 Jon Hazeldine1,2*
Jon Hazeldine1,2* Janet M. Lord1,2,3
Janet M. Lord1,2,3- 1MRC-Versus Arthritis Centre for Musculoskeletal Ageing Research, Institute of Inflammation and Ageing, University of Birmingham, Birmingham, United Kingdom
- 2National Institute for Health Research Surgical Reconstruction and Microbiology Research Centre, Queen Elizabeth Hospital Birmingham, Birmingham, United Kingdom
- 3National Institute for Health Research Birmingham Biomedical Research Centre, University Hospital Birmingham NHS Foundation Trust and University of Birmingham, Birmingham, United Kingdom
Whilst the majority of individuals infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative pathogen of COVID-19, experience mild to moderate symptoms, approximately 20% develop severe respiratory complications that may progress to acute respiratory distress syndrome, pulmonary failure and death. To date, single cell and high-throughput systems based analyses of the peripheral and pulmonary immune responses to SARS-CoV-2 suggest that a hyperactive and dysregulated immune response underpins the development of severe disease, with a prominent role assigned to neutrophils. Characterised in part by robust generation of neutrophil extracellular traps (NETs), the presence of immature, immunosuppressive and activated neutrophil subsets in the circulation, and neutrophilic infiltrates in the lung, a granulocytic signature is emerging as a defining feature of severe COVID-19. Furthermore, an assessment of the number, maturity status and/or function of circulating neutrophils at the time of hospital admission has shown promise as a prognostic tool for the early identification of patients at risk of clinical deterioration. Here, by summarising the results of studies that have examined the peripheral and pulmonary immune response to SARS-CoV-2, we provide a comprehensive overview of the changes that occur in the composition, phenotype and function of the neutrophil pool in COVID-19 patients of differing disease severities and discuss potential mediators of SARS-CoV-2-induced neutrophil dysfunction. With few specific treatments currently approved for COVID-19, we conclude the review by discussing whether neutrophils represent a potential therapeutic target for the treatment of patients with severe COVID-19.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), an enveloped, single-stranded positive-sense RNA virus, is the causative pathogen of Coronavirus disease 2019 (COVID-19), a respiratory tract infection that was declared a global pandemic by The World Health Organisation on March 11th 2020 (1). Respiratory droplets and direct contact with infected individuals are the main routes of transmission for SARS-CoV-2, which was first identified in bronchoalveolar-lavage fluid (BALF) samples obtained from patients admitted to the Wuhan Jinyuntan Hospital in the Hubei Province of China with severe pneumonia of unknown aetiology in December 2019 (2). As of 10th May 2021, the virus has infected over 157 million people worldwide, resulting in over 3.2 million deaths (3). Whilst the majority of individuals (~80%) infected with SARS-CoV-2 experience a self-resolving mild to moderate respiratory disease, 10-20% of patients diagnosed with COVID-19 develop a severe viral pneumonia (4). In approximately 5% of cases, disease progresses to a critical illness characterised by acute respiratory distress syndrome (ARDS) and respiratory failure that requires ventilatory support and specialised treatment in intensive care units (ICU) (4–8). Through systematic reviews and meta-analyses, a number of demographic and host risk factors have been identified that influence the clinical outcomes of COVID-19 patients. Linked to reduced physiological reserve, a heightened basal inflammatory state or systemic endothelial dysfunction, advanced age, male gender and pre-existing co-morbidities [e.g. diabetes, malignancy, hypertension, cardiovascular disease and chronic kidney disease (CKD)] have been shown to be associated with increased disease severity, admission to ICU and mortality (9–12). However, as adults aged 20-44 years account for 20% and 12% of COVID-19-associated hospitalisations and ICU admissions respectively (13), and >5% of hospitalised younger adults (aged 18-34 years) with no underlying health conditions experience respiratory failure (14), additional host and/or virus-associated factors must contribute to disease pathogenesis. A dysregulated physiological response to SARS-CoV-2 infection is one such factor, with striking differences observed in the immune response of COVID-19 patients when graded by disease severity, with a profound inflammatory and dysregulated myeloid response emerging as a key driver of severe COVID-19 (15–21).
