


Abstract
Dasatinib was approved in 2006 for the treatment of imatinib-resistant chronic myelogenous leukemia and functions primarily through the inhibition of BCR-ABL and Src kinase. Dasatinib is extensively metabolized in humans by CYP3A4. In this study, we report that the bioactivation of dasatinib by CYP3A4 proceeds through a reactive intermediate that leads to CYP3A4 inactivation with KI = 6.3 μM and kinact = 0.034 min–1. The major mechanism of inactivation proceeds through hydroxylation at the para-position of the 2-chloro-6-methylphenyl ring followed by further oxidation, forming a reactive quinone-imine, similar to the reactive intermediates formed by acetaminophen and diclofenac. Formation of a reactive imine-methide was also detected but appears to be a minor pathway. When glutathione was added to human liver microsomal incubations, dasatinib-glutathione adducts were detected. Numerous dasatinib analogs were synthesized in an effort to understand what modifications would block the formation of reactive intermediates during dasatinib metabolism. It is interesting to note that blocking the site of hydroxylation with a methyl group was not effective because a reactive imine-methide was formed, nor was blocking the site with fluorine because the fluorine was removed through an oxidative defluorination mechanism and the reactive quinone-imine was still formed. Numerous analogs are presented that did effectively block the formation of glutathione adducts and prevent the inactivation of CYP3A4.
Chronic myeloid leukemia (CML) is associated with a cytogenic abnormality referred to as the Philadelphia chromosome, resulting from the translocation of the c-abl oncogene from chromosome 9 with the breakpoint cluster region (bcr) on chromosome 22 (de Klein et al., 1982; Shtivelman et al., 1985). This results in the generation of the bcr-abl fusion gene, which when translated yields a constitutively active form of abl kinase referred to as BCR-ABL, leading to increased proliferation and survival of myeloid progenitor cells (Calabretta and Perrotti, 2004). Imatinib, a kinase inhibitor targeting BCR-ABL, was first introduced in phase 1 trials in mid-1998 and was a breakthrough in the treatment of CML, greatly increasing patient life expectancy (Druker et al., 2001). Despite the impressive results of imatinib treatment, approximately 30% of patients receiving imatinib as first-line therapy will discontinue treatment by 5 years because of disease resistance or drug toxicity (Druker et al., 2006). The major driver of imatinib resistance is point mutations in the BCR-ABL gene (Deininger et al., 2005), which decrease or eliminate imatinib efficacy.
Dasatinib was approved by the U.S. Food and Drug Administration in June 2006; for the treatment of imatinib-resistant acute myeloid leukemia. Because of the absence of an effective alternative treatment for imatinib-resistant CML, dasatinib was granted accelerated approval before the completion of phase III clinical trials. Dasatinib is structurally diverse from imatinib and is a highly potent inhibitor of BCR-ABL with biochemical potency between 0.1 and 3 nM for BCR-ABL and most of the common BCR-ABL mutations (Gambacorti-Passerini et al., 2005; O'Hare et al., 2005).
Dasatinib has five primary phase I metabolites. Three of these are catalyzed by CYP3A4: hydroxylation at the para-position of the chloromethylphenyl ring (major metabolite), hydroxylation of the C5-methyl of the chloromethylphenyl ring, and N-dealkylation of the hydroxyethyl moiety. Flavin-containing monooxygenase can catalyze the formation of an N-oxide on the piperazine ring, and an unidentified cytosolic oxidoreductase converts the hydroxyethyl group to an acid. Additional secondary metabolites were detected as phase II metabolites due to sulfation or glucuronidation (Christopher et al., 2008a,b; Kamath et al., 2008; Wang et al., 2008).
The product information sheet for dasatinib states that hepatotoxicity is a possible side effect of treatment, and dasatinib is a possible mechanism-based inactivator of CYP3A4; however, no details are given, and there is nothing in the literature to allow quantitation of the risk. Details of a case of dasatinib-induced acute hepatitis published in July 2008 (Bonvin et al., 2008) were similar to reports of immune-mediated idiosyncratic hepatotoxicity for compounds with similar functional groups such as diclofenac (Schapira et al., 1986). An additional report of dasatinib-induced lupus was reported in August 2008 and was concluded to be immune-mediated (Rea et al., 2008). Evaluation of the structure of dasatinib and the known CYP3A4-catalyzed oxidations on the ortho-methyl and the para-position of the chloromethylphenyl ring would make the formation of reactive quinone-imine and imine-methide intermediates plausible. Quinone-imine and imine-methides are reactive electrophiles that may adduct to cysteine and lysine residues of proteins and may act as haptens and initiate immunological responses (Pirmohamed et al., 2002).

