
The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis

Abstract
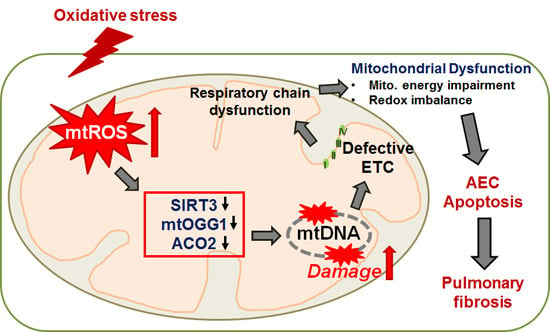
:
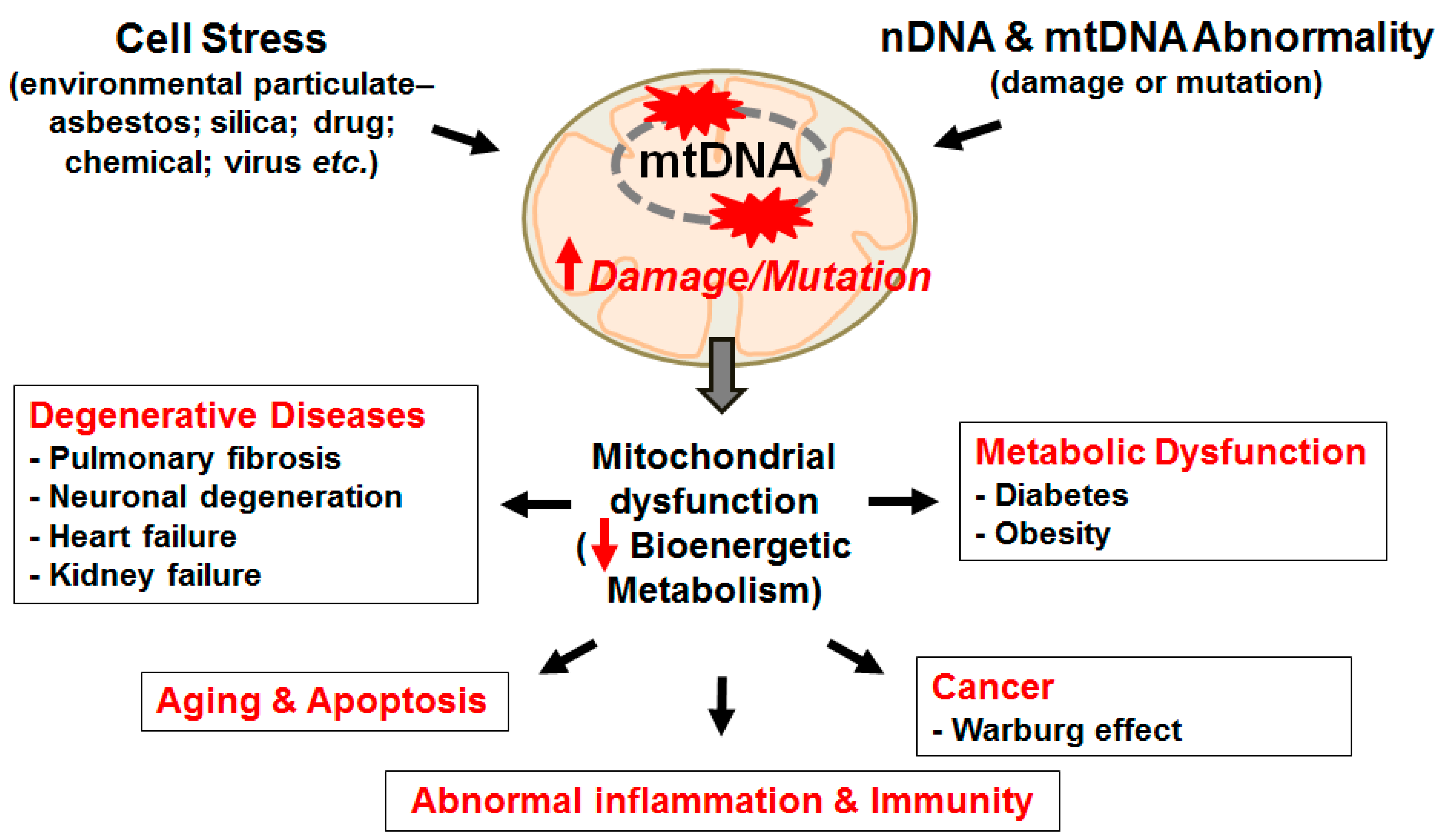
{kind=link}
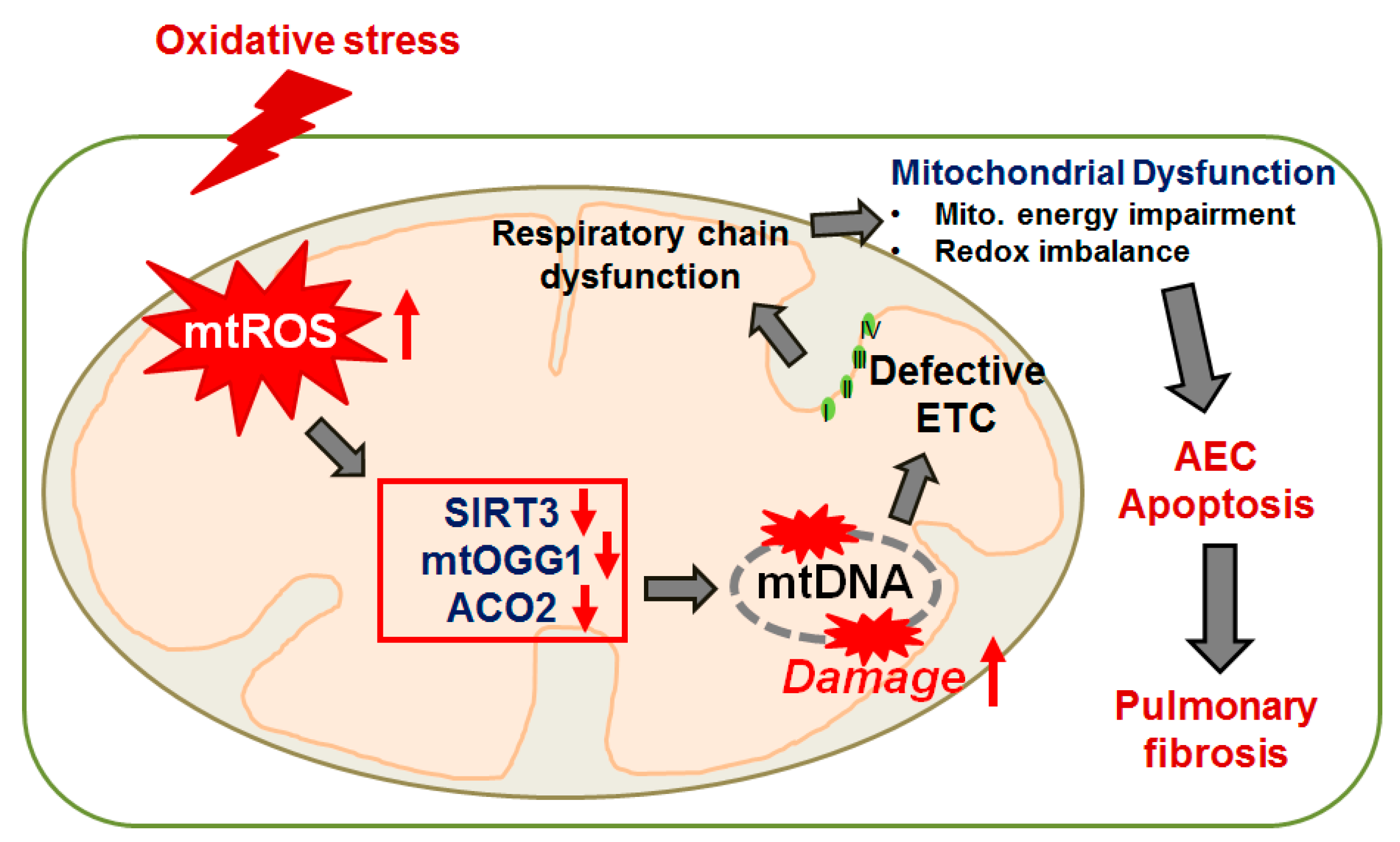
{kind=link}
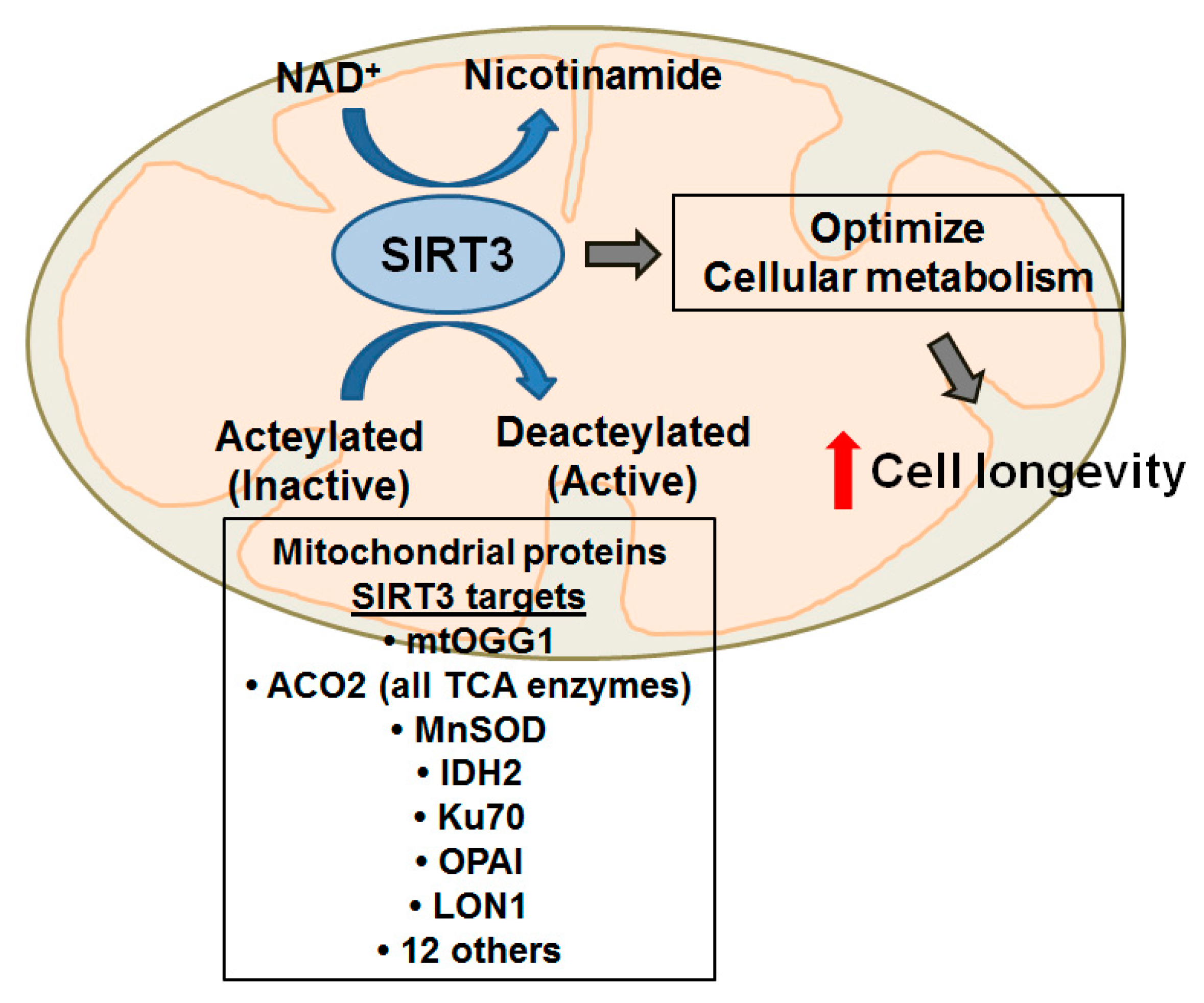
{kind=link}
{kind=link}
1. Introduction


2. The Mitochondria, mtDNA, and ROS—The Basics
3. AEC Apoptosis and Lung Fibrosis—Role of the Mitochondria
3.1. AEC Aging, Apoptosis and Pulmonary Fibrosis
3.2. Mitochondria-Regulated AEC Apoptosis
3.3. mtDNA Damage and Repair—Role in Cancer and Lung Fibrosis
3.4. Animals Models of Pulmonary Fibrosis—Role of Mitochondrial ROS, mtDNA Damage, and Mitochondrial Dysfunction
4. The Role of Sirtuins in Mitochondrial Integrity, mtDNA Damage Repair, and Aging
4.1. Overview of the Sirtuin (SIRT) Family Members
4.2. The Role of SIRTs and Mitochondria; Normal and General Diseases

4.3. The Role of SIRT3 in mtDNA Damage, Aging and Diseases Including Lung
4.4. Therapeutic Approach: Resveratrol/Viniferin/Honokiol
5. MtDNA and Metabolism in Mitochondria
5.1. Mitochondrial Metabolism—The Basics
5.2. Role of Mitochondria-Derived ROS, mtDNA Damage, and Mitochondrial Metabolism
5.3. Role of Sirtuins and Energy Metabolism in Mitochondria
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays 2015, 37, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J.; Rosenfeldt, F.L.; Zhang, C.; Linnane, A.W.; Nagley, P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a pcr-based assay: Lack of change of copy number with age. Nucleic Acids Res. 2003, 31, e61. [Google Scholar] [CrossRef] [PubMed]
- Bell, O.; Tiwari, V.K.; Thoma, N.H.; Schubeler, D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 2011, 12, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial bioenergetic etiology of disease. J. Clin. Investig. 2013, 123, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Cheresh, P.; Williams, D.; Cheng, Y.; Ridge, K.; Schumacker, P.T.; Weitzman, S.; Bohr, V.A.; Kamp, D.W. Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J. Biol. Chem. 2014, 289, 6165–6176. [Google Scholar] [CrossRef] [PubMed]
- Panduri, V.; Liu, G.; Surapureddi, S.; Kondapalli, J.; Soberanes, S.; de Souza-Pinto, N.C.; Bohr, V.A.; Budinger, G.R.; Schumacker, P.T.; Weitzman, S.A.; et al. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic. Biol. Med. 2009, 47, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y. Regulation of intracellular localization of human MTH1, OGG1, and MYH proteins for repair of oxidative DNA damage. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 68, 75–94. [Google Scholar] [PubMed]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 2006, 103, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in lung biology and pathology: More than just a powerhouse. Am. J. Physiol. Lung. Cell Mol. Physiol. 2014, 306, L962–L974. [Google Scholar] [CrossRef] [PubMed]
- Chouteau, J.M.; Obiako, B.; Gorodnya, O.M.; Pastukh, V.M.; Ruchko, M.V.; Wright, A.J.; Wilson, G.L.; Gillespie, M.N. Mitochondrial DNA integrity may be a determinant of endothelial barrier properties in oxidant-challenged rat lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L892–L898. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.K.; van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Patterson, C.; Yan, C.N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res. 2000, 86, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Bohr, V.A.; Stevnsner, T.; de Souza-Pinto, N.C. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 2002, 286, 127–134. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M. The contribution of mitochondria to common disorders. Mol. Genet. Metab. 2003, 80, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 2012, 1819, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a source of reactive oxygen and nitrogen species: From molecular mechanisms to human health. Antioxid. Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Aaij, R.; Abellan Beteta, C.; Adeva, B.; Adinolfi, M.; Adrover, C.; Affolder, A.; Ajaltouni, Z.; Albrecht, J.; Alessio, F.; Alexander, M.; et al. First observation of CP violation in the decays of mesons. Phys. Rev. Lett. 2013, 110, 221601. [Google Scholar] [CrossRef] [PubMed]
