Article Text

Abstract
AIM Achondroplasia can result in respiratory difficulty in early infancy. The aim of this study was to document lung growth during infancy, together with the cause of any cardiorespiratory and sleep dysfunction.
PATIENTS AND METHODS Seventeen prospectively ascertained infants (14 boys and three girls) with respiratory symptoms starting before 1 year of age underwent clinical, sleep, and lung function studies.
RESULTS Three distinct groups were identified. Group 1 (n = 6) were the least symptomatic and only had obstructive sleep apnoea. Group 2 (n = 6) had obstructive sleep apnoea of muscular aetiology and, neurologically, hydrocephalus and a small foramen magnum were common. Group 3 (n = 5), the most severely affected group, all developed cor pulmonale, with three deaths occurring as a result of terminal cardiorespiratory failure. All five had obstructive sleep apnoea with a muscular aetiology (a small foramen magnum predominated) with severe or moderately severe gastro-oesophageal reflux. Initially, lung function studies found no evidence of restriction or reduced lung volumes standardised according to weight. However, with growth these infants had worsening function, with raised airway resistance and severe reductions in respiratory compliance.
CONCLUSIONS These groups appear to be distinct phenotypes with distinct anatomical aetiologies: “relative” adenotonsillar hypertrophy, resulting from a degree of midfacial hypoplasia (group 1); muscular upper airway obstruction along with progressive hydrocephalus, resulting from jugular foramen stenosis (group 2); and muscular upper airway obstruction, but without hydrocephalus, resulting from hypoglossal canal stenosis with or without foramen magnum compression and no jugular foramen stenosis (group 3). The aetiology of these abnormalities is consistent with localised alteration of chondrocranial development: rostral, intermediary and caudal in groups 1, 2, and 3, respectively.
- achondroplasia
- breathing
- respiratory function
Statistics from Altmetric.com
Achondroplasia is an autosomal dominant syndrome of short limbed stature, most commonly caused by a mutation in fibroblast growth factor receptor-3 (FGFR-3).1 2 Between 10% and 85% of these infants and children present for treatment because of major respiratory difficulty such as obstructive sleep apnoea, waking cyanotic episodes, and chronic respiratory insufficiency and failure.3-8 Although such problems are not uncommon in other forms of dwarfism, there are specific developmental abnormalities in achondroplasia that might predispose to these sometimes life threatening symptoms, and these include: midfacial hypoplasia and upper airway obstruction; dysplasia of the basiocciput, exoccipital bone, and craniovertebral junction with foramen magnum stenosis and cervicomedullary cord compression; and possible thoracic cage restriction.
Recently published series and reviews of sleep physiology7-10 and neurology11-15 in children with achondroplasia have helped to elaborate further the previously reported hypotheses concerning the pathophysiology of disordered breathing in achondroplasia.3-6 Upper airway obstruction alone is an important component of the “achondroplasia respiratory difficulty syndrome”, although there is often an interaction between this factor and underlying brainstem dysfunction. In relation to the other possible major patho-anatomical factor in infancy—thoracic development—there has been little but qualitative, cross sectional data describing chest size in limited numbers of symptomatic achondroplastic infants.5 16 17 A small thoracic cavity has been noted, especially in those less than 2 years of age. In contrast, by later childhood and adult life, major primary respiratory problems are unusual, and Stokes and colleagues18 19 have shown that above 7 years of age, the vital capacity falls within the expected range for body mass, and functionally the lungs and airways are normal. This discrepancy between descriptions of chest size and symptoms in infancy and “normal” lung volumes later in development, although unresolved, is important prognostically: it either reflects a selection bias, in that achondroplastic infants with small chests do not survive to adulthood because of a restrictive defect or pulmonary hypoplasia, or catch up thoracic growth, such that pulmonary problems become less significant with age.
In practice, it is difficult to establish which of the above anatomical and physiological abnormalities is the most significant. Often, the clinician has to resolve the relative contribution that each abnormality might have in producing a given child’s symptomatology. Therefore, thorough, systematic, clinical, neurological, and respiratory investigation is recommended.5 14 20 In attempting to identify treatable problems in children referred because of respiratory difficulty complicating achondroplasia, and to improve the understanding of lung growth during infancy, we have undertaken serial comprehensive clinical, sleep physiology, and lung function studies. Three distinct, ascending patterns of severity of early respiratory difficulty in achondroplasia with their respective underlying pathophysiologies are described.
Subjects and methods
SUBJECTS
Between 1987 and 1996, 17 young achondroplastic children with ages ranging from 3 months to 3.5 years (median, 11 months; interquartile range, 7–24) were referred to our paediatric respiratory service at Great Ormond Street Hospital for Children NHS Trust in London, UK. All patients were reviewed by the genetics service at this institution and were evaluated comprehensively by the same respiratory team because of referring physicians’ concerns about breathing difficulties.
ASSESSMENT
Based on a previously reported approach to systematic clinical review of children with achondroplasia,5 we have devised a clinical investigative protocol, which has been approved by our hospital and institution ethics committee, for clinicophysiological assessment at three to six monthly intervals.
Clinical data and investigation
Clinical history and a general physical examination were noted on each patient visit. The clinical history was taken from the parents and covered a general multisystem review, as well as specific questioning focusing on any noisy breathing, respiratory distress, cyanotic episodes, apnoea, sleep disturbance, feeding problems, hypotonia, abnormal movements, and developmental delay. The physical examination was thorough and documented growth parameters as well as abnormal cardiorespiratory and neurological signs.
