Article Text

Abstract
Although the activity of nitric oxide (NO) synthases are increased in lung tissue of patients with cystic fibrosis, the concentrations of nasal and exhaled NO have recently been found to be decreased in cystic fibrosis. This could either be due to reduced NO formation or metabolism of NO within airway fluids. In this study, the stable NO metabolites, nitrate and nitrite, were determined in the saliva and sputum of 18 stable cystic fibrosis patients, 21 cystic fibrosis patients during a pulmonary exacerbation, and in saliva and endotracheal secretions of normal controls. Median saliva concentrations of NO metabolites (nitrate plus nitrite) were 704 μmol/l (95% confidence interval (CI) 419 to 1477) in stable cystic fibrosis patients, 629 μmol/l (95% CI 382 to 1392) in cystic fibrosis patients presenting with pulmonary exacerbation, and 313 μmol/l (95% CI 312 to 454) in controls. Median sputum NO metabolite concentration in stable cystic fibrosis was 346 μmol/l (95% CI 311 to 504). This was not significantly different from cystic fibrosis patients presenting with pulmonary exacerbation (median 184 μmol/l, 95% CI 249 to 572), but significantly higher than in endotracheal secretions of controls (median 144 μmol/l, 95% CI 96 to 260). Sputum NO metabolite concentration in cystic fibrosis pulmonary exacerbation significantly increased during antibiotic treatment. A positive correlation was observed between sputum NO metabolites and lung function in stable cystic fibrosis, suggesting less airway NO formation in cystic fibrosis patients with more severe lung disease. These data indicate that decreased exhaled NO concentrations in cystic fibrosis patients may be due to retention and metabolism of NO within the airway secretions. However, sputum NO metabolites are not a useful marker of airway inflammation in cystic fibrosis lung disease.
- cystic fibrosis
- lung inflammation
- nitric oxide
- nitrate
Statistics from Altmetric.com
Nitric oxide (NO) is enzymatically produced within the human airways and is detectable in the exhaled air of healthy individuals.1 2 The physiological role of airway NO is thought to include modulation of ciliary activity, mediation of inflammation, vasodilation, and bronchodilation.3Induction of NO synthases during airway inflammation results in increased concentrations of exhaled NO, as demonstrated in asthma, bronchiectasis, or upper respiratory tract infection.4-6
Lung disease in cystic fibrosis is characterised by chronic airway inflammation even in stable patients, as reflected by high airway fluid concentrations of proinflammatory cytokines.7-9 The cytokines tumour necrosis factor-α and interleukin-1β (IL-1β) have been shown to induce NO synthases in airway epithelial cells,10 11 and increased NO synthases activity has recently been found in lung tissue of cystic fibrosis patients.12 While these data would suggest increased NO formation in cystic fibrosis, lower than normal concentrations of NO have been found in the upper airways of cystic fibrosis patients.13-15 Lower airway NO in cystic fibrosis patients was either decreased or not different from normal individuals.13-16 It remains unclear, whether decreased airway NO concentrations in cystic fibrosis lung disease result from reduced NO formation or clearance of NO within the airway fluids. Since NO is a free radical, it is rapidly converted to distinct oxides of nitrogen (NOx) in aqueous solutions and at air-aqueous interfaces. Interconversion among the NOx species occurs, resulting in formation of the stable NO degradation products nitrate and nitrite.17It is possible that NO produced by airway tissues is partially metabolised to stable NO products at diffusion through the air-liquid interface. In a preliminary study involving a small number of patients, higher concentrations of both nitrite and IL-8 were found in cystic fibrosis patients during pulmonary exacerbation as compared with clinically stable cystic fibrosis patients.18 To assess whether NO degradation products in airway liquids could be used as markers of airway inflammation in cystic fibrosis lung disease, we assayed nitrate and nitrite in airway fluids of stable cystic fibrosis patients and cystic fibrosis patients with pulmonary exacerbation.
Patients and methods
CYSTIC FIBROSIS PATIENTS
Saliva and expectorated sputum were obtained from 39 cystic fibrosis patients. The diagnosis of cystic fibrosis had been previously confirmed in all patients. The study was approved by the ethics committee of our institution.
