.24 pmol). Mice treated with 17-DMAG after the bleomycin challenge showed lower MMP activity (2.11±1.44 pmol in the 10 mg·kg−1 group and 0.99±0.66 pmol in the 25 mg·kg−1 group) (figure 7a–c).

17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin) suppresses pulmonary matrix metalloproteinase (MMP) activity in bleomycin-challenged mice. MMP activity in mouse lungs was measured in vivo using quantitative fluorescence molecular tomography (FMT) at day 21 after instillation of bleomycin (bleo) or saline (sham), in the absence or presence of 17-DMAG treatment (10 mg·kg−1 or 25 mg·kg−1 every 2 days from day 8 to day 21). The MMP-activatable fluorescent probe, MMPSense680 was injected via the tail vein 24 h before the FMT measurement. a) Representative images of MMP activity in mouse lungs; b) virtual three-dimensional rendering of skeleton, lungs and MMP activity in bleomycin-challenged lung acquired using FMT in combination with microcomputed tomography; c) quantitative FMT measurement of MMP activity in mouse lungs. Data are presented as mean±sem of n=5 mice. ***: p<0.001 versus sham; ###: p<0.001 versus bleo.
Discussion
To our knowledge, this study provides for the first time direct experimental proof that HSP90 plays a pathological role in pulmonary fibrosis. HSP90 inhibition results in antifibrotic effects in vitro by preventing myofibroblast transdifferentiation from both fibroblasts and AECs. Furthermore, treatment with a HSP90 inhibitor reduces TGF-βRII expression in fibrotic lungs and attenuates bleomycin-induced pulmonary fibrosis in mice.
It is well documented that TGF-β is a master switch in the initiation and progression of IPF [8, 21, 22]; therefore, inhibition of aberrant TGF-β signalling has long been pursued as a potential strategy for the treatment of IPF, among other fibrotic diseases. Despite intensive investigations in preclinical studies or in clinical trials, attempts to block TGF-β directly using either TGF-β neutralising antibodies [23, 24] or TGF-βR kinase inhibitors [25] have thus far been ineffective or are still in the experimental or early clinical stage. To overcome this challenge, it is of great interest to identify other mediators of TGF-β signalling that can be targeted pharmacologically. Based on the results of our study, we propose HSP90 as such a mediator and HSP90 inhibition as a novel therapeutic approach for IPF.
To better understand the pathophysiology of IPF, many studies have tried to identify differences in the transcriptome [26, 27] or proteome [16] of healthy lungs and IPF lungs. Herein, we show higher protein expression of both HSP90α and HSP90β in ILFBs derived from patients with IPF compared with donors. This finding is in line with a previous proteomic study reporting elevated HSP90α and HSP90β in IPF lungs, along with other stress-related proteins [16]. HSP90 chaperone machinery maintains homeostasis of a certain set of client proteins either by assisting protein folding and maturation or by stabilising protein complexes [14, 28, 29]. HSP90 has a broad range of pathophysiological functions depending on cell types and stresses. However, there is no available report on the role of HSP90 in IPF. By using TGF-β1-induced primary ILFB activation as an in vitro model, we show that the HSP90 inhibitor 17-AAG prevents TGF-β1-induced transdifferentiation, collagen production and migration of pulmonary interstitial fibroblasts. Similarly, in a study on systemic sclerosis, 17-DMAG was shown to block TGF-β1-induced mRNA expression of collagen and fibronectin in skin fibroblasts [11]. Moreover, HSP90 inhibition suppresses tumour growth and metastasis by abrogating mobility [30]. These findings are in support of the possibility that HSP90 plays a critical role in sensitising the interstitial fibroblasts to transdifferentiation in response to TGF-β during the development of pulmonary fibrosis.
Based on the close association of HSP90 with TGF-β and the role of TGF-β in EMT, we postulated that 17-AAG may prevent TGF-β1-induced EMT. Our results confirmed this theory, that 17-AAG reversed the switch from epithelial to mesenchymal markers induced by TGF-β1 in A549 AECII, at the expense of TGF-βRII levels and Smad2 activation. Another heat shock protein, HSP27, was recently reported to contribute to EMT in IPF, and genetic blockade of HSP27 prevented pulmonary fibrosis in a TGF-β-overexpression rat model [31].