Depending on the findings of the clinical review, specific investigations were directed as follows. Barium swallow and 24 hour oesophageal pH probe studies were ordered if there was a history of feeding difficulty, recurrent pneumonia, sleep disturbance, regurgitation, or gastro-oesophageal reflux. Gastro-oesophageal reflux was defined, using the pH probe results, as: severe, when > 20% of the time the pH was < 4; moderate, when 10–20% of the time the pH was < 4; and mild, when 4–9% of the time the pH was < 4.21-23 x Rays of the chest and postnasal space were used to assess the lung parenchyma and upper airway, and electrocardiography and echocardiography were used for cardiac assessment. Somatosensory evoked potentials and cranial computed tomography or magnetic resonance imaging were carried out in patients with abnormal neurology and possible hydrocephalus or foramen magnum stenosis.
Assessment of respiratory patterns during sleep
To assess the patient’s respiratory patterns, overnight cardiorespiratory sleep studies were performed during hospital admission in all patients. These studies were not planned to assess the response to intervention; therefore, patients were studied even if they had an intercurrent illness. These studies comprised between 3 and 9.5 hours (mean (SD), 6.9 (1.9)) of continuous and simultaneous recording during natural sleep of the following variables: (1) rib cage and abdominal respiratory movements (using uncalibrated strain gauges); (2) arterial oxygen saturation (SpO2) by pulse oximetry (Ohmeda Biox 4700, averaging over 3.0 seconds); (3) electrocardiography; (4) video and sound recording. A computer based sleep system (CARDAS, Oxford, UK) was used for recording and analysing data. Breathing patterns and behavioural sleep stage were evaluated by observation using standard criteria.24 Quiet sleep (QS), which approximates to non-rapid eye movement deep sleep, was identified as periods during which there were no facial movements, only occasional startles and extremity movements, constant heart rate, and regular breathing. Conversely, active sleep (AS), which approximates to rapid eye movement sleep, was defined as periods where there were more frequent movements of the body and extremities, facial grimaces, rapid eye movements, variable heart rate, and irregular breathing.
Periods of artefactual signal caused by gross body movement were excluded by visual inspection of the raw data signals and by reference to the video record and observation notes. Mean (SD) values for SpO2 and heart rate were calculated for each behavioural sleep state. The patterns of the recorded signals were analysed in three minute epochs to allow visualisation of individual breaths and to identify both central and obstructive respiratory anomalies25:
• Central respiratory pauses were defined as cessation of respiratory movements in both rib cage and abdominal signals. These pauses were described in terms of their frequency (number/hour of sleep), duration (in seconds), and their impact on SpO2(the number of dips in SpO2 greater than 4% for each hour of sleep (“4% SpO2 dip rate”) and the lowest SpO2 values consequent to the respiratory pause (“min SpO2”).
• Obstructive apnoeas were diagnosed by using a combination of clinical and polygraphic criteria. Inspection of the polygraphic data was used to define the relation between rib cage and abdominal signals (which varied between being completely synchronous and completely paradoxical or “out of phase”), changes in SpO2 caused by the periods of obstruction (4% SpO2 dip rate, min SpO2), and percentage of sleep time spent with SpO2 below 92% (“< 92% SpO2”).
• Mixed apnoeas were defined as periods during which both central and obstructive apnoeic components were evident. The order and sequence of obstructive efforts and central respiratory pauses were not taken into account. The effects on SpO2 were measured as described above (4% SpO2 dip rate, min SpO2, and < 92% SpO2).
The upper limit of the normal 4% SpO2 dip rate is three dips/hour.26 Values of four to 10 dips/hour are classified as mildly abnormal, between 11 and 25 dips/hour as moderately abnormal, and > 25 dips/hour as severe. Both SpO2 and heart rate data were recorded at a frequency of 1 Hz and, by using interactive computer analysis in which periods of QS and AS were determined from behavioural criteria, the overall mean and SD for each sleep state were determined.
Measurement of infant respiratory function
Infant respiratory function parameters were measured using previously described and validated methods27 in those infants and young children where sedation before measurements was possible and whose weight was less than 12 kg. Because we intended to assess lung growth and development by measuring respiratory function, and the results of these tests would be distorted by current respiratory infections,28 these studies were deferred by three weeks if patients presented with intercurrent illness.
Measurements were made following the onset of quiet sleep24 28 aided by sedation with triclofos sodium 100 mg/kg given orally (equivalent to 66 mg/kg chloral hydrate). Resting lung volume at functional residual capacity and airway resistance were measured using a whole body plethysmograph as described previously.27 29 Passive respiratory compliance (the combined compliance of the lungs, tissues, and chest wall) was then assessed using the multiple occlusion technique.27 30
Data display, recording, and analysis, including that of tidal breathing parameters (respiratory rate and tidal volume), were performed using a computer assisted system (Respiratory Analysis Program; PhysioLogic Ltd, Berks, UK) with interactive operator quality control on an IBM compatible computer, according to previously established criteria.27 Lung volume was reported as the mean of three to five end inspiratory occlusions, with results being corrected to functional residual capacity. Airway resistance was reported as the mean of between five and 25 technically acceptable breaths at 50% maximum flow during initial inspiration and end expiration.27 31 Respiratory compliance was calculated from the least squares linear regression through the volume pressure data.30
CLINICAL INTERVENTIONS
Specific treatment interventions in these patients included adenotonsillectomy in nine cases, ventriculoperitoneal shunt in five cases, and foramen magnum decompression in two cases. The approach to intervention was as follows.
Adenotonsillectomy
Adenotonsillectomy was performed in patients with a postnasal space x ray showing airway encroachment by the tonsils and adenoids and at least moderately severe airway obstruction on sleep study. Before surgery, a four week trial of nasal steroids was undertaken to exclude the possibility that inflamed soft tissue or anterior nasal obstruction was the more important component.
Ventriculoperitoneal shunt
Ventriculoperitoneal shunt was performed in patients whose head circumference was inappropriately expanding and crossing centiles according to achondroplasia specific charts,32 and who had periventricular oedema changes on cranial computed tomography or magnetic resonance imaging.