Clinical characteristics of the cystic fibrosis patients are displayed in table 1. Patients were divided into two groups according to the following criteria: stable cystic fibrosis patients who attended the outpatient department for scheduled visits. None of these patients presented with exacerbation of respiratory symptoms at the time of study. Lung function was assessed by spirometry in all patients (table1). Sputum was obtained from all 18 and saliva from 10 of these patients. Cystic fibrosis patients who were included in the other group presented with symptoms of pulmonary exacerbation according to widely accepted criteria.19 Lung function, assessed at the last scheduled visit before exacerbation, did not differ significant from the stable patients (table 1). Sputum from 21 patients with symptoms of pulmonary exacerbation was obtained on the day of admission to hospital and, in 11 of these patients, after 14 days of antibiotic treatment. Saliva was obtained from 17 of these patients at admission.
Age, pulmonary function, and sputum colonisation status in the cystic fibrosis patients
CONTROLS
Saliva was obtained from 43 non-smoking, healthy children and adults with an age range of 6–37 years (mean (SD) 22.7 (7.8) years). None had a history of respiratory or cardiovascular disease or was receiving any medication.
As sputum is not produced by healthy individuals cystic fibrosis sputum concentrations were compared with endotracheal secretions obtained from 10 additional controls, undergoing surgery for non-pulmonary diseases. The age range was 17–64 years (mean (SD) 37.7 (20.5) years). Endotracheal secretions were collected from the mucus layer coating a freshly removed endotracheal tube in these patients. These samples were stored at −80°C before analysis.
LUNG FUNCTION
Forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) were measured by spirometry (Volugraph, Mijnhardt, Bunnik, The Netherlands). Results were expressed as percentages of reference values.20 21
MEASUREMENT OF NITRATE AND NITRITE
Airway fluid and serum samples were collected in sterile containers and stored at −20°C. Analyses were performed within 24 hours. In preliminary measurements we observed no changes in nitrate or nitrite concentrations during three days of storage at −20°C. Airway fluids were diluted with an equal volume of double distilled water and then homogenised in an ultrasonic waterbath for 10 minutes. Samples were centrifuged at 10 × g for 10 minutes. The clear supernatant was used for analysis. Serum samples were assayed undiluted. All samples were blinded before analysis. Nitrate and nitrite were determined by a colorimetric assay based on the Griess reaction, as described previously.22 Briefly, nitrite was assayed directly after 10 minutes of incubation with Griess reagent by measuring the absorbance at 546 nm versus a reference cuvette containing aqua bidest and airway fluid or serum. Nitrate was measured after its enzymatical reduction to nitrite. Concentrations of nitrate were determined from a linear standard curve obtained from sodium nitrate under the same experimental conditions. Concentrations of nitrate or nitrite were expressed in μmol/l. The detection limit of the assay was 3 μmol/l.
STATISTICS
NO metabolite concentrations were expressed as median and 95% confidence interval (CI). Since the data were not normally distributed, comparison between the groups was performed with the Wilcoxon test. Correlations between NO metabolite concentrations in cystic fibrosis were assessed by linear regression analysis. A p value of less than 0.05 was considered statistically significant.
Results
Nitrate and nitrite concentrations in cystic fibrosis and control airway secretions are shown in tables 2 and 3. NO metabolite concentrations in stable cystic fibrosis sputum were significantly higher than in endotracheal secretions of controls (p<0.005), but did not differ significantly from cystic fibrosis patients presenting with pulmonary exacerbation (fig 1). Nitrite was not detectable in any of the control endotracheal secretions but present in all cystic fibrosis sputum samples (table2).