- Kenney, M.C.; Chwa, M.; Atilano, S.R.; Falatoonzadeh, P.; Ramirez, C.; Malik, D.; Tarek, M.; Caceres-del-Carpio, J.; Nesburn, A.B.; Boyer, D.S.; et al. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: Insights into mitochondrial-nuclear interactions. Hum. Mol. Genet. 2014, 23, 3537–3551. [Google Scholar] [CrossRef] [PubMed]
- Kamp, D.W.; Graceffa, P.; Pryor, W.A.; Weitzman, S.A. The role of free radicals in asbestos-induced diseases. Free Radic. Biol. Med. 1992, 12, 293–315. [Google Scholar] [CrossRef]
- Turci, F.; Tomatis, M.; Lesci, I.G.; Roveri, N.; Fubini, B. The iron-related molecular toxicity mechanism of synthetic asbestos nanofibres: A model study for high-aspect-ratio nanoparticles. Chemistry 2011, 17, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Faner, R.; Rojas, M.; Macnee, W.; Agusti, A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Kliment, C.R.; Oury, T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Gazdhar, A.; Lebrecht, D.; Roth, M.; Tamm, M.; Venhoff, N.; Foocharoen, C.; Geiser, T.; Walker, U.A. Time-dependent and somatically acquired mitochondrial DNA mutagenesis and respiratory chain dysfunction in a scleroderma model of lung fibrosis. Sci. Rep. 2014, 4, 5336. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cheresh, P.; Kamp, D.W. Molecular basis of asbestos-induced lung disease. Annu. Rev. Pathol. 2013, 8, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Oury, T.D.; Thakker, K.; Menache, M.; Chang, L.Y.; Crapo, J.D.; Day, B.J. Attenuation of bleomycin-induced pulmonary fibrosis by a catalytic antioxidant metalloporphyrin. Am. J. Respir. Cell Mol. Biol. 2001, 25, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Osborn-Heaford, H.L.; Ryan, A.J.; Murthy, S.; Racila, A.M.; He, C.; Sieren, J.C.; Spitz, D.R.; Carter, A.B. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J. Biol. Chem. 2012, 287, 3301–3312. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.; Ryan, A.; He, C.; Mallampalli, R.K.; Carter, A.B. Rac1-mediated mitochondrial H2O2 generation regulates MMP-9 gene expression in macrophages via inhibition of SP-1 and AP-1. J. Biol. Chem. 2010, 285, 25062–25073. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.; Adamcakova-Dodd, A.; Perry, S.S.; Tephly, L.A.; Keller, R.M.; Metwali, N.; Meyerholz, D.K.; Wang, Y.; Glogauer, M.; Thorne, P.S.; et al. Modulation of reactive oxygen species by rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L846–L855. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Murthy, S.; McCormick, M.L.; Spitz, D.R.; Ryan, A.J.; Carter, A.B. Mitochondrial Cu, Zn-superoxide dismutase mediates pulmonary fibrosis by augmenting H2O2 generation. J. Biol. Chem. 2011, 286, 15597–15607. [Google Scholar] [CrossRef] [PubMed]
- Mossman, B.T.; Marsh, J.P.; Sesko, A.; Hill, S.; Shatos, M.A.; Doherty, J.; Petruska, J.; Adler, K.B.; Hemenway, D.; Mickey, R.; et al. Inhibition of lung injury, inflammation, and interstitial pulmonary fibrosis by polyethylene glycol-conjugated catalase in a rapid inhalation model of asbestosis. Am. Rev. Respir. Dis. 1990, 141, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Kinnula, V.L.; Myllarniemi, M.; Oury, T.D. Extracellular superoxide dismutase in pulmonary fibrosis. Antioxid. Redox Signal. 2008, 10, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Hubbard, R.C.; Crystal, R.G. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 1989, 139, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Waghray, M.; Cui, Z.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005, 19, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.I.; Ishii, Y.; Kitamura, S. Aerosolized administration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am. J. Respir. Crit. Care Med. 2000, 162, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; de Andrade, J.A.; Anstrom, K.J.; King, T.E., Jr.; Raghu, G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2093–2101. [Google Scholar] [PubMed]