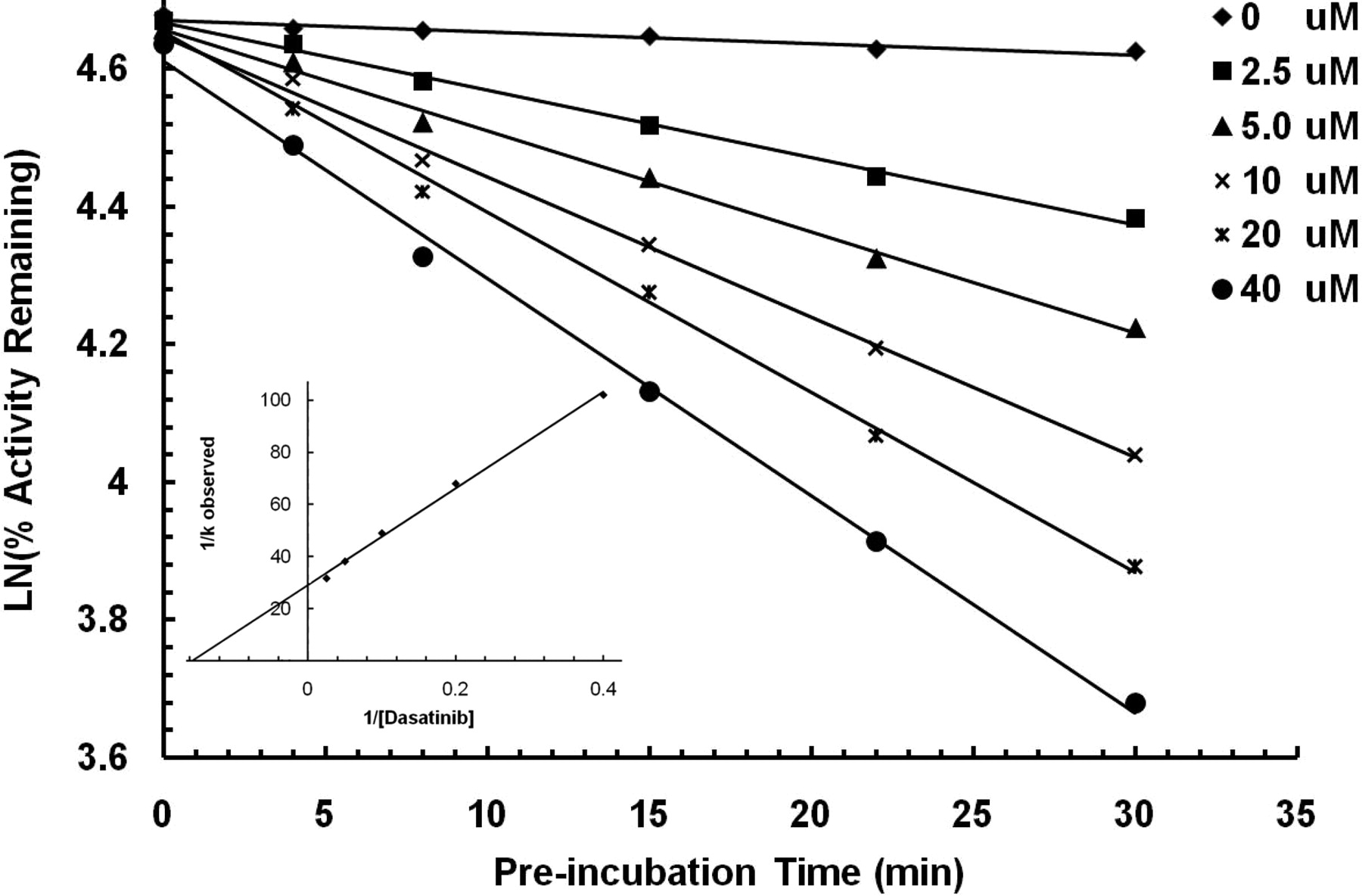
Time- and concentration-dependent inactivation of CYP3A4 by dasatinib. Six concentrations of dasatinib (0, 2.5, 5, 10, 20, and 40 μM) were incubated with human liver microsomes. Aliquots were removed and assayed for remaining CYP3A4 activity at various time points. Each point represents the mean from three separate replicates that do not differ by more than 10%. The remaining activity versus incubation time was plotted, and the slopes for the individual dasatinib concentrations were fit to a Kitz-Wilson plot (inset). The calculated KI and kinact for CYP3A4 inactivation were 6.3 μM and 0.034 min–1, respectively.
Materials and Methods
Chemicals Used. Midazolam, carbamazepine, ketoconazole, and glutathione were purchased from Sigma-Aldrich (St. Louis, MO). Dasatinib and its structural analogs were synthesized at Scripps Florida. All solvents used for LC-mass spectrometry were of chromatographic grade. HLM (pooled) were purchased from XenoTech, LLC (Lenexa, KS). All solutions were prepared from Milli-Q-treated water with a specific resistance ≥17.8 MΩ.
Synthesis of Dasatinib Analogs. Dasatinib was synthesized using the published method of Lombardo et al. (2004) in five linear steps beginning with the condensation of the lithium anion of 2-chlorothiazole with 2-chloro-6-methylphenyl isocyanate. A modified method was used for the synthesis of dasatinib analogs. The analogs bearing the (hydroxyethyl)piperazine group were synthesized in two linear steps beginning with the coupling of 2-bromothiazole-5-carboxylic acid and the corresponding anilines followed by a Buchwald-Hartwig amination reaction with 2-(4-(6-amino-2-methylpyrimidin-4-yl)piperazin-1-yl)ethanol. The analogs 984, 1920, and 1235 were synthesized using similar methods.