Nitrate and nitrite concentrations (μmol/l) in cystic fibrosis sputum and control endotracheal secretions
Nitrate and nitrite concentrations (μmol/l) in saliva of cystic fibrosis patients and controls
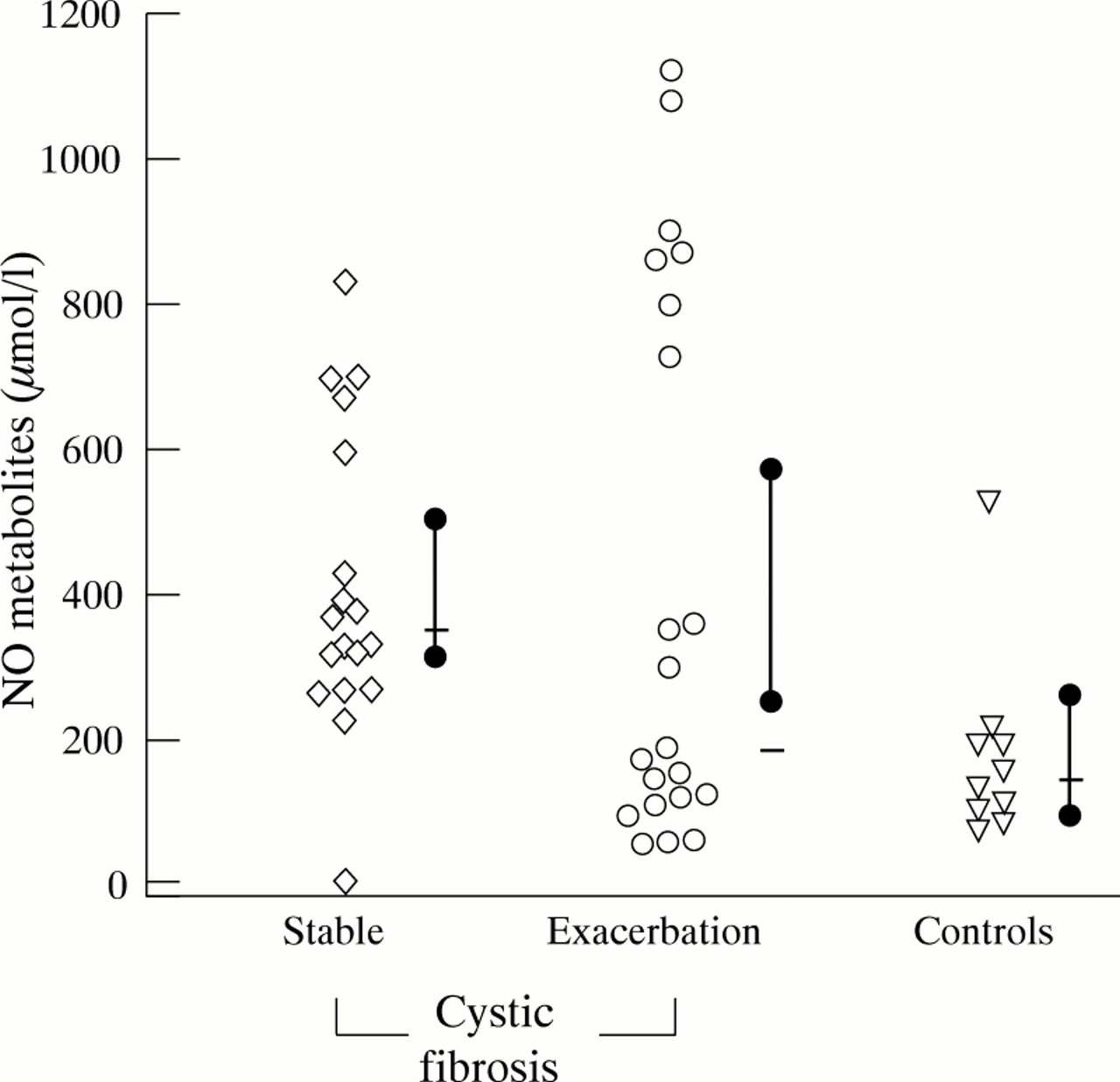
NO metabolite concentrations (μmol/l) in sputum of stable cystic fibrosis patients, cystic fibrosis patients presenting with a pulmonary exacerbation, and in endotracheal secretions of controls. Also shown are the median (bar) with 95% CI (solid dot). Sputum concentrations in stable cystic fibrosis patients were significantly higher than in control endotracheal secretions (p<0.005).