- Crestani, B.; Besnard, V.; Boczkowski, J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2011, 43, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Cheng, J.; Thannickal, V.J. Targeting nox enzymes in pulmonary fibrosis. Cell Mol. Life Sci. 2012, 69, 2365–2371. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra247. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J. Mechanistic links between aging and lung fibrosis. Biogerontology 2013, 14, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Uhal, B.D.; Nguyen, H. The Witschi Hypothesis revisited after 35 years: Genetic proof from SP-C BRICHOS domain mutations. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L906–L911. [Google Scholar] [CrossRef] [PubMed]
- Mossman, B.T.; Lippmann, M.; Hesterberg, T.W.; Kelsey, K.T.; Barchowsky, A.; Bonner, J.C. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J. Toxicol. Environ. Health B Crit. Rev. 2011, 14, 76–121. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Blackwell, T.S.; Lawson, W.E. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L721–L729. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.H.; Budinger, G.R.; Mutlu, G.M.; Jain, M. Proteasomal regulation of pulmonary fibrosis. Proc. Am. Thorac. Soc. 2010, 7, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Brody, A.R.; Overby, L.H. Incorporation of tritiated thymidine by epithelial and interstitial cells in bronchiolar-alveolar regions of asbestos-exposed rats. Am. J. Pathol. 1989, 134, 133–140. [Google Scholar] [PubMed]
- Shukla, A.; Jung, M.; Stern, M.; Fukagawa, N.K.; Taatjes, D.J.; Sawyer, D.; van Houten, B.; Mossman, B.T. Asbestos induces mitochondrial DNA damage and dysfunction linked to the development of apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L1018–L1025. [Google Scholar] [CrossRef] [PubMed]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Pritchett, J.M.; Zoz, D.F.; Crossno, P.F.; Markin, C.; Garnett, E.T.; Degryse, A.L.; Mitchell, D.B.; Polosukhin, V.V.; Rickman, O.B.; et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Cogan, J.D.; Kropski, J.A.; Zhao, M.; Mitchell, D.B.; Rives, L.; Markin, C.; Garnett, E.T.; Montgomery, K.H.; Mason, W.R.; McKean, D.F.; et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2015, 191, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Med. 2013, 5, a021220. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Jaurand, M.C.; Kamp, D.W.; Whysner, J.; Hei, T.K. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J. Toxicol. Environ. Health B Crit. Rev. 2011, 14, 179–245. [Google Scholar] [CrossRef] [PubMed]
- Anathy, V.; Roberson, E.C.; Guala, A.S.; Godburn, K.E.; Budd, R.C.; Janssen-Heininger, Y.M. Redox-based regulation of apoptosis: S-glutathionylation as a regulatory mechanism to control cell death. Antioxid. Redox Signal. 2012, 16, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Anathy, V.; Roberson, E.; Cunniff, B.; Nolin, J.D.; Hoffman, S.; Spiess, P.; Guala, A.S.; Lahue, K.G.; Goldman, D.; Flemer, S.; et al. Oxidative processing of latent fas in the endoplasmic reticulum controls the strength of apoptosis. Mol. Cell. Biol. 2012, 32, 3464–3478. [Google Scholar] [CrossRef] [PubMed]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M. Telomerase and idiopathic pulmonary fibrosis. Mutat. Res. 2012, 730, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Xu, X.C.; Newman, J.L.; Mitchell, D.B.; Tanjore, H.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Phillips, J.A., 3rd; et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 2012, 38, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Hunakova, L.; Chen, Y.; Bortner, C.; van Houten, B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J. Biol. Chem. 2003, 278, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Budinger, G.R.; Mutlu, G.M.; Eisenbart, J.; Fuller, A.C.; Bellmeyer, A.A.; Baker, C.M.; Wilson, M.; Ridge, K.; Barrett, T.A.; Lee, V.Y.; et al. Proapoptotic bid is required for pulmonary fibrosis. Proc. Natl. Acad. Sci. USA. 2006, 103, 4604–4609. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. Pink1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and pink1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [PubMed]