Foramen magnum decompression
Foramen magnum decompression was performed in patients who had paucity of upper limb movement and/or lower limb clonus and evidence of cord compression on magnetic resonance imaging. A small foramen magnum was identified by minimal cerebrospinal fluid space around the cord. In isolation, this imaging finding was not taken as an indication for surgery.
Results
Table 1 is a summary of the clinical, cardiorespiratory, upper airway, and neurological findings from our structured assessment. This series included 14 boys and three girls who were all first symptomatic before the age of 12 months, although they were not necessarily referred to our institution for assessment at that time. Based on the dynamic clinical picture seen over the period of follow up at our institution (median, 7 months; interquartile range, 5–17), these patients have been categorised broadly into three readily identifiable groups. Group 1 patients (n = 6; follow up between 4 and 17 months; median, 7) were the least symptomatic with only sleep disturbance or night time heavy breathing reported, and were generally well with no central nervous system or lower respiratory problems. Group 2 patients (n = 6; follow up between 5 and 19 months; median, 13) were symptomatic with reported episodes of cyanosis, apnoea, and noisy breathing, and had both upper airway obstruction and central neurological problems. Group 3 patients (n = 5; follow up between 4 months and 2 years; median, 6 months) had progressive respiratory problems from infancy, which culminated in the development of chronic cardiorespiratory failure and dependency on supplemental oxygen treatment.
Clinical summary of 17 young children with achondroplasia
CLINICAL ASSESSMENT AND CARDIORESPIRATORY SLEEP STUDIES
Thirty two sleep studies were fully assessable by the CARDAS system and are summarised in table 2. Because the three patient groups were distinguishable clinically (group 1, airway obstruction only; group 2, airway obstruction and central neurological problems; group 3, oxygen dependency), results from sleep studies and respiratory function tests are presented with respect to this stratification.
Summary of serial sleep monitoring studies in 13 young children with achondroplasia
Group 1 patients
Group 1 patients were the least symptomatic, but did raise sufficient parental and physician concern to warrant referral. Neither central neurological problems nor gastro-oesophageal reflux were evident on screening these patients.
Ten sleep studies were analysed in five children in this group. All patients had obstructive sleep apnoea. The standard scores for obstructive sleep apnoea assessed before adenotonsillectomy classified the patients as three mild, one moderate, and one severe. The objective measures of sleep are presented in table 2. Total sleep time was between 7.1 and 9.5 hours, with QS accounting for 29–55% of that time (mean (SD), 41% (9%); normal value at this age is 59% (7%)33). SpO2 readings below 92% are considered abnormal. In this group the mean SpO2 was in the range 88–97%. Seven of the 10 studies showed abnormally high 4% SpO2 dip rate (> 4/hour) and unstable SpO2 readings (SD range, 0.9–9.4).
Analysis of QS and AS revealed that the variability of SpO2during QS was generally less than that seen during AS, as would be expected in obstructive sleep apnoea (data not shown). In the four patients who had adenotonsillectomy (cases 5, 9, 12, and 16) because of radiological evidence of airway encroachment and failure to respond to medical therapy, or for severe obstructive sleep apnoea, none had severe obstructive sleep apnoea after surgery (two had moderate and two had mild problems). These patients all had marked symptomatic improvement after surgery.
Group 2 patients
Group 2 patients did not require supplemental oxygen to maintain their SpO2. These patients did not have severe gastro-oesophageal reflux, but neurologically both hydrocephalus and a small foramen magnum were common, albeit usually with normal somatosensory evoked potentials.
Sixteen sleep studies were analysed. All patients had obstructive sleep apnoea with varying degrees of central apnoea. The standard scores for obstructive sleep apnoea in the five patients who had studies before airway intervention classified three as moderate and two as severe. Total sleep time was between 3 and 8.9 hours, with QS accounting for 13–59% of that time (mean (SD), 40% (12%)). The mean SpO2 was in the range 91–99%. The 4% SpO2dip rate was between 5 and 10 dips/hour in seven studies, between 11 and 25 dips/hour in four, and exceeded 25 dips/hour in the remaining two studies.
Analysis of QS and AS revealed that the variability of SpO2during QS was generally less than that seen during AS, as would be expected in obstructive sleep apnoea (data not shown). Four of these six children had adenotonsillectomy. Despite this intervention, in contrast to group 1 patients, they all continued to have breathing abnormalities reported by their parents. In two patients who had sleep studies both before and after adenotonsillectomy (patients 8 and 10), moderate or severe obstructive sleep apnoea was still evident in both QS and AS after surgery.
Group 3 patients
Group 3 patients all had severe desaturation without supplemental oxygen. They each developed evidence of cor pulmonale with either right ventricular hypertrophy or right axis deviation, and three of the five children in this group died because of severe cardiorespiratory failure. All patients in this group had severe or moderately severe gastro-oesophageal reflux based on barium swallow studies and lower oesophageal pH probe monitoring. Four of these patients had a small foramen magnum with one infant proceeding to foramen magnum decompression. After surgery this patient developed hydrocephalus and required ventriculoperitoneal shunting.
Six sleep studies in three of these children were analysable. All patients had obstructive sleep apnoea and central apnoea. The standard scores for obstructive sleep apnoea in this group were at best moderately severe, even with airway intervention such as nasopharyngeal airway. Total sleep time was between 3.5 and 9.1 hours. Restless and disrupted sleep with frequent arousals is evidenced by a decreased duration of QS in all the cases: QS accounted for 22–56% of total sleep time (mean (SD), 36% (12%)), well below the normal range. The baseline SpO2 was low in most cases, with an overnight range of 88–97% despite supplemental fractional inspired oxygen. In all cases, the SpO2 was unstable with frequent dips of ⩾ 4% and with minimum SpO2 values < 85%. This is reflected in both the SD of SpO2 for each individual and the average 4% SpO2 dip rate, which were abnormally high, 1.1–6.7 and > 4/hour, respectively.