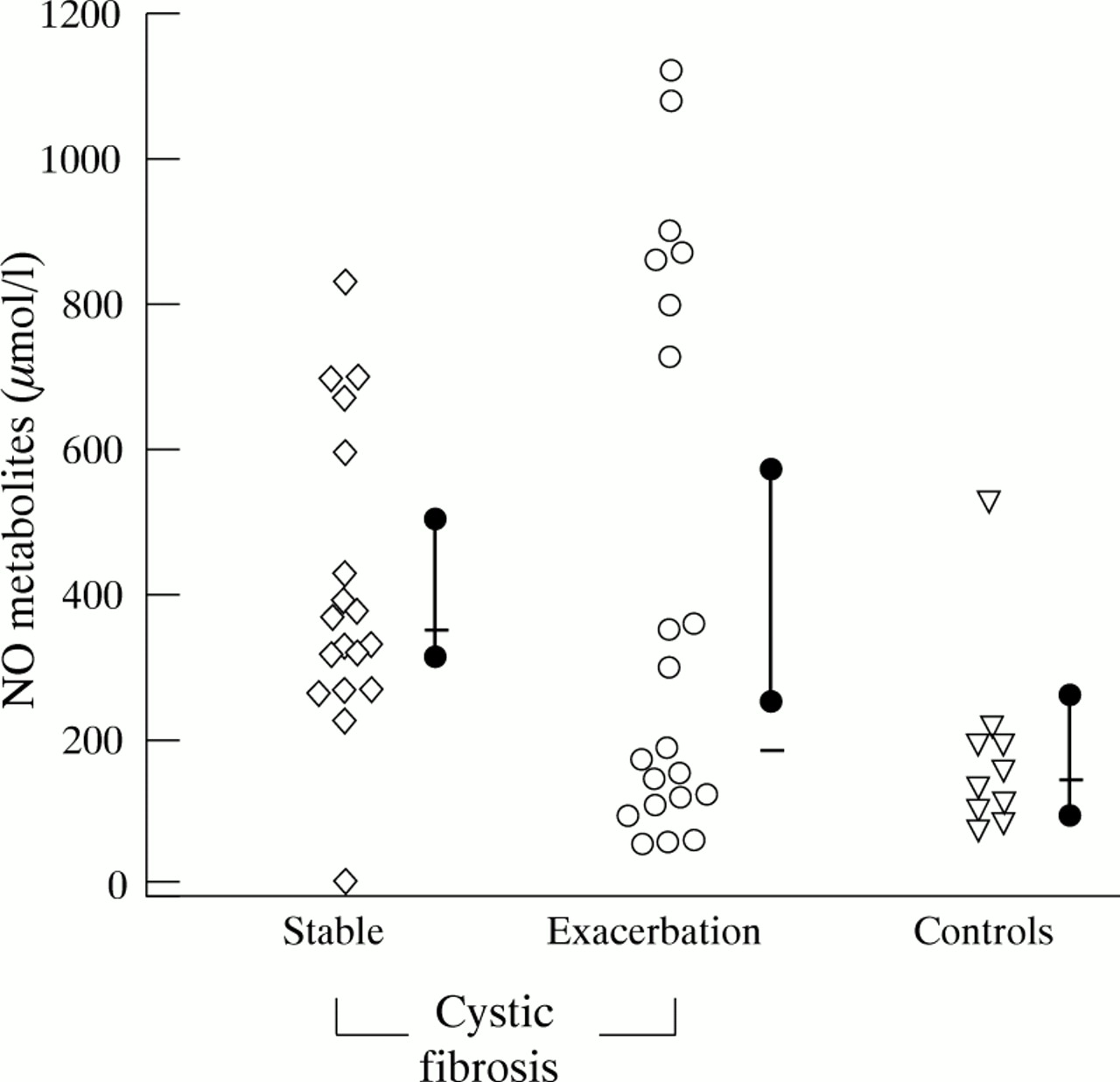
Salivary NO metabolite concentrations in stable cystic fibrosis were also significantly higher than in controls (p<0.01), and not different from cystic fibrosis patients presenting with pulmonary exacerbation (fig 2).

NO metabolite concentrations (μmol/l) in saliva of stable cystic fibrosis patients, cystic fibrosis patients presenting with a pulmonary exacerbation, and controls. Also shown are the median (bar) with 95% CI (solid dot). Saliva concentrations in stable cystic fibrosis patients were significantly higher than in controls (p<0.01).