Integral to the initiation of anti-viral immune responses is the production of type I (α/β) and type III (γ) interferons (IFNs) (22). Compared to those with mild/moderate disease, patients with critical COVID-19 exhibit an impaired peripheral type I IFN response (23). Shown to precede the onset of respiratory failure, this weakened IFN response results in an increased plasma viral load (23) that triggers excessive nuclear factor kappa β-driven inflammatory responses, a hallmark of severe COVID-19 (23–25). Indeed, significantly elevated concentrations of interleukin (IL)-2, IL-6, IL-7, IL-8, IL-10, IL-18, tumour necrosis factor-alpha (TNF-α), granulocyte colony stimulating factor (G-CSF), monocyte chemoattractant protein 1 (MCP-1) and macrophage inflammatory protein 1 alpha (MIP-1α) have been measured in the circulation of ICU patients when compared to their non-ICU counterparts (5, 26–29), with additional studies reporting significantly higher concentrations of IL-6 in non-survivors versus survivors (30, 31). Reminiscent of a cytokine storm, this hyperinflammatory state would be exacerbated by the systemic dysregulation in lipid metabolism that occurs in patients with severe COVID-19 (32). Compared to non-ICU patients, significantly reduced circulating concentrations of the proresolving lipid mediator resolvin E3 have been detected in ICU patients, who also present with increased concentrations of pro-inflammatory lipids generated by the enzymes ALOX5 and cytochrome p450 (32). Linked to this state of systemic hyperinflammation are aberrant SARS-CoV-2-induced innate and adaptive immune responses. Lymphopenia, T cell exhaustion, impaired activity of cytotoxic lymphocytes, emergency granulopoiesis and increased neutrophil activation are just some of the examples of immune dysregulation that have been reported in patients with severe COVID-19 (28, 33–35). Of these, it is the systemic hyperactivation of neutrophils, and their infiltration into pulmonary tissues, that has been assigned a prominent role in the immunopathology that underpins the transition from mild to severe COVID-19 (35–37).
Renowned for providing immediate frontline protection against rapidly dividing bacteria, fungi and yeast, there is a growing body of evidence implicating neutrophils, via their generation of reactive oxygen species (ROS), neutrophil extracellular traps (NETs) and ability to act as antigen presenting cells, in the host response to viral infections (38, 39). However, their involvement must be tightly regulated as exaggerated neutrophil responses to viral challenge result in tissue injury, vascular leakage and organ dysfunction (38, 39). In the context of SARS-CoV-2, cross-sectional and prospective studies analysing peripheral blood, BALF and/or lung tissue samples from COVID-19 patients of differing disease severities have demonstrated significant virus-induced changes in the composition, phenotype and/or function of circulating and pulmonary neutrophil pools (Figure 1). Neutrophilia, robust NET generation and the presence of immature, immunosuppressive and pro-inflammatory neutrophil subsets are examples of some of the consequences of SARS-CoV-2 infection described thus far, with data suggesting that signatures of neutrophil activation are enriched in patients with severe COVID-19 and associated with poor clinical outcome (17–21). Combined with the results of rodent and non-human primate based studies, these data demonstrate striking similarities between SARS-CoV-2-induced changes in neutrophil biology and the neutrophil response triggered by other members of the Coronaviridae family such as Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV (Table 1).