- Lounsbury, K.M.; Stern, M.; Taatjes, D.; Jaken, S.; Mossman, B.T. Increased localization and substrate activation of protein kinase Cδ in lung epithelial cells following exposure to asbestos. Am. J. Pathol. 2002, 160, 1991–2000. [Google Scholar] [CrossRef]
- Buder-Hoffmann, S.A.; Shukla, A.; Barrett, T.F.; MacPherson, M.B.; Lounsbury, K.M.; Mossman, B.T. A protein kinase Cδ-dependent protein kinase D pathway modulates ERK1/2 and JNK1/2 phosphorylation and Bim-associated apoptosis by asbestos. Am. J. Pathol. 2009, 174, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Choi, S.; Lee, J.; Huang, Y.T.; Chen, F.; Zhao, Y.; Lin, X.; Neuberg, D.; Kim, J.; Christiani, D.C. Mitochondrial variations in non-small cell lung cancer (NSCLC) survival. Cancer Inform. 2015, 14, 1–9. [Google Scholar] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. Ros-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.P.; Attardi, G. Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell 1988, 52, 811–819. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, S.; Radicella, J.P.; Levalois, C.; Lebeau, J.; Poupon, M.F.; Oudard, S.; Dutrillaux, B.; Boiteux, S. Mutations in OGG1, a gene involved in the repair of oxidative DNA damage, are found in human lung and kidney tumours. Oncogene 1998, 16, 3083–3086. [Google Scholar] [CrossRef] [PubMed]
- Youn, C.K.; Song, P.I.; Kim, M.H.; Kim, J.S.; Hyun, J.W.; Choi, S.J.; Yoon, S.P.; Chung, M.H.; Chang, I.Y.; You, H.J. Human 8-oxoguanine DNA glycosylase suppresses the oxidative stress induced apoptosis through a p53-mediated signaling pathway in human fibroblasts. Mol. Cancer Res. 2007, 5, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Ruchko, M.; Gorodnya, O.; LeDoux, S.P.; Alexeyev, M.F.; Al-Mehdi, A.B.; Gillespie, M.N. Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L530–L535. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, K.; Stuart, J.A.; de Souza-Pinto, N.C.; Bohr, V.A. The C-terminal aO helix of human Ogg1 is essential for 8-oxoguanine DNA glycosylase activity: The mitochondrial β-Ogg1 lacks this domain and does not have glycosylase activity. Nucleic Acids Res. 2004, 32, 5596–5608. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Ikeda-Saito, M.; Szweda, L.I. Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry 2003, 42, 14846–14855. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Wang, X.; Kaufman, B.A.; Butow, R.A. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 2005, 307, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, P.; Morales-Nebreda, L.; Kim, S.J.; Yeldandi, A.; Williams, D.B.; Cheng, Y.; Mutlu, G.M.; Budinger, G.R.; Ridge, K.; Schumacker, P.T.; et al. Asbestos-induced pulmonary fibrosis is augmented in 8-oxoguanine DNA glycosylase knockout mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Park, L.C.; Albers, D.S.; Xu, H.; Lindsay, J.G.; Beal, M.F.; Gibson, G.E. Mitochondrial impairment in the cerebellum of the patients with progressive supranuclear palsy. J. Neurosci. Res. 2001, 66, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Przybylowska, K.; Kabzinski, J.; Sygut, A.; Dziki, L.; Dziki, A.; Majsterek, I. An association selected polymorphisms of XRCC1, OGG1 and MUTYH gene and the level of efficiency oxidative DNA damage repair with a risk of colorectal cancer. Mutat. Res. 2013, 745–746, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.X.; Hua, R.X.; Yi, W.; Shen, L.J.; Jin, Z.X.; Zhao, Y.H.; Yi, D.H.; Chen, W.S.; Yu, S.Q. The association between OGG1 Ser326Cys polymorphism and lung cancer susceptibility: A meta-analysis of 27 studies. PLoS ONE 2012, 7, e35970. [Google Scholar] [CrossRef] [PubMed]