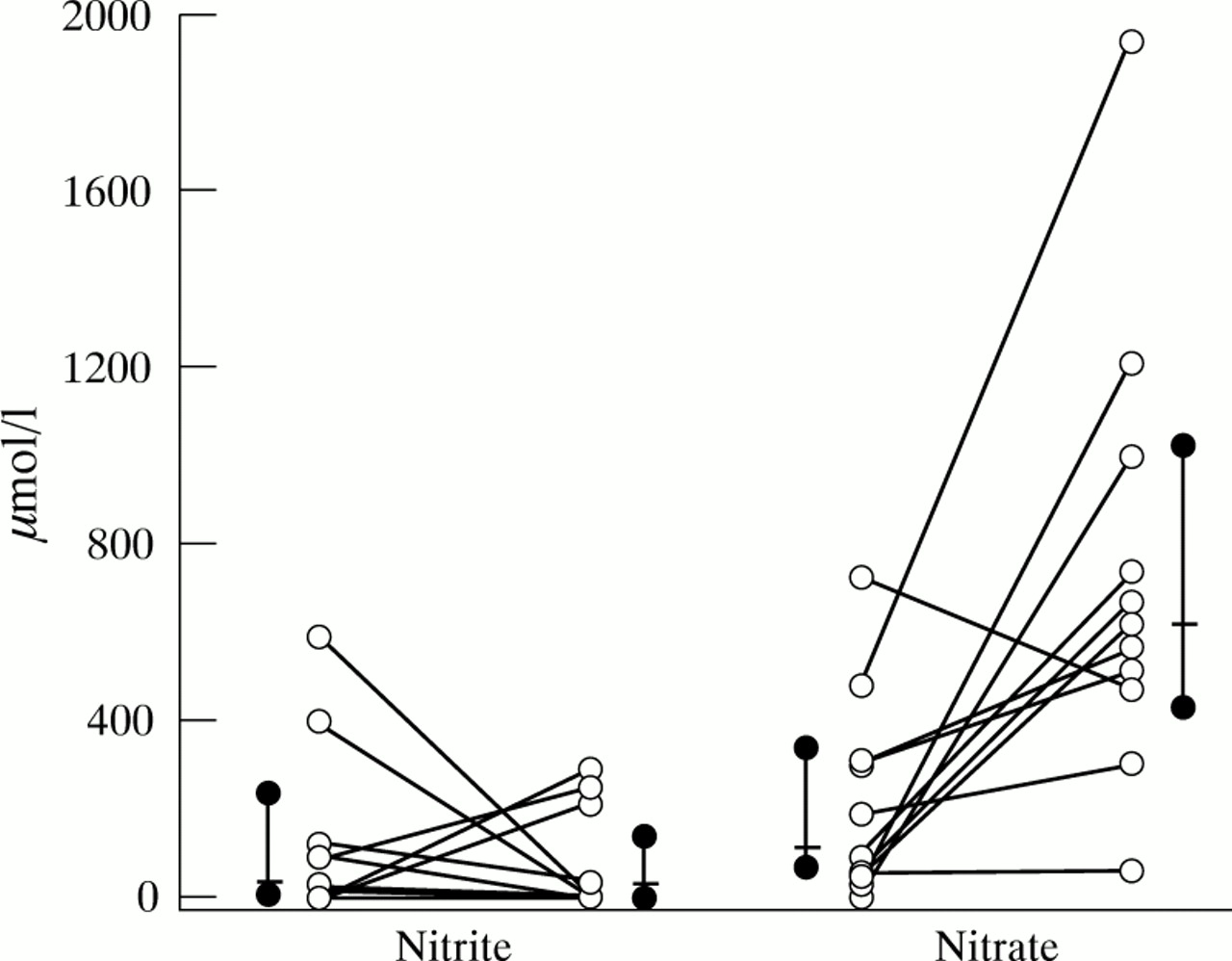
Saliva or sputum NO metabolite concentrations in cystic fibrosis patients presenting with symptoms of pulmonary exacerbation did not differ significantly from saliva or endotracheal secretions of controls (figs 1 and 2). However, sputum nitrate concentration increased significantly during 14 days of intravenous antibiotic treatment for a pulmonary exacerbation (p<0.005), while no significant changes were observed in sputum nitrite concentrations (fig 3).