Human Liver Microsomal Incubations: CYP3A4 Inactivation. The CYP3A4 activity in HLM incubations was assessed using a selective marker reaction, oxidation of midazolam to 1′-hydroxymidazolam. Time-dependent inhibition was assessed using a two-step procedure. Primary incubations (250 μl, containing 2 mM NADPH, 0.5 mg/ml HLM, and various concentrations of dasatinib or its analogs in 0.1 M potassium phosphate buffer, pH 7.4) were incubated with human liver microsomes. At various time points, e.g., 0, 4, 8, 15, 22, and 30 min, 10 μl of the primary reaction was removed and added into a secondary reaction containing midazolam, NADPH, and buffer. The final reaction volume and concentrations in the secondary reactions were 200 μl with 20 μM midazolam, 1 mM NADPH, 0.05 mg/ml HLM, and 0.1 M potassium phosphate buffer, pH 7.4. Both reactions were performed in 96-well plates on a shaking incubator maintained at 37°C. The secondary incubation was stopped after 5 min by adding acetonitrile in a 1:1 ratio (v/v) containing 0.2 μM carbamazepine as an internal standard. At the end of the assay, the incubations were centrifuged and passed through a Millipore Multiscreen Solvinter 0.45-μm low-binding poly(tetrafluoroethylene) hydrophilic filter plate and analyzed by LC-MS/MS for the formation of 1′-hydroxymidazolam using an API 4000 mass spectrometer (Applied Biosystems, Foster City, CA) interfaced with an Agilent 1200 high-performance liquid chromatograph (Agilent Technologies, Palo Alto, CA). Chromatographic separation was achieved by using a Phenomenex Synergi Fusion-RP C18 column (2.0 × 50 mm, 4 μm). Mobile phases consisted of 0.1% aqueous formic acid (solvent A) and acetonitrile with 0.1% formic acid (solvent B) run at a constant flow rate of 0.375 ml/min. A 2.5-min HPLC method was used with percent B equal to 2% at t = 0 min, 80% at t = 1.35 to 1.6 min, and 2% at t = 1.61–2.5 min (all gradients were linear). 1′-Hydroxymidazolam was detected using the transition from m/z 342.2 to m/z 203.1, DP = 90, and CE = 38. All inhibition studies were conducted in triplicate.
HLM Incubations for GSH Adduct Identification. Incubations to test for the formation of GSH adducts contained 40 μM dasatinib or analog (from dimethyl sulfoxide stock resulting in 0.2% dimethyl sulfoxide, v:v), and 1 mg/ml HLM in 0.1 M potassium phosphate buffer, pH 7.4, in the presence or absence of NADPH (1 mM) and reduced glutathione (5 mM). The total incubation volume for each sample was 0.5 ml. After a 3-min preincubation at 37°C, the incubations were initiated by adding NADPH. Incubations were quenched after 1 h with 1 ml of ice-cold acetonitrile and vortexed for 1 min. These mixtures were centrifuged at 14,000 rpm for 10 min, and the supernatants were transferred to clean 1.5-ml microcentrifuge tubes and dried at 25°C in a temperature-controlled SpeedVac for up to 4 h. Once dried, extracts were reconstituted in 100 μl of a 30% acetonitrile solution and transferred to HPLC autosampler vials for analysis. Experiments to show the time-dependent production of dasatinib-glutathione adducts and to evaluate the effect of the CYP3A4 inhibitor ketoconazole were done using similar procedures except that dasatinib was lowered to 20 μM. Ketoconazole (1 μM), an inhibitor of P450 3A4, was from a methanol stock and resulted in 0.1% (v:v) methanol in the final reaction.
LC-MS/MS Setting for Detection of Glutathione Adducts. LC-MS/MS analyses were performed on an API4000 Q-Trap mass spectrometer (Applied Biosystems) interfaced with an Agilent 1200 high-performance liquid chromatograph (Agilent Technologies). Chromatographic separation was achieved by using a ZORBAX RX-C18 column (2.1 × 150 mm, 5 μm; Agilent Technologies). Mobile phases consisted of 0.1% aqueous formic acid (solvent A) and acetonitrile with 0.1% formic acid (solvent B) run at a constant flow rate of 0.4 ml/min. A 35-min HPLC method was used with percent B equal to 5% at t = 0–3 min, 10% at t = 3.5 min, 50% at t = 23 min, 80% at t = 28–29 min, and 5% at t = 29.5–35 min (all gradients were linear).
MS/MS analyses incorporated polarity switching, using conditions similar to those reported in recent publications by Bo Wen and colleagues (Wen and Fitch, 2009; Wen et al., 2008a,b). The polarity switching method uses improved specificity and lower background of the negative ion precursor scan to detect the presence of GSH adducts and then obtains a more informative positive ion fragmentation pattern in a single injection. In brief, a negative ion precursor ion scan (precursor = m/z 272, DP = –75, and CE = –30) was used with a range from m/z 350 to m/z 900 followed by a positive ion enhanced resolution scan and information-dependent acquisition-triggered positive ion enhanced product ion spectra with m/z 80 to 900 (DP = 75 and CE = 40). A settling time of 700 ms was required to accommodate the switch in polarity and the total cycle time was 4.6 s.