- Elahi, A.; Zheng, Z.; Park, J.; Eyring, K.; McCaffrey, T.; Lazarus, P. The human OGG1 DNA repair enzyme and its association with orolaryngeal cancer risk. Carcinogenesis 2002, 23, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Q.; Watson, L.J.; Jones, S.P.; Epstein, P.N. Cardiac overexpression of 8-oxoguanine DNA glycosylase 1 protects mitochondrial DNA and reduces cardiac fibrosis following transaortic constriction. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2073–H2080. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H. Oxidative stress in heart failure: The role of mitochondria. Intern. Med. 2001, 40, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Mehta, J.L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep. 2013, 3, 1077. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Jung, B.; Liang, F.; Azuelos, I.; Hussain, S.; Goldberg, P.; Godin, R.; Danialou, G.; Chaturvedi, R.; Rygiel, K.; et al. Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am. J. Respir. Crit. Care Med. 2012, 186, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Mouner, M.; Chouteau, J.M.; Gorodnya, O.M.; Ruchko, M.V.; Potter, B.J.; Wilson, G.L.; Gillespie, M.N.; Parker, J.C. Mitochondrial-targeted DNA repair enzyme 8-oxoguanine DNA glycosylase 1 protects against ventilator-induced lung injury in intact mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L287–L297. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.H.; Feng, T.H.; Lin, Y.F.; Chang, P.L.; Juang, H.H. P53 downregulates the gene expression of mitochondrial aconitase in human prostate carcinoma cells. Prostate 2011, 71, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Koczor, C.A.; Torres, R.A.; Fields, E.J.; Boyd, A.; Lewis, W. Mitochondrial matrix P53 sensitizes cells to oxidative stress. Mitochondrion 2013, 13, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Marudamuthu, A.S.; Bhandary, Y.P.; Shetty, S.K.; Fu, J.; Sathish, V.; Prakash, Y.; Shetty, S. Role of the urokinase-fibrinolytic system in epithelial-mesenchymal transition during lung injury. Am. J. Pathol. 2015, 185, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Marudamuthu, A.S.; Shetty, S.K.; Bhandary, Y.P.; Karandashova, S.; Thompson, M.; Sathish, V.; Florova, G.; Hogan, T.B.; Pabelick, C.M.; Prakash, Y.S.; et al. Plasminogen activator inhibitor-1 suppresses profibrotic responses in fibroblasts from fibrotic lungs. J. Biol. Chem. 2015, 290, 9428–9441. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Fu, J.; Pinson, B.M.; Levin, J.; Shetty, S. Role of p53-fibrinolytic system cross-talk in the regulation of quartz-induced lung injury. Toxicol. Appl. Pharmacol. 2015, 283, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.T.; Akhter, H.; Jiang, C.; MacEwen, M.; Ding, Q.; Antony, V.; Thannickal, V.J.; Liu, R.M. Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp. Gerontol. 2015, 61, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in Nlrp3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, V.; Siler, J.T.; Ramadass, M.; Ge, L.; Davis, J.; Grant, G.; Nathan, S.D.; Jarai, G.; Trujillo, G. Sustained activation of toll-like receptor 9 induces an invasive phenotype in lung fibroblasts: Possible implications in idiopathic pulmonary fibrosis. Am. J. Pathol. 2015, 185, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wu, G.; Yao, Y.; Zeng, J.; Shi, D.; Lv, T.; Luo, L.; Song, Y. Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the TLR9-p38 mapk pathway. Free Radic. Biol. Med. 2015, 83, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kuck, J.L.; Obiako, B.O.; Gorodnya, O.M.; Pastukh, V.M.; Kua, J.; Simmons, J.D.; Gillespie, M.N. Mitochondrial DNA damage-associated molecular patterns mediate a feed-forward cycle of bacteria-induced vascular injury in perfused rat lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1078–L1085. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, G.; Rogel, M.R.; Baker, M.A.; Troken, J.R.; Urich, D.; Morales-Nebreda, L.; Sennello, J.A.; Kutuzov, M.A.; Sitikov, A.; Davis, J.M.; et al. Vimentin regulates activation of the Nlrp3 inflammasome. Nat. Commun. 2015, 6, 6574. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A. The conserved role of sirtuins in chromatin regulation. Int. J. Dev. Biol. 2009, 53, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Hirschey, M.D.; Finley, L.W.; Haigis, M.C. Sirtuin regulation of mitochondria: Energy production, apoptosis, and signaling. Trends Biochem. Sci. 2010, 35, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegue, L.; Vaquero, A. Sirtuins in stress response: Guardians of the genome. Oncogene 2014, 33, 3764–3775. [Google Scholar] [CrossRef] [PubMed]