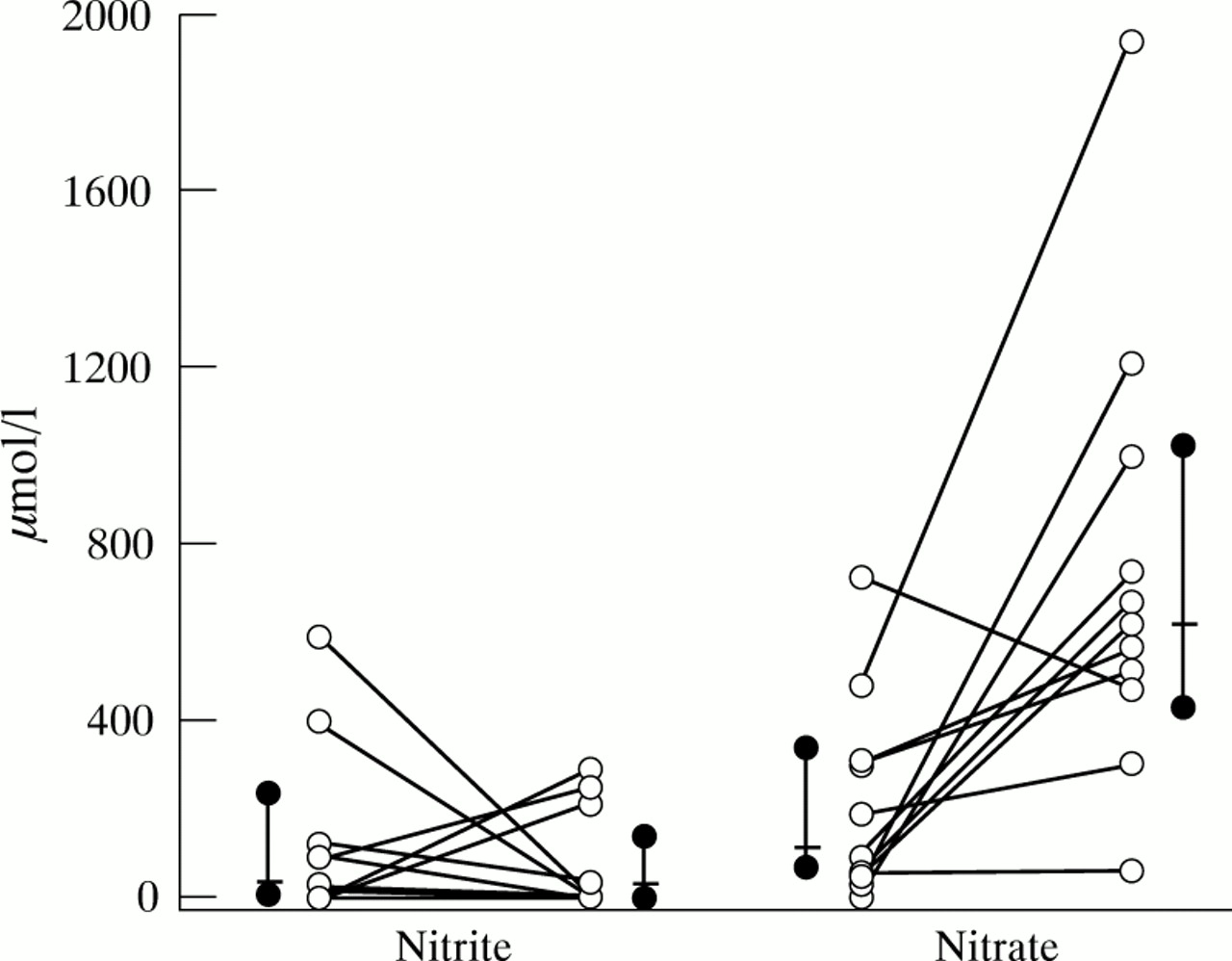
Sputum nitrite and nitrate concentrations (μmol/l) in 11 cystic fibrosis patients before and after 14 days of antibiotic treatment for a pulmonary exacerbation. Each coupled symbol represents one individual. Also shown are the median (bar) with 95% CI (solid dot). A significant increase was observed in sputum nitrate concentrations (p<0.005).
All patients were colonised with either Pseudomonas aeruginosa or Staphylococcus aureus, 10 with both organisms. There was no difference in sputum NO metabolite concentrations between P aeruginosa positive or negative as well as S aureus positive or negative cystic fibrosis patients.
Median serum concentration of nitrate in cystic fibrosis patients was 36 μmol/l (95% CI 29 to 55), similar to previously reported levels in healthy children and adults.22 23 However, the variability was considerably higher in cystic fibrosis patients. Nitrite was not detectable in any of the serum samples. Serum nitrate did not show any correlation with other serum markers of inflammation such as white cell count, percentage of neutrophils, C reactive protein, or erythrocyte sedimentation rate (data not shown).
Significant correlations were observed between nitrate concentrations in sputum and serum from patients with cystic fibrosis (n=30) (p=0.001;r=0.66), saliva and serum (p<0.01, r=0.54), and sputum and saliva (n=55) (p<0.001, r=0.5) (data not shown).