Although HSP90 is essential for the viability of healthy cells, cancer cells tend to have a higher level of HSP90-dependent clients and therefore a higher required level of HSP90 activity than normal tissues, which may provide a favourable therapeutic window [9, 14]. This may also hold true for fibrotic diseases. In either fibroblast activation or EMT, no noticeable change of HSP90 expression after 17-AAG treatment in vitro was observed, suggesting that HSP90 inhibitor blocks the chaperone ability of HSP90 rather than inhibiting HSP90 expression [32]. We found that the antifibrotic effects of HSP90 inhibition were a consequence of significant depletion of TGF-βRs on the post-translational level, but not on the transcriptional level (mRNA data not shown), followed by decreased Smad2/3 activation. Indeed, our results demonstrate that the response to TGF-β1 in terms of fibroblast activation and ECM production was greater in ILFBs derived from patients with IPF than in ILFBs from donors. However, HSP90 inhibition shows similar magnitude of effect on cells from both sources, suggesting that HSP90 is accountable for the higher sensitivity of the canonical TGF-β/Smad pathway in ILFBs derived from patients with IPF compared with donor-derived cells. Furthermore, endogenous co-immunoprecipitation proves that TGF-βRII is in the same protein complex with HSP90β, but not HSP90α. TGF-β1 enhances co-immunoprecipitation of TGF-βRII and HSP90β in ILFBs derived from IPF patients, whereas HSP90 inhibition disrupts the co-immunoprecipitation, indicating that TGF-βRII is a client of HSP90β and can be stabilised by HSP90β. The result with MG-132, a proteasome inhibitor, indicates that the compromised complex formation of HSP90 and TGF-βRII leads to proteasome degradation of TGF-βRs. In agreement with our findings, recent studies using epithelial cell lines have demonstrated an interaction of TGF-βRs with HSP90 [12, 13]. Additionally, the investigators showed that the HSP90 inhibitor 17-AAG disrupts this interaction and recruits Smurf2 to TGF-βRs which promotes TGF-βR ubiquitination and subsequent degradation [12].
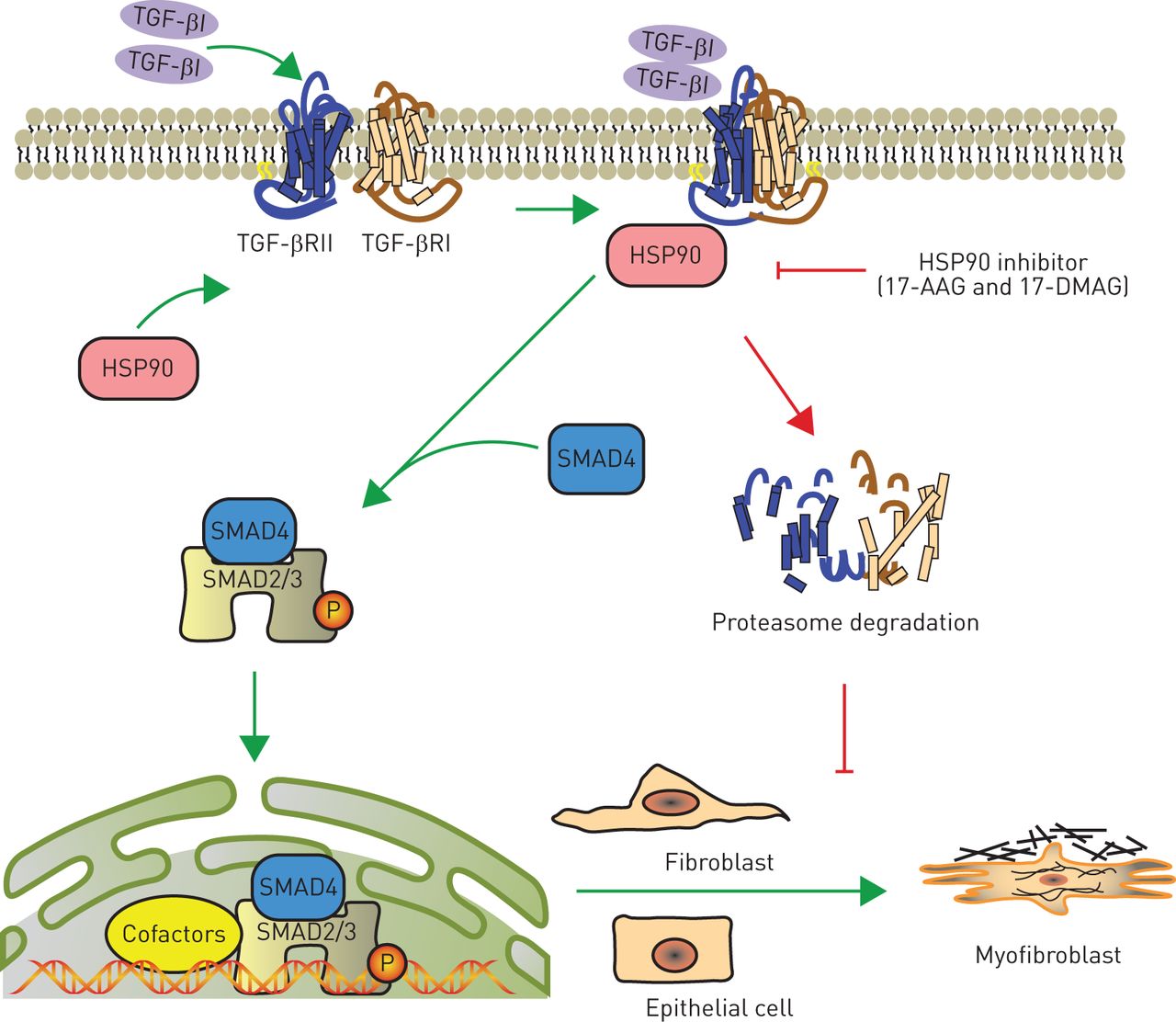
We summarise our working hypothesis in figure 8, in which HSP90 protects TGF-βRs from degradation in proteasomes and therefore stabilises the complex of TGF-β1 and TGF-βRs on the cell membrane. Consequently, Smad2/3 is constitutively activated and translocates to the nucleus to switch on transcription of either profibrotic genes in fibroblasts or mesenchymal markers in epithelial cells. The phenotypic switch to myofibroblast-like cells is responsible for the excessive ECM accumulation in IPF.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram for the proposed mechanism by which heat shock protein (HSP)90 promotes pulmonary fibrosis. Excessive transforming growth factor (TGF)-β1, a highly profibrotic cytokine, binds to TGF-β receptor II (TGF-βRII) and forms a complex together with TGF-βRI, which is stabilised by HSP90 interaction. The activated TGF-βRI kinase phosphorylates Smad2/3, which subsequently recruits Smad4 and translocates into the nucleus to initiate expression of fibrotic genes. HSP90 inhibition (e.g. with 17-allylamino-17-demethoxygeldanamycin (17-AAG) or 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG)) prevents the formation of HSP90/TGF-βR complexes, and thus promotes the degradation of TGF-βRs in proteasomes. This terminates the TGF-β signalling pathway and blocks the transdifferentiation of myofibroblasts from fibroblasts or epithelial cells in the lung.