Results
Initial incubations of dasatinib with human liver microsomes verified that oxidation of the chloromethylphenyl ring yielded two major hydroxylated metabolites and that the addition of ketoconazole inhibited their formation (Wang et al., 2008). Preliminary incubations using 10 μM dasatinib also determined that dasatinib caused the NADPH- and time-dependent inactivation of CYP3A4 (data not shown). Based on these results, more detailed experiments were used to determine the kinetic constants for dasatinib-mediated loss of CYP3A4 activity.
Dasatinib-Mediated Time-Dependent Inactivation of CYP3A4. A series of samples with a range of dasatinib concentrations were allowed to incubate with HLM and NADPH. At various time points, the remaining CYP3A4 activity was determined by the marker reaction of midazolam conversion to 1′-hydroxymidazolam. Appropriate controls, lacking dasatinib, were included to assure that the loss in activity was not due to thermal inactivation. The data are represented graphically in Fig. 1. The observed first-order rate constants (kobs) for the inactivation of CYP3A4 by individual concentrations of dasatinib were obtained from the slope of the individual lines. These slopes were fit to a Kitz-Wilson plot, which is shown in the inset of Fig. 1. The calculated KI was 6.3 μM and the kinact was 0.034 min–1. The addition of 2 mM reduced glutathione to the reaction did not protect the enzyme, and the calculated KI and kinact values were virtually identical, suggesting that bioactivated dasatinib inactivates CYP3A4 before release from the active site into the bulk solution. Incubations of dasatinib with recombinant CYP3A4 also showed time-dependent inhibition (KI = 21 μM and kinact = 0.095 min–1).
Removal or modification of the piperazine or methylpyrimidine rings of dasatinib did not eliminate the time-dependent inactivation properties of the molecule. Kinetic constants for three dasatinib analogs are shown in Table 1. Additional analogs were made with modifications on the chloromethylphenyl ring of dasatinib. Time-dependent CYP3A4 inactivation was detected for six of the seven analogs tested. The one compound that did not show CYP3A4 inactivation had the methyl removed and the para-position of the chloromethylphenyl ring blocked with chlorine (Table 2).
Kinetic constants for CYP3A4 time-dependent inactivation by dasatinib analogs
Kinetic constants for CYP3A4 time-dependent inactivation by dasatinib analogs
Dasatinib-Glutathione Adduct Formation. Glutathione has a free sulfhydral group that is nucleophilic and can spontaneously react with electrophiles. The addition of glutathione to microsomal incubations containing dasatinib and NADPH resulted in the detection of dasatinib-glutathione adducts. Six distinct adducts were observed (Fig. 2): two dasatinib-glutathione adducts [D-GSH(a) and (b)], two hydroxy-dasatinib-GSH adducts [D-OH-GSH(a) and D-OH-GSH(b)], and two dihydroxy-dasatinib-GSH adducts [D-2OH-GSH(a) and D-2OH-GSH(b)]. The D-OH-GSH(a) adduct appeared to be the major adduct. If we assume that all of the glutathione adducts have similar ionization efficiency, then the relative concentrations of D-2OH-GSH/D-OH-GSH/D-GSH were 25%:67%:8%. The addition of 1 μM ketoconazole to the incubations inhibited the formation of all adducts (Fig. 2B), implying that the formation of all of the glutathione adducts was predominantly catalyzed by CYP3A4. This result was further verified with the detection of D-2OH-GSH, D-OH-GSH, and D-GSH in incubations using recombinant CYP3A4 (data not shown). Repeating the incubation with both rat and mouse microsomes gave the same profile of glutathione adducts with only minor alterations in the relative concentrations (data not shown). No adducts were observed in any of the microsomal incubations if either glutathione or NADPH was eliminated from the reaction.

Chromatographic separation of dasatinib-glutathione adducts generated in incubations with HLM and GSH. The incubation had 40 μM dasatinib, 1 mM NADPH, 5 mM GSH, and 1 mg/ml human liver microsomes (A); 1 μM ketoconazole, was added to incubation B. D-2OH-GSH (m/z 825.2 → 347.2) corresponds to a dihydroxylated dasatinib-glutathione adduct. D-OH-GSH (m/z 809.2 → 347.2) corresponds to a hydroxylated dasatinib-glutathione adduct. D-GSH (m/z 793.2 → 347.2) corresponds to a dasatinib-glutathione adduct. The internal standard (IS) is 100 nM carbamazepine (m/z 237.0 → 194.0).
The product ion spectra of dasatinib has characteristic fragment ions at m/z 319 and 347, corresponding to loss of the chloromethylphenyl ring through fragmentation on both sides of the carboxyl group of the amide bond (Fig. 3). Because dasatinib contains chlorine, the 35Cl and 37Cl isotopes were useful in fragment pattern evaluation of dasatinib and the glutathione adducts (data not shown). All of the detected dasatinib-glutathione adducts had characteristic m/z 319 and 347 fragments, indicating that glutathione was not bound to that portion of the molecule and that all adducts were located on the chloromethylphenyl ring.