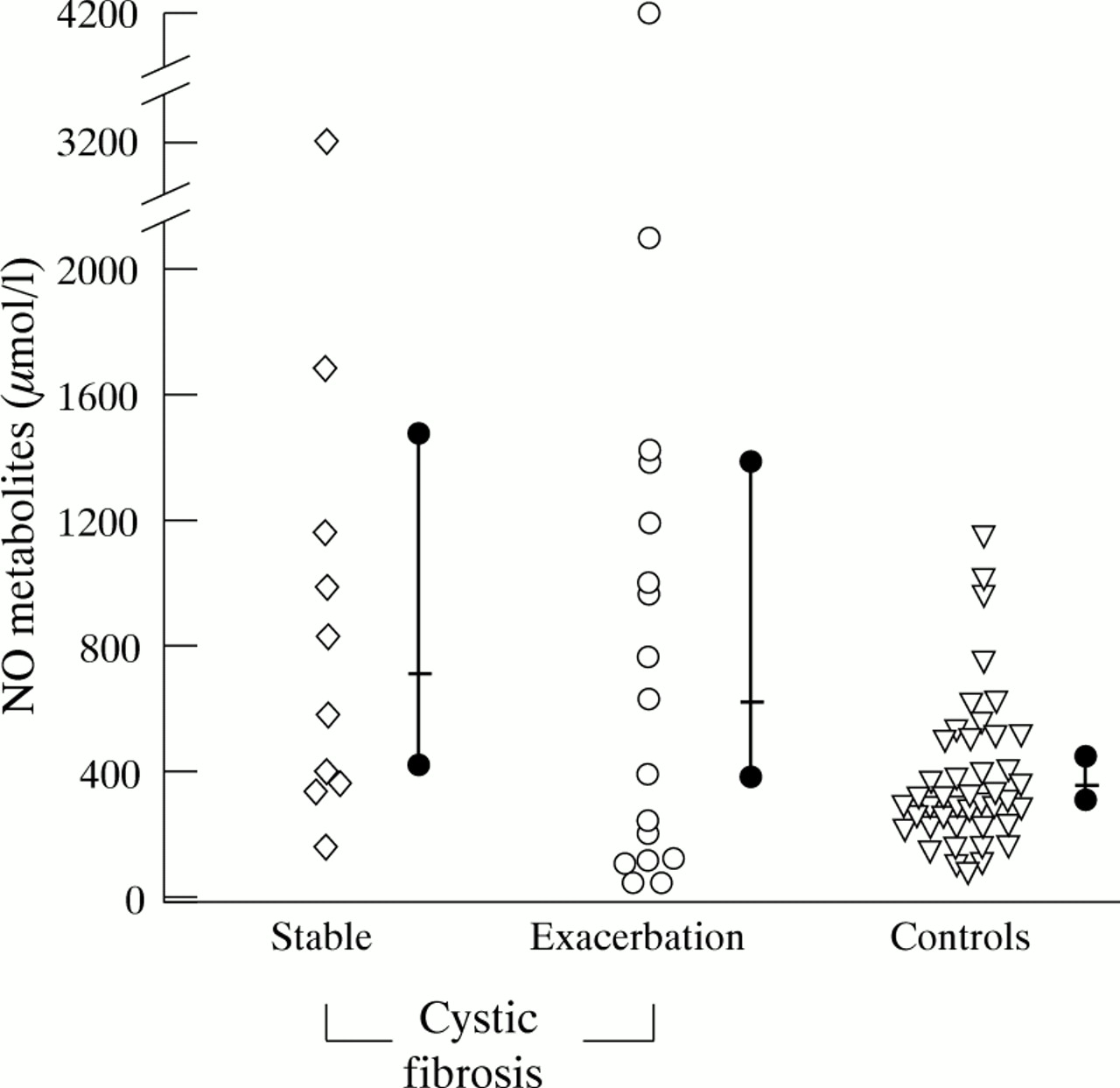
Sputum NO metabolite concentrations were compared with pulmonary function parameters in stable cystic fibrosis patients. NO metabolites were significantly correlated with FEV1 (p<0.02,r=0.58) (fig 4), as well as FVC (p<0.02,r=0.55) in these patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between sputum concentration of NO metabolites (nitrite plus nitrate) in μmol/l and FEV1 in % predicted in cystic fibrosis patients. Each diamond symbol represents one individual. NO metabolite concentrations increase with increasing FEV1 (r=0.58, p<0.02, linear regression analysis).