Analysis of QS and AS revealed that the variability of SpO2during QS was generally less than that seen during AS (data not shown). In the two patients who had some form of upper airway intervention (adenotonsillectomy in patient 14 and nasopharyngeal tube in patient 17), moderate or severe obstructive sleep apnoea remained evident in both QS and AS following the intervention.
INFANT RESPIRATORY FUNCTION
Ten infants had complete lung function measurements, these being obtained on more than one occasion in eight infants. Table 3 summarises the results. Results were interpreted with respect to the 95% confidence limits of a cohort of healthy infants who were studied prospectively as part of an ongoing epidemiological study using identical methods and equipment.34 Most of the infants were tachypnoeic (respiratory rate > 50/minute) but tidal volume fell within the range for normal infants (7–10 ml/kg).35
Summary of serial lung function studies in 10 young children with achondroplasia
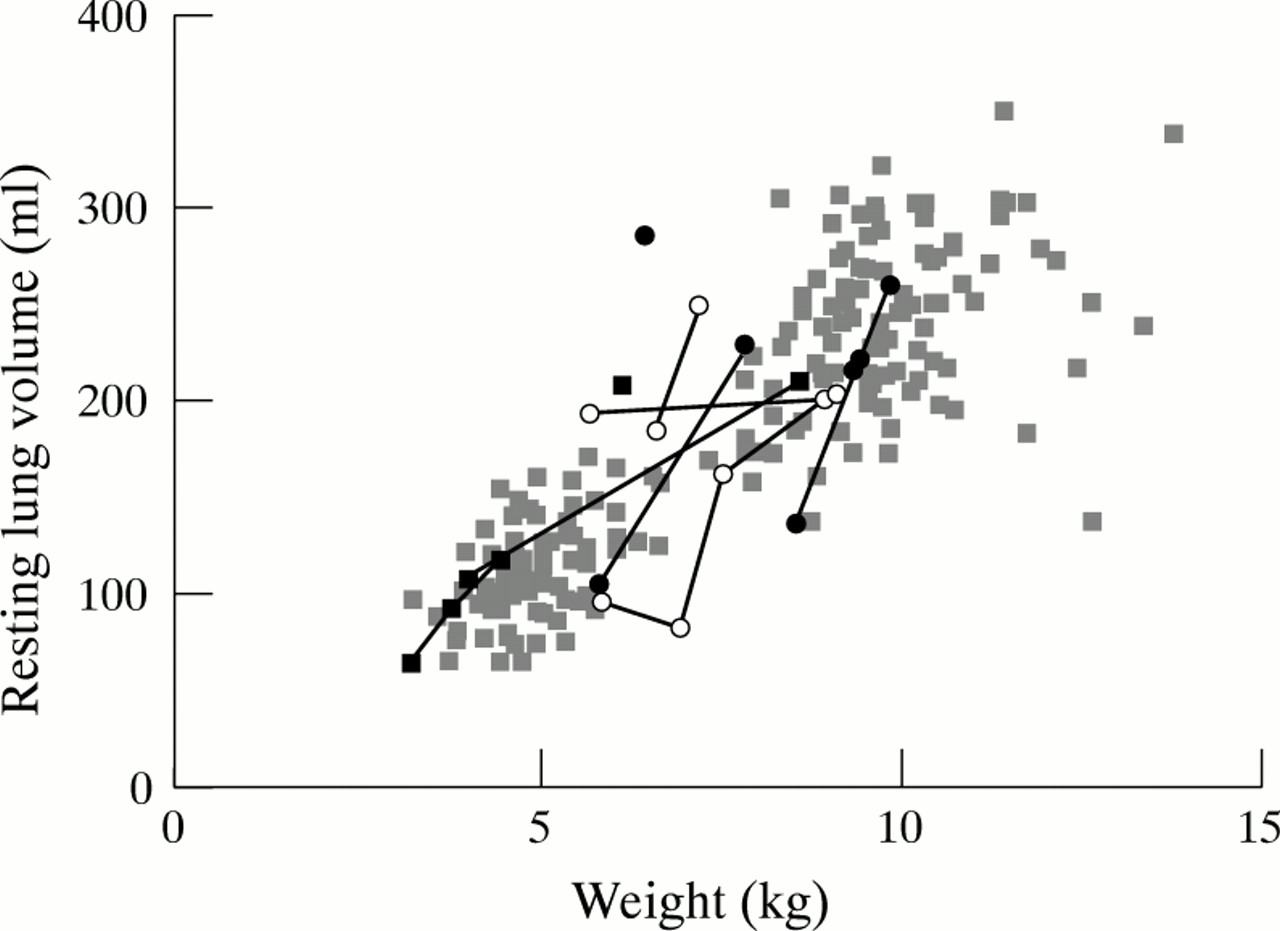
In general, group 1 patients had normal lung volume and compliance, although lung volume was very high in patient 4. In this patient, the weight for age was very low, which complicates further the expression of lung volume. Inspiratory resistance was raised in two patients but normalised after adenotonsillectomy in patient 5. Two of the group 2 patients had raised lung volumes (patients 8 and 10) and two had moderately reduced values of compliance (patients 3 and 8). Expiratory resistance was within normal ranges, but raised inspiratory resistance, suggestive of upper airway obstruction, was noted in patient 8 on both measurement occasions. Patients in group 3 had normal or high lung volume with very low compliance, and raised inspiratory and expiratory airway resistance. The former is indicative of upper airway obstruction and the latter of small airway disease. Serial measurements of lung volume and compliance for the 10 children with achondroplasia, together with the group of healthy children34 are shown in figs 1 and 2, respectively. Values for lung volume in the children with achondroplasia were generally similar to those obtained from healthy infants based on weight (fig 1). The relation between respiratory compliance and lung volume for both the children with achondroplasia and the healthy infants is shown in fig 2, and illustrates that the oxygen dependent infants with achondroplasia (group 3) had stiffer than normal lungs, which worsened with growth, suggesting either hyperinflation or reduced compliance secondary to gastro-oesophageal reflux.