- Kaniak-Golik, A.; Skoneczna, A. Mitochondria-nucleus network for genome stability. Free Radic. Biol. Med. 2015, 82, 73–104. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, D.; Rose, G.; Cavalcante, P.; Covello, G.; Dato, S.; de Rango, F.; Greco, V.; Maggiolini, M.; Feraco, E.; Mari, V.; et al. A novel vntr enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics 2005, 85, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, L.L.; Wen, X.; Wang, X.Y.; Liu, J.; Cheng, Y.; Huang, J. Sirtuin-3 (sirt3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014, 5, e1047. [Google Scholar] [CrossRef] [PubMed]
- Shulga, N.; Pastorino, J.G. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-d mediated by sirtuin-3. J. Cell Sci. 2010, 123, 4117–4127. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Banck, M.; Mujtaba, S.; Zhou, M.M.; Sugrue, M.M.; Walsh, M.J. P53-induced growth arrest is regulated by the mitochondrial sirt3 deacetylase. PLoS ONE 2010, 5, e10486. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Uchijima, Y.; Horike, N.; Tonami, K.; Nishiyama, K.; Amano, T.; Asano, T.; Kurihara, Y.; Kurihara, H. Sirt3 protects in vitro-fertilized mouse preimplantation embryos against oxidative stress-induced p53-mediated developmental arrest. J. Clin. Investig. 2010, 120, 2817–2828. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, P.; Rose, G.; Panno, M.L.; Passarino, G.; Bellizzi, D. SIRT3 gene expression: A link between inherited mitochondrial DNA variants and oxidative stress. Gene 2012, 497, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Lee, H.C.; Liao, C.C.; Wei, Y.H. Regulation of mitochondrial F0F1ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977bp deletion of mitochondrial DNA. Biochim. Biophys. Acta 2013, 1832, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Bause, A.S.; Matsui, M.S.; Haigis, M.C. The protein deacetylase SIRT3 prevents oxidative stress-induced keratinocyte differentiation. J. Biol. Chem. 2013, 288, 36484–36491. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S. Mitochondrial DNA, aconitase “wraps” it up. Trends Biochem. Sci. 2005, 30, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; de Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Xu, J.; Kondo, K.; Ding, D.; Salvi, R.J.; Yamasoba, T.; Rabinovitch, P.S.; Weindruch, R.; Leeuwenburgh, C.; Tanokura, M.; et al. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 19432–19437. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.C.; Olivier, A.K.; Jacobus, J.A.; Mapuskar, K.A.; Mao, G.; Martin, S.M.; Riley, D.P.; Gius, D.; Spitz, D.R. Superoxide mediates acute liver injury in irradiated mice lacking sirtuin 3. Antioxid. Redox Signal. 2014, 20, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, J.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Zeng, M.; Zhang, Y.; Bu, P. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H424–H434. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Peritore, C.; Ginsberg, J.; Kayhan, M.; Donmez, G. SIRT3 attenuates MPTP-induced nigrostriatal degeneration via enhancing mitochondrial antioxidant capacity. Neurochem. Res. 2015, 40, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Shao, Y.; Ma, C.Y.; Chen, W.; Sun, L.; Liu, W.; Zhang, D.Y.; Fu, B.C.; Liu, K.Y.; Jia, Z.B.; et al. Decreased SIRT3 in aged human mesenchymal stromal/stem cells increases cellular susceptibility to oxidative stress. J. Cell Mol. Med. 2014, 18, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Kang, X.; Wang, X.; Wu, S.; Xiao, J.; Li, Z.; Wu, X.; Zhang, W. Conversion