Discussion
We have shown that high concentrations of the stable NO metabolites nitrate and nitrite are detectable in cystic fibrosis airway secretions. In previous studies concentrations of exhaled NO in cystic fibrosis were either not significantly different or lower than in controls.13-16 The present study suggests that a significant amount of cystic fibrosis airway NO metabolites are retained in airway fluids. Whether lower airway NO metabolite concentrations are higher in cystic fibrosis than in controls cannot yet be answered. Since sputum is not produced by healthy individuals, cystic fibrosis sputum was compared with control endotracheal secretions. The components of these two airway secretions are different, and so might be the NO metabolite concentrations. However, sputum NO metabolite concentrations in cystic fibrosis did not seem to correlate with the activity of airway inflammation since no significant differences were observed between stable cystic fibrosis patients and cystic fibrosis patients presenting with symptoms of pulmonary exacerbation. Therefore, as previously demonstrated for exhaled NO, sputum nitrate or nitrite are unsuitable markers of cystic fibrosis airway inflammation.
Metabolites of NO are detectable in bronchoalveolar lavage fluid and airway aspirates from normal subjects.23 24 It is conceivable that increased airway tissue NO formation will result in increased NO metabolite concentrations in airway fluids. This has recently been confirmed by the observation of increased nitrate concentrations in bronchoalveolar lavage fluid of children with pneumonia, compared with healthy children.23 However, nitrate and nitrite concentrations in airway secretions are also influenced by bacterial infection. The majority of the airways in the cystic fibrosis study population were chronically colonised withP aeruginosa. Pseudomonas, a common pathogen in cystic fibrosis lung disease, is able to metabolise both nitrate and nitrite by enzymatic reduction.25 Furthermore, P aeruginosa synthesises a phenazine pigment, pyocyanin, that interacts with NO and might thereby reduce nitrate or nitrite concentrations in pseudomonas infected cystic fibrosis patients.26 The increase in cystic fibrosis sputum nitrate concentration during antibiotic treatment of a pulmonary exacerbation could thus be explained by a reduction in the bacterial load, resulting in decreased bacterial metabolism of nitrate and in decreased pyocyanin sputum concentrations. Interestingly, in a recent study, serum NO metabolite concentrations in children with bacterial infections showed a comparable pattern as they were normal at the time of diagnosis but increased during the treatment period.27
Diffusion of NO metabolites from the airways across the alveolar-capillary membrane into the circulation or the inflammatory response to pulmonary inflammation could increase serum NO metabolite concentrations. We therefore assayed nitrate or nitrite in cystic fibrosis serum. However, mean nitrate concentration in cystic fibrosis patients did not differ from previously reported concentrations in healthy controls,22 23 and serum nitrate did not correlate with serological markers of inflammation.
We observed a positive correlation between sputum NO metabolite concentrations and lung function in stable cystic fibrosis patients, indicating decreased NO formation in the lower airways of cystic fibrosis patients with more advanced lung disease. This coincides with our recent findings of low exhaled NO concentration in cystic fibrosis patients with decreased lung function.16 As NO is a potent bronchodilator, low NO formation in advanced cystic fibrosis lung disease may be of physiological relevance as it could contribute to the bronchial obstruction in these patients.
The highest NO metabolite concentrations in our study were observed in saliva. Although NO synthases are expressed in epithelial cells from the upper and lower respiratory tract,17 exhaled NO is mainly derived from the upper airway.28 Therefore, one could speculate, that high NO metabolite concentrations in saliva reflect upper airway NO formation. Saliva NO metabolites could also be influenced by nitrate and nitrite formation in salivary glands or by colonisation with oral bacteria.29 Expectorated sputum is inevitably contaminated with saliva and it is conceivable that this artificially raised sputum NO metabolite concentrations in our study.
In conclusion, concentrations of the NO metabolites nitrate and nitrite are high in cystic fibrosis, despite the fact that both lower airway and nasal NO concentrations are low in these patients. Sputum nitrate or nitrite concentrations appear not to be useful markers of cystic fibrosis airway inflammation. However, our results give further evidence that cystic fibrosis patients with more advanced pulmonary disease produce less airway NO than patients with better lung function. Further studies will address the question whether augmentation of NO formation has a positive effect on lung function in cystic fibrosis patients with severe lung disease.