Scatter plot of resting lung volume and weight. Pale grey squares, healthy infants34; closed circles, group 1 (achondroplastic infants with mild respiratory symptoms only); open circles, group 2 (achondroplastic infants with airway obstruction and central neurological problems); closed squares, group 3 (oxygen dependent achondroplastic infants).
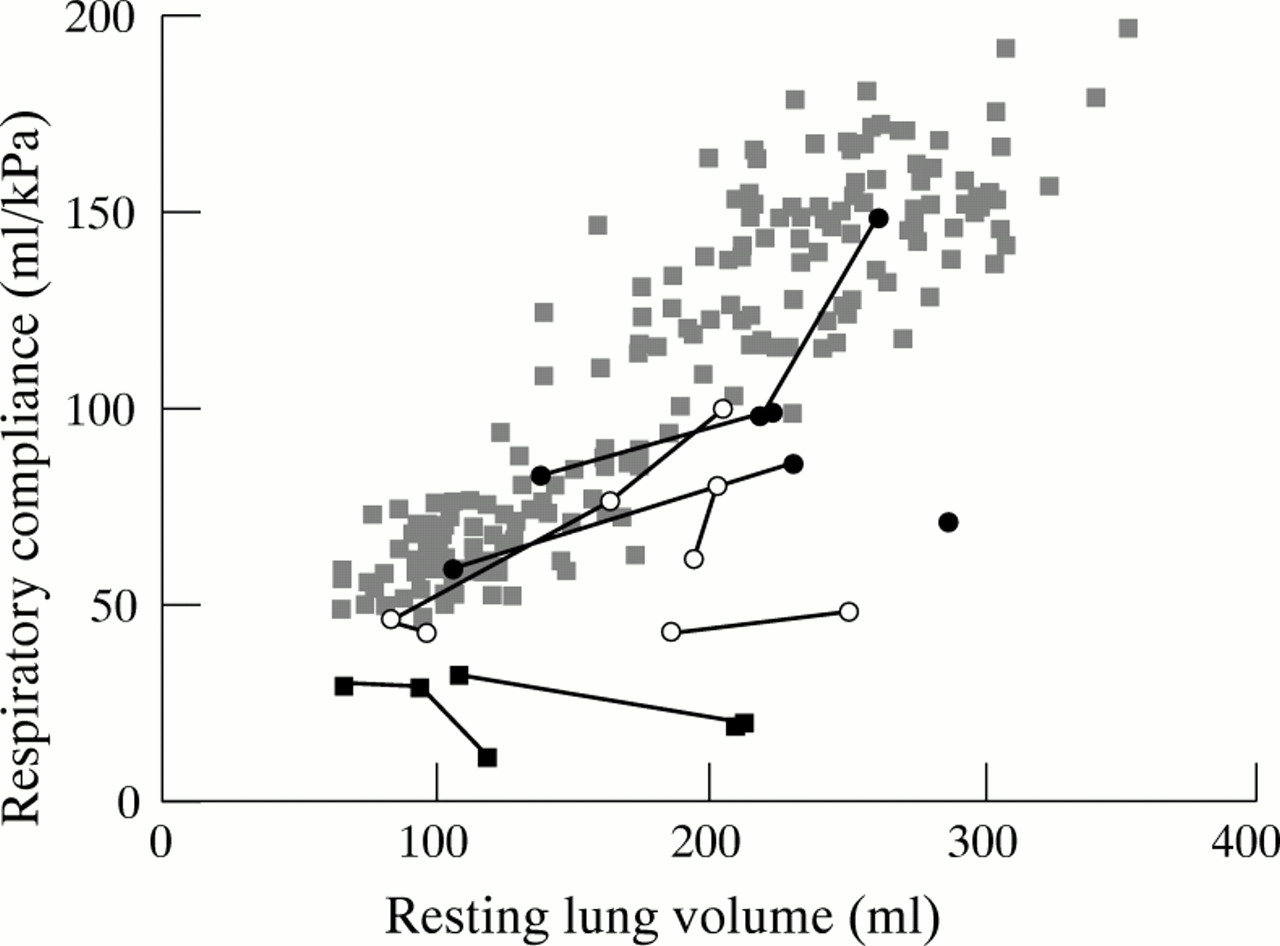
Scatter plot of respiratory compliance and resting lung volume. Pale grey squares, healthy infants34; closed circles, group 1 (achondroplastic infants with mild respiratory symptoms only); open circles, group 2 (achondroplastic infants with airway obstruction and central neurological problems); closed squares, group 3 (oxygen dependent achondroplastic infants).
Taken together, these data provide little support for the notion that the most severely affected infants have pulmonary hypoplasia, although it is possible that a raised lung volume and increased resistance could result from small lungs with decreased alveolar numbers combined with gas trapping and overinflation of existing alveoli as a result of airway obstruction. However, in this case one would not expect normal tidal volumes for weight. To illustrate these findings, representative examples of lung function and sleep study results from each group are presented in figs 3 and 4. These provide evidence of increasing severity of airway problems between groups 1 and 2, and groups 2 and 3.