Based on earlier evidence from others and our present in vitro results, it is reasonable to speculate that HSP90 may be more than a cancer target; it may also be a therapeutic target for IPF. Consistent with expression patterns in human ILFBs, we observed increased HSP90 expression in ILFBs from bleomycin-challenged mice compared with sham controls. The small molecule HSP90 inhibitor 17-DMAG, an analogue of 17-AAG, is water-soluble and displays high oral bioavailability and low toxicity [33], which make it desirable for therapeutic purposes. In the bleomycin-challenged mouse model, we demonstrated for the first time that therapeutic dosing of 17-DMAG (10 mg·kg−1 or 25 mg·kg−1 every 2 days) can attenuate experimental pulmonary fibrosis and improve lung function. Furthermore, the therapeutic effects are probably due to the loss of TGF-βRII exclusively in the mesenchymal cell populations of bleomycin-challenged lungs.
MMP is a key element in tissue repair, cell invasion and epithelial basement membrane destruction [19, 34]. Several studies have suggested that there is an increase in MMPs, rather than a loss of MMPs, in IPF [20]. In this context, we examined the total MMP activity in bleomycin-challenged lungs using FMT imaging, a state-of-art in vivo imaging technology, and observed significantly reduced MMP activity in 17-DMAG treated lungs compared with control lungs.
The bleomycin-induced pulmonary fibrosis model is initiated by a severe acute injury and inflammatory response, followed by chronic inflammation and fibrosis [35]. An increased total number of inflammatory cells in BALF was observed 21 days after bleomycin challenge, which was consistent with studies demonstrating a sustained inflammation up to day 28 [36, 37]. 17-DMAG partially restored the inflammatory cell number in BALF, suggesting a favourable effect of HSP90 inhibition on sustained pulmonary inflammation in bleomycin-challenged mice. Several studies have shown that HSP90 inhibition suppressed acute lung inflammation induced by endotoxin [38, 39]. Nevertheless, we avoid further speculation about whether the pronounced antifibrotic effects observed in the bleomycin model may be partially due to the influence of pulmonary inflammation without experimental evidence.
Data from the experimental animal model with the support of the in vitro data focusing on canonical TGF-β/Smad signalling are intriguing; however, we also recognise two limitations of this study. First, given that fact that HSP90 regulates a wide range of proteins, further studies following other possible targets are required to better understand the involvement of HSP90 in IPF. Second, because epithelial injury is one of the first events leading to pulmonary fibrosis, it is extremely appealing to explore whether HSP90s modulate apoptosis of alveolar epithelial cells at the onset of the disease, for example by applying HSP90 inhibitor to mice in a preventive manner. We consider it to be beyond the scope of the present study, but regard this study as a “gate-opener” which invites further investigation of HSP90's contribution to IPF, including but not limited to the initial epithelial injury and the following interstitial fibrogenesis.
In conclusion, we describe for the first time, to our knowledge, a pathological role of HSP90 in pulmonary fibrosis (via manipulation of TGF-β-Smad2/3 signalling) in vitro and in vivo. The clinical implications of the findings are profound: the profibrotic phenotype features are sensitised by HSP90 machinery in pulmonary fibrosis, which may present therapeutic advantages of HSP90 inhibitors in treating IPF.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figures ERJ-01941-2015_figures_online_supplement
Supplementary material ERJ-01941-2015_supplement_information
Acknowledgements
We acknowledge Christina Vroom (Universities of Giessen & Marburg Lung Center, Giessen, Germany) for her valuable technical assistance with the animal experiments.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: None declared.
- Received November 20, 2015.
- Accepted September 17, 2016.
- Copyright ©ERS 2017
References