of bone marrow mesenchymal stem cells into type II alveolar epithelial cells reduces pulmonary fibrosis by decreasing oxidative stress in rats. Mol. Med. Rep. 2015, 11, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Irving, B.A.; Lanza, I.R.; Vendelbo, M.H.; Konopka, A.R.; Robinson, M.M.; Henderson, G.C.; Klaus, K.A.; Morse, D.M.; Heppelmann, C.; et al. Differential effect of endurance training on mitochondrial protein damage, degradation, and acetylation in the context of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.H.; Chen, T.; Wang, Y.H.; Zhu, J.; Luo, P.; Rao, W.; Yang, Y.F.; Fei, Z.; Jiang, X.F. Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int. J. Mol. Sci. 2014, 15, 14591–14609. [Google Scholar] [CrossRef] [PubMed]
- Rato, L.; Duarte, A.I.; Tomas, G.D.; Santos, M.S.; Moreira, P.I.; Socorro, S.; Cavaco, J.E.; Alves, M.G.; Oliveira, P.F. Pre-diabetes alters testicular PGC1-α/SIRT3 axis modulating mitochondrial bioenergetics and oxidative stress. Biochim. Biophys. Acta 2014, 1837, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.S.; Oh, J.E.; Nan, J.; Lee, H.; Jung, H.S.; Chung, S.S.; Park, K.S. SIRT3 overexpression attenuates palmitate-induced pancreatic β-cell dysfunction. PLoS ONE 2015, 10, e0124744. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Deng, C.X.; Finley, L.W.; Kim, H.S.; Gius, D. SIRT3 is a mitochondrial tumor suppressor: A scientific tale that connects aberrant cellular ROS, the warburg effect, and carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed]
- Koentges, C.; Pfeil, K.; Schnick, T.; Wiese, S.; Dahlbock, R.; Cimolai, M.C.; Meyer-Steenbuck, M.; Cenkerova, K.; Hoffmann, M.M.; Jaeger, C.; et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res. Cardiol. 2015, 110, 493. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014, 20, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yang, Y.; Hu, Y.; Sun, Y.; Du, Z.; Xie, Z.; Zhou, T.; Kong, W. Age-related decrease in the mitochondrial sirtuin deacetylase Sirt3 expression associated with ROS accumulation in the auditory cortex of the mimetic aging rat model. PLoS ONE 2014, 9, e88019. [Google Scholar] [CrossRef] [PubMed]
- Marques-Aleixo, I.; Santos-Alves, E.; Mariani, D.; Rizo-Roca, D.; Padrao, A.I.; Rocha-Rodrigues, S.; Viscor, G.; Torrella, J.R.; Ferreira, R.; Oliveira, P.J.; et al. Physical exercise prior and during treatment reduces sub-chronic doxorubicin-induced mitochondrial toxicity and oxidative stress. Mitochondrion 2015, 20, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Desquiret-Dumas, V.; Gueguen, N.; Leman, G.; Baron, S.; Nivet-Antoine, V.; Chupin, S.; Chevrollier, A.; Vessieres, E.; Ayer, A.; Ferre, M.; et al. Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J. Biol. Chem. 2013, 288, 36662–36675. [Google Scholar] [CrossRef] [PubMed]
- Royce, S.G.; Dang, W.; Yuan, G.; Tran, J.; El Osta, A.; Karagiannis, T.C.; Tang, M.L. Resveratrol has protective effects against airway remodeling and airway hyperreactivity in a murine model of allergic airways disease. Pathobiol. Aging Age Relat. Dis. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Haohao, Z.; Guijun, Q.; Juan, Z.; Wen, K.; Lulu, C. Resveratrol improves high-fat diet induced insulin resistance by rebalancing subsarcolemmal mitochondrial oxidation and antioxidantion. J. Physiol. Biochem. 2015, 71, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Sener, G.; Topaloglu, N.; Sehirli, A.O.; Ercan, F.; Gedik, N. Resveratrol alleviates bleomycin-induced lung injury in rats. Pulm. Pharmacol. Ther. 2007, 20, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Zghonda, N.; Yoshida, S.; Ezaki, S.; Otake, Y.; Murakami, C.; Mliki, A.; Ghorbel, A.; Miyazaki, H. Epsilon-viniferin is more effective than its monomer resveratrol in improving the functions of vascular endothelial cells and the heart. Biosci. Biotechnol. Biochem. 