The MS/MS spectra and proposed fragments of D-OH-GSH and D-2OH-GSH are shown in Fig. 3. Only the spectra for the most intense D-OH-GSH(a) and D-2OH-GSH(a) adducts are shown because the mass and fragmentation are identical for the corresponding lower abundance hydroxylated adducts. We have drawn the structures with glutathione adducted at the 3-position because we believe this to be the major site of adduction for a para-quinone-imine owing to the electron-withdrawing effect of the chlorine and the electron-donating properties of the methyl group. The MS/MS spectra are shown for both of the D-GSH adducts (Fig. 4) because the two had different fragmentation patterns. D-GSH(a) was drawn as having glutathione bound at the C5 position and has a fragment at m/z 520 corresponding to dasatinib plus sulfur caused by fragmentation on the glutathione side of the thioether bond. Binding of sulfur to the aromatic ring should result in a stronger bond than adduction through the methyl. D-GSH(b) has a fragment at 486 corresponding to fragmentation on the dasatinib side of the thioether bond and was assigned as glutathione bound directly to the benzylic methyl group. The proposed structures are based on expected sites of adduction for an ortho-imine-methide intermediate.
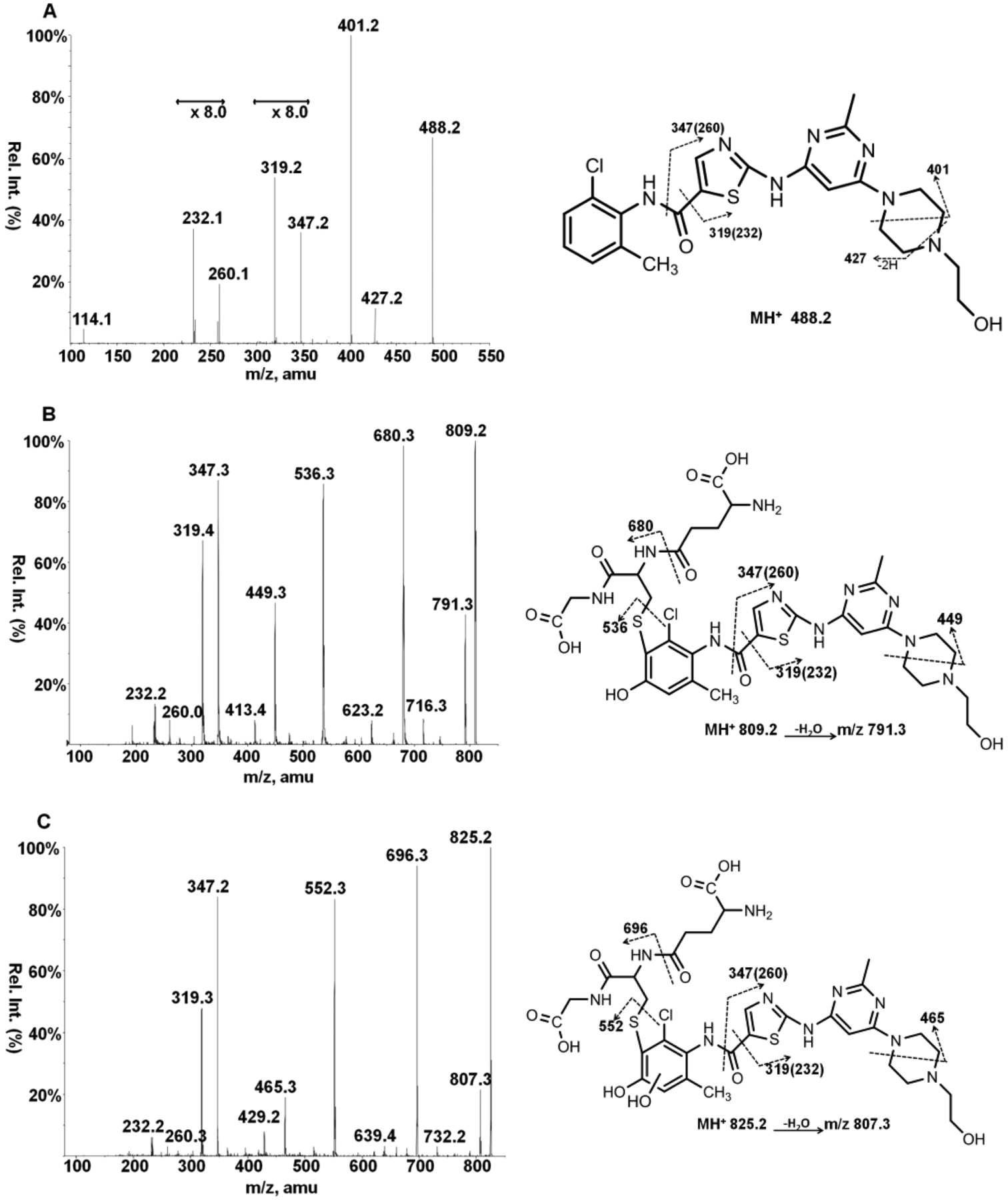
Fragmentation patterns of dasatinib and hydroxylated-glutathione adducts. Dasatinib-glutathione adducts were generated in an incubation mixture containing 40 μM dasatinib, 1 mM NADPH, 5 mM GSH, and 1 mg/ml human liver microsomes. The enhanced product ion scan is shown for dasatinib (A), D-OH-GSH (B), and D-2OH-GSH (C). The structure depicted represents one possible regioisomer. Rel. Int., relative intensity; amu, atomic mass units.