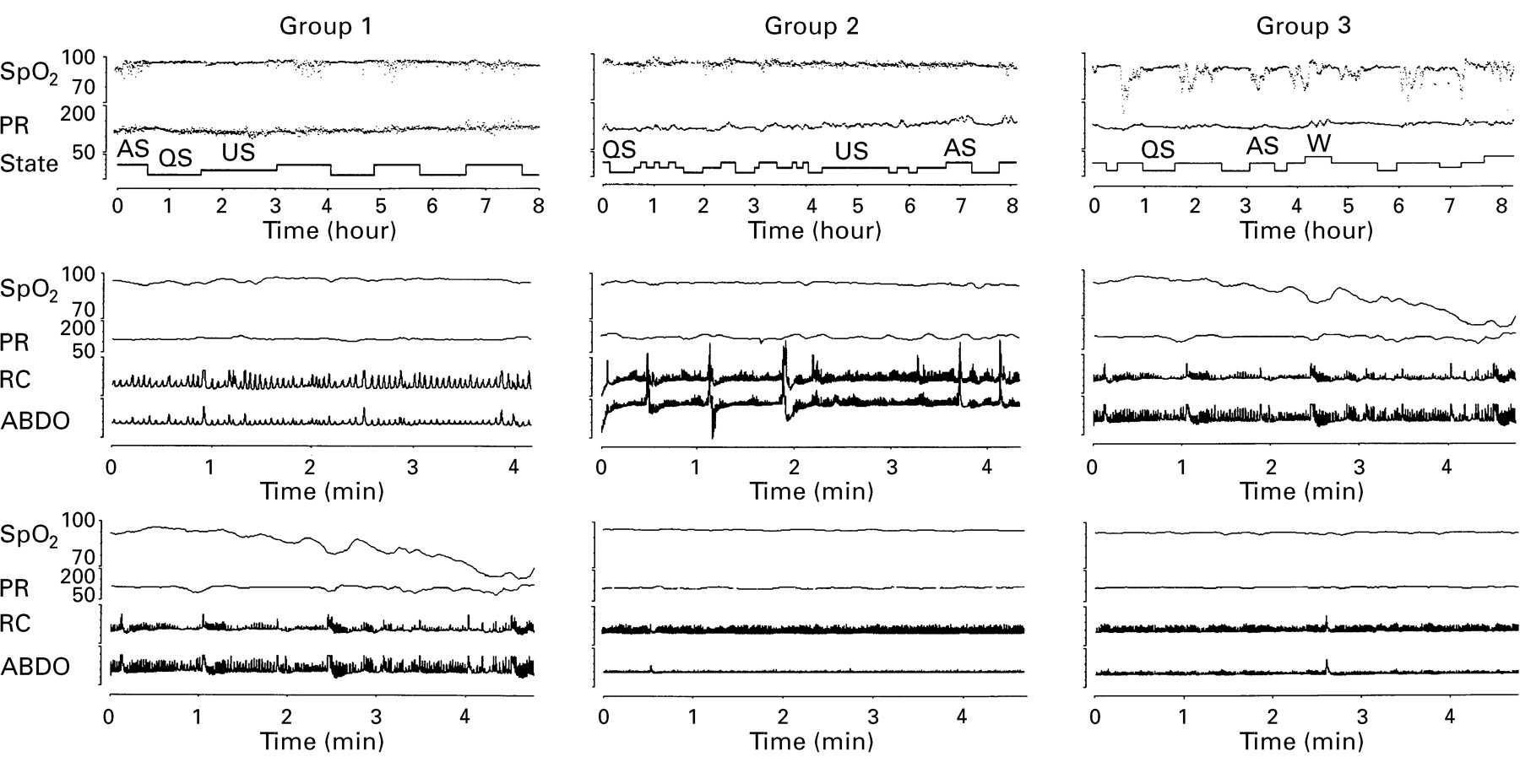
Airway resistance and tidal flow–volume curves of patients in (top) group 1 (achondroplastic infants with mild respiratory symptoms only) showing normal curves; (middle) group 2 (achondroplastic infants with airway obstruction and central neurological problems) showing a flattening of the inspiratory part of both airway resistance and tidal curves, suggesting upper airway obstruction; and (bottom) group 3 (oxygen dependent achondroplastic infants) showing high pressures in the airway resistance curve during late expiration and concavity of the late expiratory part of the tidal curve, suggesting small airway disease. High inspiratory pressures suggest that an upper airways obstructive component is also present. RawI, airway resistance during inspiration; RawE, airway resistance during expiration.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sleep study records from representative patients in group 1 (achondroplastic infants with airway obstruction only); group 2 (achondroplastic infants with airway obstruction and central neurological problems); and group 3 (oxygen dependent achondroplastic infants). Top panels show approximately eight hours of sleep data with arterial oxygen saturation (SpO2, percentage units), pulse rate (PR, beats/minute units), and behavioural sleep state hypnogram (AS, active sleep; QS, quiet sleep; US, unknown sleep; W, wakefulness). Middle panels show typical AS segments of approximately four minutes duration for each representative patient with SpO2, PR, and rib cage and abdominal movements (RC and ABDO, respectively, no units). The lower panels show typical QS segments of approximately four minutes duration with the same format as the AS panels.
Discussion
In this group of infants with achondroplasia and respiratory difficulty we have seen three clinically significant, ascending patterns of severity, where the respective underlying pathophysiologies appear to be of increasing complexity. These identifiable groups comprise: (1) those patients who are the least symptomatic (and then only during sleep), who at worst have obstructive sleep apnoea that appears to be caused by adenotonsillar hypertrophy, because there is a marked clinical improvement with adenotonsillectomy; (2) those patients who continue to have episodes of apnoea and airway obstruction despite adenotonsillectomy, as well as hydrocephalus requiring treatment; (3) the most severely affected patients who have progressive respiratory problems from infancy, which culminate in chronic cardiorespiratory failure and dependency on supplemental oxygen treatment; these patients all appear to have gastro-oesophageal reflux, pulmonary small airway pathology, and obstructive and central sleep apnoea.