2012, 76, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Jin, J.; Cichewicz, R.H.; Hageman, S.A.; Ellis, T.K.; Xiang, L.; Peng, Q.; Jiang, M.; Arbez, N.; Hotaling, K.; et al. Trans-(–)-ε-viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of huntington disease. J. Biol. Chem. 2012, 287, 24460–24472. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Tocchetti, C.G.; Bhatt, N.; Paolocci, N.; Cortassa, S. Protective mechanisms of mitochondria and heart function in diabetes. Antioxid. Redox Signal. 2015, 22, 1563–1586. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, Q.; Bai, L.; Chen, W.; Wang, X.; Tellez, C.S.; Leng, S.; Padilla, M.T.; Nyunoya, T.; Belinsky, S.A.; et al. RIP1 maintains DNA integrity and cell proliferation by regulating PGC-1α-mediated mitochondrial oxidative phosphorylation and glycolysis. Cell Death Differ. 2014, 21, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.R.; Larrick, J.W. Partial reversal of skeletal muscle aging by restoration of normal NAD+ levels. Rejuv. Res. 2014, 17, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Metodiev, M.D.; Gerber, S.; Hubert, L.; Delahodde, A.; Chretien, D.; Gerard, X.; Amati-Bonneau, P.; Giacomotto, M.C.; Boddaert, N.; Kaminska, A.; et al. Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J. Med. Genet. 2014, 51, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [PubMed]
- Van Waveren, C.; Sun, Y.; Cheung, H.S.; Moraes, C.T. Oxidative phosphorylation dysfunction modulates expression of extracellular matrix—Remodeling genes and invasion. Carcinogenesis 2006, 27, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Vachharajani, V.; Millet, P.; Bharadwaj, M.S.; Molina, A.J.; McCall, C.E. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J. Biol. Chem. 2015, 290, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Pena, S. Role of oxidative DNA damage in mitochondrial dysfunction and huntington’s disease pathogenesis. Free Radic. Biol. Med. 2013, 62, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [PubMed]
- De Cavanagh, E.M.; Inserra, F.; Ferder, L. Angiotensin II blockade: A strategy to slow ageing by protecting mitochondria? Cardiovasc Res. 2011, 89, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Koltai, E.; Taylor, A.W.; Higuchi, M.; Kumagai, S.; Ohno, H.; Goto, S.; Boldogh, I. Redox-regulating sirtuins in aging, caloric restriction, and exercise. Free Radic. Biol. Med. 2013, 58, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Boyette, L.B.; Tuan, R.S. Adult stem cells and diseases of aging. J. Clin. Med. 2014, 3, 88–134. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MNSoD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. Mitochondrial complex I: A central regulator of the aging process. Cell Cycle 2011, 10, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Peek, C.B.; Affinati, A.H.; Ramsey, K.M.; Kuo, H.Y.; Yu, W.; Sena, L.A.; Ilkayeva, O.; Marcheva, B.; Kobayashi, Y.; Omura, C.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013, 342, 1243417. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, M.; Zeng, X.; Yang, J.; Deng, H.; Yi, L.; Mi, M.T. Resveratrol regulates mitochondrial reactive oxygen species homeostasis through Sirt3 signaling pathway in human vascular endothelial cells. Cell Death Dis. 2014, 5, e1576. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hu, Q.; Ma, Q.; Liu, C.; Wang, G. The protease Omi regulates mitochondrial biogenesis through the GSK3β/PGC-1α pathway. Cell Death Dis. 2014, 5, e1373. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Williams, D.B.; Kamp, D.W. The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis. Int. J. Mol. Sci. 2015, 16, 21486-21519. https://doi.org/10.3390/ijms160921486
Kim S-J, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis. International Journal of Molecular Sciences. 2015; 16(9):21486-21519. https://doi.org/10.3390/ijms160921486
Chicago/Turabian StyleKim, Seok-Jo, Paul Cheresh, Renea P. Jablonski, David B. Williams, and David W. Kamp. 2015. "The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis" International Journal of Molecular Sciences 16, no. 9: 21486-21519. https://doi.org/10.3390/ijms160921486