Bioactivation of Dasatinib Analogs. Two mass spectroscopy methods were used to detect glutathione adducts for microsomal incubations containing dasatinib analogs. Polarity switching, as described under Materials and Methods, uses a diagnostic glutathione fragment and is suitable for detecting glutathione bound to any part of the molecule. A second enhanced product ion scan using a characteristic m/z 347 ion was particularly sensitive because of excellent signal intensity, reduced background, and shorter cycle time for the acquisition without the need to switch from negative to positive ion mode during each scan. This second method is limited to detecting adducts in which glutathione is bound to the chloromethylphenyl ring. Although we did not use the common methodology of scanning for loss of the pyroglutamate group (neutral loss of 129), all detected adducts contained the characteristic loss (Table 3).
Glutathione-dasatinib/analog adducts
The modification in boldface represents the most intense signal.

Fragmentation patterns of dasatinib-glutathione adducts (A) and (B). Dasatinib-glutathione adducts were generated in an incubation mixture containing 40 μM dasatinib, 1 mM NADPH, 5 mM GSH, and 1 mg/ml human liver microsomes. The enhanced product ion scan is shown for D-GSH (A) and D-GSH (B). The structures depicted are proposed adducts. Rel. Int., relative intensity; amu, atomic mass units.
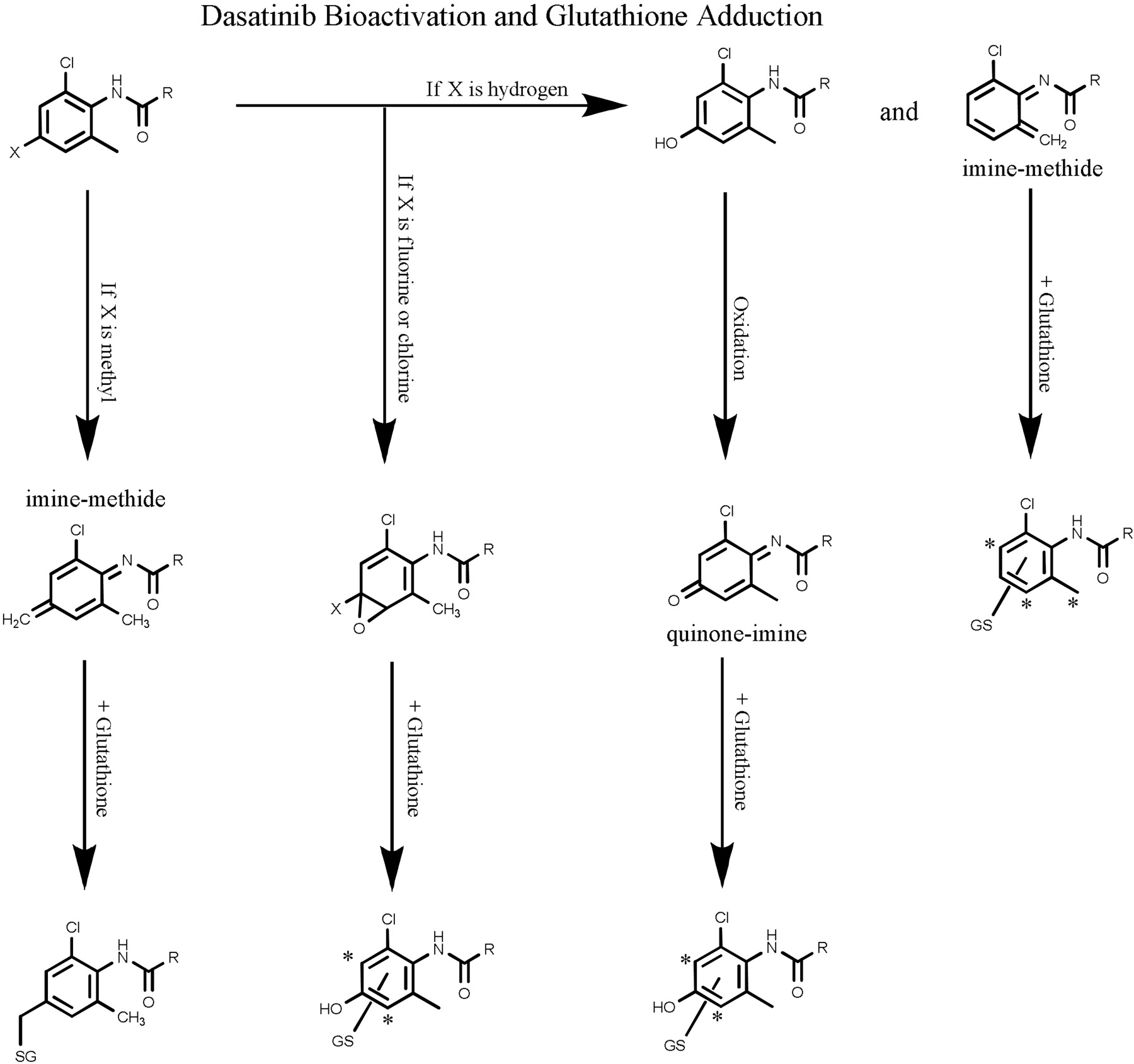
Twelve dasatinib structural analogs on the chloromethylphenyl ring were evaluated for their potential to form adducts with glutathione (Table 3). The role of the methyl group was investigated by replacing the methyl group with fluorine (1698), and 1698 retains the ability to inactivate CYP3A4 (Table 2). As expected, the fluorine did not prevent hydroxylation at the para-position, and 1698-OH-GSH and 1698-2OH-GSH adducts were detectable, whereas 1698-GSH adducts were not. The +GSH (nonhydroxylated) adducts were hypothesized to be generated through oxidation of the methyl group to form an imine-methide that would then undergo nucleophilic attack on the methyl group and on the adjacent C5 carbon (Fig. 5). When tested, all five analogs that retained the methyl group had detectable +GSH adducts. On the contrary, +GSH adducts were not observed for any of the six analogs that lacked the methyl group.