Identification of these distinct patient groups was based on the dynamic clinical picture seen over the period of follow up. However, a full description was only possible with a systematic neurological, radiological, and physiological investigation, which was clearly enhanced by serial studies. We did not find that a single test alone could reliably stratify patients into one of the three groups. Rather, our study supports the value of coordinated investigation.5 Sleep study is effective in confirming reported symptoms and identifies obstructive sleep apnoea, which was common to all of our patients. In young children however, one should be aware of practical limitations in monitoring oronasal airflow during sleep, so that these studies do not exclude completely the possibility of confounding hypopnoea. Nevertheless, characteristic patterns of change in SpO2, heart rate, and respiratory movements are indicative of increased upper airway resistance, as a result of either partial or total obstruction. The indices of severity of obstructive sleep apnoea that we adopted failed to follow objectively the reported clinical course, and this was particularly noticeable in group 1 patients, whose carers reported a favourable clinical and behavioural response to adenotonsillectomy. This discrepancy might have been a result of our practice of not deferring admission for sleep studies in patients with intercurrent upper respiratory infection. Alternatively, had we used measures of sleep quality and architecture,33 such as electroencephalography and electro-oculography, we might have been able to monitor improvement in sleep more sensitively. In contrast, serial respiratory function testing identified our most severely affected patients and confirmed a normal lower respiratory tract in all but the group 3 children.
The various developmental and acquired abnormalities in achondroplasia, which might predispose to these three patterns of respiratory difficulty could include: midfacial hypoplasia and adenotonsillar hypertrophy, resulting in upper airway obstruction; dysplasia of the basiocciput, exoccipital bone, and craniovertebral junction, resulting in foramen magnum stenosis and jugular foramen compression with consequent cervicomedullary cord compression, cerebral venous hypertension, and hydrocephalus; and possible thoracic cage restriction. Other confounding factors are gastro-oesophageal reflux and pulmonary parenchymal damage exhibited by small airway obstruction and, finally, upper airway obstruction with obstructive sleep apnoea as a result of central causes leading to pharyngeal hypotonia, particularly marked during rapid eye movement or AS.
ACHONDROPLASIA AND RESPIRATORY FUNCTION
The hypothesis that infants and children with achondroplasia and respiratory difficulty are “children with small chests”, implying pulmonary hypoplasia,5 16 17 is not borne out by our cross sectional and longitudinal data. Even in severely affected individuals, we found no evidence for reduced lung volumes, standardised according to weight or restriction. The hyperinflation noted in patient 4 might partially reflect the difficulty in relating lung volume to body weight in this patient, who had remarkably low weight for age in comparison with the rest of the series. (In addition, in the presence of such marked upper airway obstruction, it is also feasible that some slight overestimation of lung volume could have occurred because of poor equilibration of pressures during airway occlusion.27) Therefore, our findings are in keeping with reports of later childhood and adult life,18 19 where lung volumes are within the expected range for body mass, which questions the idea that the more severe infants have pulmonary hypoplasia. Rather, our data suggest that there is an association between lung injury, cor pulmonale, and gastro-oesophageal reflux. Importantly only two of our most severely affected patients have survived, they were both seen late in the series (patients 14 and 17) and, in contrast to the others, have received aggressive antireflux treatment.
Gastro-oesophageal reflux has been little reported in achondroplasia.36 However, it might result in problems as severe as sudden death and pulmonary parenchymal disease,37 both of which have been well documented in achondroplasia.3-6 11 Our report defines better the significance of severe gastro-oesophageal reflux in the “achondroplasia respiratory difficulty syndrome”. Our data do not identify a causal pathophysiological process, such as gastro-oesophageal reflux leading to lung injury and hyperinflation, or hyperinflation leading to gastro-oesophageal reflux by a mechanical effect on the lower oesophageal sphincter pressures.38However, the time course in some of our more severely affected patients indicates that respiratory compliance worsens with lung growth in the presence of gastro-oesophageal reflux. That being the case, gastro-oesophageal reflux could lead to hyperinflation and small airway disease by at least three mechanisms: (1) frank aspiration of gastric contents into the lower respiratory tract39; (2) aspiration of gastric contents as far as the pharynx, causing bronchoconstriction by stimulation of irritant receptors40; and (3) reflux of gastric contents limited to the lower oesophagus causing symptoms either by increasing bronchial reactivity,41 or by bronchoconstriction secondary to a vagal reflex.42 In general, our patients with severe gastro-oesophageal reflux and lung injury did not have hydrocephalus (group 3), and patients with hydrocephalus did not have gastro-oesophageal reflux (group 2). Jugular venous hypertension with compression at the jugular foramen is an important cause of hydrocephalus in achondroplasia.43 44 This foramen also contains fibres from the nucleus ambiguus forming cranial nerves IX, X, and XI. It is plausible that in patients with jugular foramen compression and hydrocephalus, there is protection against vagally mediated bronchoconstriction, whereas in those without compression there is not.
ACHONDROPLASIA AND UPPER AIRWAY OBSTRUCTION
The occurrence of obstructive sleep apnoea in achondroplasia is well described.3-8 10 Such obstruction might be caused by associated, fixed anatomical abnormalities such as midfacial hypoplasia and/or adenotonsillar hypertrophy, or variable pathophysiological changes occurring in nasopharyngeal or glossal muscle tone. Of the fixed abnormalities, adenotonsillar hypertrophy is the most amenable to treatment intervention, and in many instances adenotonsillectomy results in clinical symptomatic improvement, as seen in our group 1 patients. However, in a significant proportion of our patients, airway obstruction could not be attributed solely to adenotonsillar hypertrophy, either because clinical problems persisted after surgery or because radiology did not confirm encroachment on the airway by adenotonsillar tissue. Rather, the airway obstruction we saw was amenable to night time nasopharyngeal prong airway or continuous positive airway pressure, which implicates pharyngeal muscle hypotonia in the pathophysiology.