Several analogs that blocked the para-position of the methylchlorophenyl ring were prepared. When a methyl group was added to the para-position (1660), no hydroxylated glutathione adducts were detected. However, the additional methyl group provided an additional site for the formation of a reactive imine-methide, and incubations of 1660 with glutathione resulted in the formation of a single +GSH adduct. The kinact of 1660 was the highest of all the analogs tested.
Two analogs were synthesized with fluorine in the para-position. We expected this modification to prevent the formation of the hydroxylated glutathione adducts and thus only the +GSH adduct arising from oxidation of the methyl group to an imine-methide would be detected. 5826 differed from dasatinib only in the presence of fluorine at the para position. 1701 had the additional modification of replacing the chlorine with a methyl group. The expected +GSH adducts was detected in both analogs. We were surprised to find that the major adducts detected for both compounds were hydroxylated glutathione adducts, and the fluorine group appeared to be removed. The hydroxylated glutathione adducts had masses, fragmentation patterns, and chromatographic retention times identical to those of dasatinib and 1695 (data not shown). Accurate mass MS/MS confirmed that the trapped adducts did not contain fluorine. Purity tests showed that 5826 and 1701 were pure, and no dasatinib or 1695 was detectable.
The presence of chlorine in the para-position (6704) gave similar results. The major glutathione adduct appeared to be due to hydroxylation and loss of chlorine. The distinctive chlorine isotope pattern seen with all of the chlorine-containing molecules was absent. No adducts were detected with four compounds. Each of these had the methyl group removed and contained two or more halogens.
Discussion
It is not our intention to imply that dasatinib is a particularly dangerous drug. Dasatinib fills an important niche in CML treatment, and several ongoing clinical trials may prove that dasatinib is effective in the treatment of other cancers as well. However, in light of the recent report of dasatinib-induced acute hepatitis (Bonvin et al., 2008) and dasatinib-induced lupus (Rea et al., 2008), further studies on the metabolic fate of dasatinib are warranted to better understand the relationship between dasatinib metabolism and adverse effects. In addition, because so many drugs used clinically today have common structural moieties or are direct analogs of existing compounds, it is important to discern what can be modified to increase drug safety.
The finding that dasatinib is a mechanism-based inhibitor of CYP3A4 and that it is bioactivated to a reactive electrophile by CYP3A4 has significant patient implications. These results imply that dasatinib may potentially have pharmacokinetic drug-drug interactions when it is coadministered with drugs that are CYP3A4 substrates. In addition, in a small percentage of individuals, immune-mediated idiosyncratic hepatotoxicity may be observed by dasatinib adducting to cellular proteins in a manner similar to that of diclofenac (Schapira et al., 1986), carbamazepine (Moore et al., 1985; Wu et al., 2006), tienilic acid (Homberg et al., 1984), or dihydralazine (Beaune et al., 1996; Masubuchi and Horie, 2007).
Pharmacokinetic drug-drug interactions are usually a result of one drug altering the metabolism or elimination of another. The prescribing information for dasatinib (Sprycel) notes a clinical drug-drug interaction study in which dasatinib was codosed with simvastatin. Single-dose administration of 100 mg of dasatinib with simvastatin in 54 healthy subjects resulted in mean simvastatin Cmax and AUC increases of 37 and 20%, respectively. Full details of the experimental conditions are not published, nor are additional studies in which dasatinib was predosed, so it is not possible to approximate what percentage of the observed simvastatin exposure is due to irreversible mechanism-based inhibition of CYP3A4 or whether the observed interaction would increase with repeated dosing. It is also not clear how the simvastatin hydroxy acid metabolite was accounted for, as its formation is not catalyzed by CYP3A4.
Pharmacokinetic drug-drug interactions with dasatinib are no doubt minimized by the high potency of dasatinib. Biochemical data for dasatinib inhibition of BCR-ABL and known BCR-ABL mutants showed IC50 values between 0.1 and 3 nM and cell-based values between 0.6 and 11 nM (O'Hare et al., 2005). Plasma dasatinib levels after a single 100-mg oral dose had a Cmax of 215 nM (105 ng/ml) (Christopher et al., 2008b). Although the plasma Cmax is significantly below the KI determined in this study, the irreversible nature of mechanism-based inhibitors leads to larger drug-drug interactions than those in a typical competitive inhibitor would. Unlike reversible inhibition, mechanism-based inhibitors may be unusually potent because the inactivation is cumulative and the enzymatic activity can only be restored after de novo protein synthesis. In addition, hepatic accumulation of dasatinib or high intestinal and hepatic exposure during absorption may lead to higher than expected rates of CYP3A4 inactivation.

Proposed scheme for CYP3A4-mediated metabolic activation of dasatinib. Dasatinib hydroxylation leads to the formation of a chemically reactive para-quinoneimine intermediate (major pathway) or hydrogen abstraction of the methyl leads to an ortho-imine-methide intermediate (minor pathway). These reactive intermediates react with glutathione to form corresponding GSH adducts or may react with CYP3A4 and lead to enzyme inactivation. *, proposed sites of glutathione adduction.
Findings in the current study that dasatinib is bioactivated through formation of both quinone-imine and imine-methide reactive intermediates indicate that dasatinib is likely to form protein adducts in the liver in addition to the adduction to glutathione shown in vitro. Quinone-imines are known to covalently modify cellular proteins and have been implicated in a number of drug-related adverse effects (Guengerich and MacDonald, 2007; Tang, 2007). The covalently modified proteins lead to the potential for idiosyncratic hepatotoxicity, sometimes referred to as immune-mediated idiosyncratic hepatotoxicity or drug-induced hepatitis. Idiosyncratic reactions are rare, occurring in a small number of patients (usually between 0.01 and 1%) and do not follow a strict dose response. Based on the hapten hypothesis, in which drug toxicity is mediated by drug-protein adduct formation (Griem et al., 1998), injured cells are internalized by phagocytes, such as Kupffer cells and dendritic cells, where they are processed, and adducted peptides are presented by major histocompatibility complex class II to helper T cells. B cells producing autoantibodies or antibodies against haptenized protein mediate antibody-dependent toxicity (Walgren et al., 2005; Masubuchi and Horie, 2007).
It is widely recognized that additional signals are needed to initiate an immune response, which has led to the “danger hypothesis.” The danger hypothesis suggests that major histocompatability complex II peptide presentation by the antigen-presenting cell alone does not cause a physiological T-cell response (Matzinger, 1998). Thus, it is not the foreignness of a compound but rather its ability to trigger “alarm” signals that determines whether it will induce an immune response. Hepatotoxic drugs may act as the source of antigen and provide the alarm by damaging the cell and causing necrotic cell death (Curtsinger et al., 1999).
Because dasatinib was able to be bioactivated through hydroxylation and oxidation to form a quinone-imine and through oxidation of the methyl to form an imine-methide intermediate, single modifications to the molecule were not successful in eliminating bioactivation or mechanism-based inhibition. Imine-methide formation requires the presence of a methyl group in the ortho-or para-position. Removal of the methyl group from these positions successfully eliminated the glutathione adducts resulting from imine-methide formation. Formation of quinone-imine intermediates required dasatinib hydroxylation. In theory, quinone-imine intermediates could be formed by hydroxyl groups at either the ortho-or para-position. However, in the analogs that we generated, glutathione adducts appeared to proceed through hydroxylations at the para-position as seen by the absence of detectable glutathione adducts in compounds such as 1250 and 1260. Perhaps hydroxylation at the ortho-position is sterically hindered. It is interesting to note that blocking the para-position with fluorine or chlorine did not prevent quinone-imine formation. Analogs containing fluorine or chlorine in the para-position were found to be dehalogenated and resulted in the same glutathione adducts as their nonfluorinated counterparts. In fact, the rates of inactivation were virtually identical and the KI values for the fluorinated analogs were lower, implying that they may be more efficiently bioactivated. We propose an oxidative mechanism proceeding via epoxide formation.
In conclusion, we have shown that dasatinib is a mechanism-based inactivator of CYP3A4 and is metabolized to generate reactive quinone-imine and imine-methide intermediates that spontaneously react with glutathione. In addition, we have demonstrated that dasatinib bioactivation can be prevented by appropriate modification of the chloromethylphenyl ring.
Acknowledgments
We thank Tomas Vojkovsky for synthesis of chemical starting materials.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.025932.
-
ABBREVIATIONS: CML, chronic myelogenous leukemia; LC, liquid chromatography; HLM, human liver microsomes; MS/MS, tandem mass spectrometry; DP, declustering potential; CE, collision energy; GSH, glutathione.
- Received December 1, 2008.
- Accepted March 11, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}