Muscles of the nasopharynx and tongue are supplied by cranial nerves IX–XII. The hypoglossal nerve (XII) originates in the medulla. The rootlets form two bundles that perforate the dura mater separately, opposite the hypoglossal canal in the occipital bone, and which then unite after traversing it. The motor portion of the nerve supplies all the muscles of the tongue except the palatoglossus. Nerve fibres from the nucleus ambiguous form cranial nerves IX, X, and XI, which supply the striated musculature of the pharynx, larynx, and upper part of the oesophagus. These three nerves leave the cranium via the jugular foramen. In general, our group 3 patients with upper airway obstruction did not have hydrocephalus, whereas group 2 patients had upper airway obstruction and hydrocephalus. The simplest “muscular” explanation consistent with these findings would be that abnormal muscle tone was exacerbated in AS: in group 3 patients, a restrictive hypoglossal canal with or without foramen magnum stenosis and medullary compression results in at least impaired hypoglossal function, but potentially also global bulbar tone; in group 2 patients, isolated jugular foramen restriction results in both cerebral venous hypertension with hydrocephalus, as well as impaired pharyngeal and laryngeal function, although in some cases associated foramen magnum and/or hypoglossal canal problems may coexist.
ACHONDROPLASIA RESPIRATORY DIFFICULTY SYNDROME IN INFANCY: AN EMBRYOLOGICAL HYPOTHESIS
This assessment of achondroplasia has concentrated on respiratory problems originating in infancy. There appears to be three distinctive phenotypes for the achondroplasia respiratory difficulty syndrome. In one form, there is solely “relative” adenotonsillar hypertrophy caused by a degree of midfacial hypoplasia, which results in obstructive sleep apnoea that clinically resolves with adenotonsillectomy (group 1). In another form there is muscular upper airway obstruction along with progressive hydrocephalus, which could have as a common aetiology jugular foramen stenosis (group 2). In a third form, there is the above muscular upper airway obstruction, but without hydrocephalus, although it is further compounded by the effects of gastro-oesophageal reflux and the development of cor pulmonale (group 3). This could be explained by hypoglossal canal stenosis with or without foramen magnum compression and no jugular foramen stenosis.
In postulating how these differences occur, it is important to consider normal chondrocranial development, current explanations of phenotypic variation as a result of mutations in FGFR-3, and the potential interaction of obstructive sleep apnoea, growth hormone secretion, and postnatal growth. Bones of the nose and basiocciput developing in cartilage from the chondrocranium are: rostrally, the ethmoid bone forming the nasal capsule; caudally, the four proximal parts of the occipital bone forming the foramen magnum; intermediately, the petrous part of the temporal bone forming the anterior border of the jugular foramen, with the posterior border being formed by the part of the occipital bone that develops in membrane. In utero, ossification of these bones progresses in a caudo–rostral direction from the beginning of the second month to the end of the third month. However, some parts of the more rostral chondrocranium still remain at birth and function as growth cartilage.
The normal function of FGFR-3 is to promote terminal differentiation of chondrocytes. Point mutations in the FGFR-3 gene result in premature closure of the growth plate from constitutively active mutant FGFR-3. Clinically, the three skeletal dysplasias attributable to distinct missense mutations in FGFR-3—hypochondroplasia, achondroplasia, and thanatophoric dysplasia45—display a graded spectrum of phenotypic severity, with thanatophoric dysplasia at the lethal end of the spectrum. Interestingly, the degree of activation of FGFR-3 has been shown to correlate closely with the above graded spectrum of phenotypic severity. Naski and colleagues46 reported that the mutation responsible for thanatophoric dysplasia was more strongly activating than the mutation causing achondroplasia, which provides a biochemical explanation for the phenotype of thanatophoric dysplasia being more severe than that of achondroplasia.
Postnatal growth of the non-ossified chondrocranium is influenced by growth hormone secretion. In achondroplasia, there have been reports of the influence of obstructive sleep apnoea on the growth hormone profile.47 48 Waters and colleagues48 found lower secretion of growth hormone in five such patients during the first two hours of slow wave sleep during the night with obstructive sleep apnoea, when compared with studies after either adenotonsillectomy or continuous positive airway pressure. This raises the possibility that phenotypic differences could merely be a result of the severity of obstructive sleep apnoea, with severe cases perpetuating a state of more restricted basioccipital growth.
However, these mechanisms alone are insufficient to account for our principal observations: the phenotypic variation cannot be explained by the degree of activation of FGFR-3 because achondroplasia results almost exclusively from the same FGFR-3 gene mutation1 2; and the pattern of infantile cardiorespiratory and central nervous system pathology with specific aetiological anatomical localisation indicates an embryological rather than postnatal growth aetiology. We propose that the phenotypic variation in infants with the achondroplasia respiratory difficulty syndrome points to a pathophysiology that, in addition to the effects of constitutively active mutant FGFR-3 on chondrocranial development, should by inference include an interactionwith either specific regions in the chondrocranium or time of development. That is to say, caudal or early developmental defects would produce severely affected patients with foramen magnum and/or hypoglossal foramen abnormalities; intermediary defects produce jugular foramen stenosis and associated hydrocephalus; and rostral or late developmental defects produce predominantly midface related problems.
CLINICAL RECOMMENDATIONS
In achondroplasia, all infants and young children with respiratory symptoms require screening for upper airway obstruction using the clinical history and sleep study. In patients with obstructive sleep apnoea, whose problems do not appear to be a result of airway encroachment by adenotonsillar tissue, further investigation of muscular causes using systematic, clinical, neurological, and respiratory physiological assessment, particularly with respect to respiratory compliance, should be undertaken. A phenotype specific pattern of pathology related to chondrocranial embryology warrants consideration because early recognition of certain problems, particularly gastro-oesophageal reflux, might limit the progressive deterioration in cardiorespiratory function seen in the most severely affected individuals.
Acknowledgments
We thank all of the families and the following for their help in the multidisciplinary care and appraisal of these children: Mr D Albert, Dr S Boyd, Professor C Brooke, Dr K Chong, Mr D Drake, Dr R Dinwiddie, Dr C Hall, Mr R Hayward, Dr P Milla, Dr R Stanhope, Dr P Sonksen, Professor P Thorogood, and Professor R Winter.