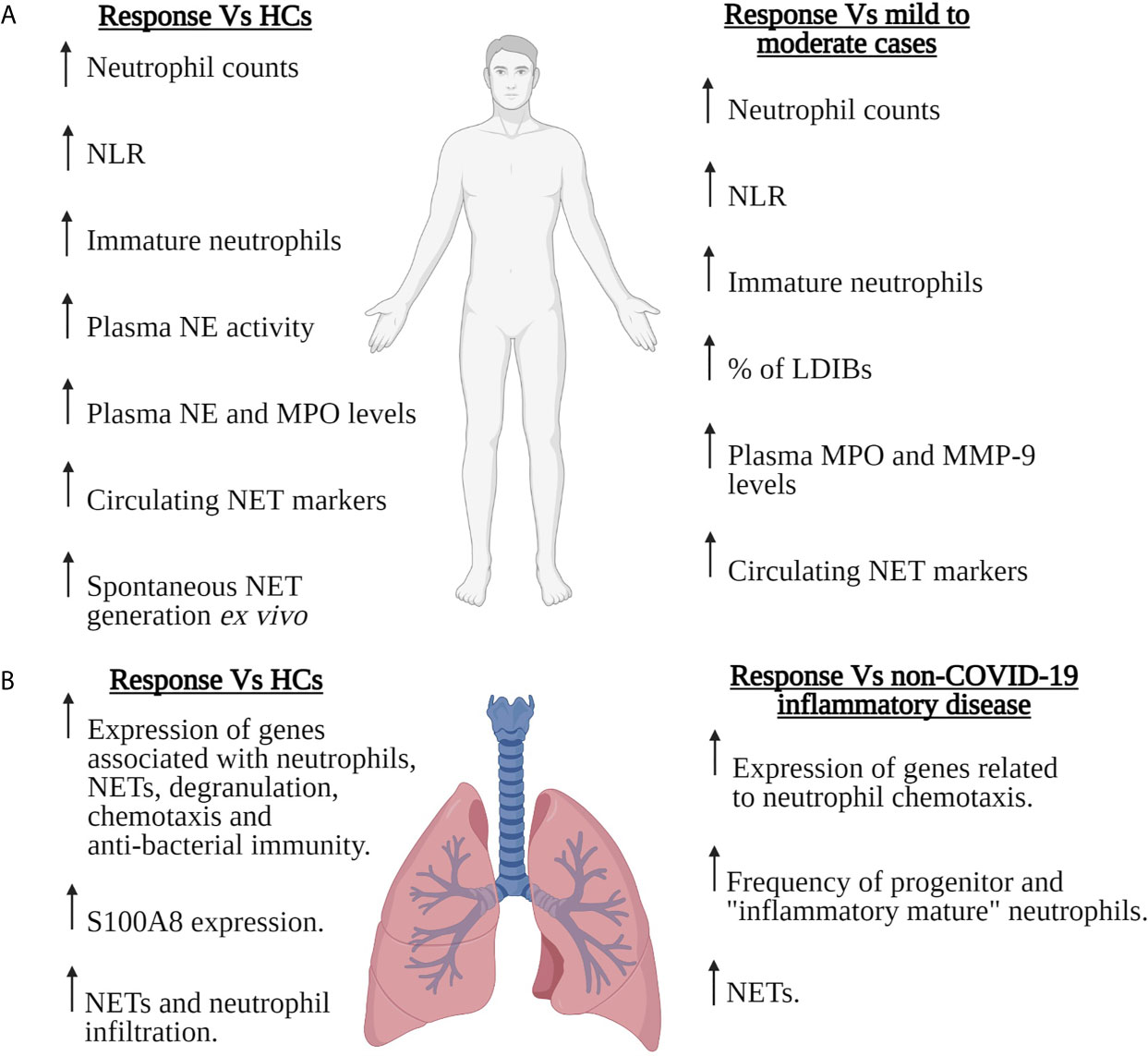
Figure 1 Neutrophils in severe COVID-19. Summary of the SARS-CoV-2-induced changes that have been reported for the number, composition and/or function of circulating (A) or pulmonary (B) neutrophils in patients with severe COVID-19. Features of the neutrophil response of severe COVID-19 patients are compared to those of healthy controls (HCs), patients with mild-to-moderate COVID-19 or patients with non-COVID-19 inflammatory disease. For the pulmonary neutrophil response summarised in (B), data was derived from the analysis of bronchoalveolar lavage fluid or post-mortem lung tissue sections. LDIBs, Low density inflammatory band cells; MMP-9, Matrix metalloproteinase-9; MPO, Myeloperoxidase; NE, Neutrophil elastase; NETs, Neutrophil extracellular traps; NLR, Neutrophil to lymphocyte ratio. Figure created with BioRender.com.
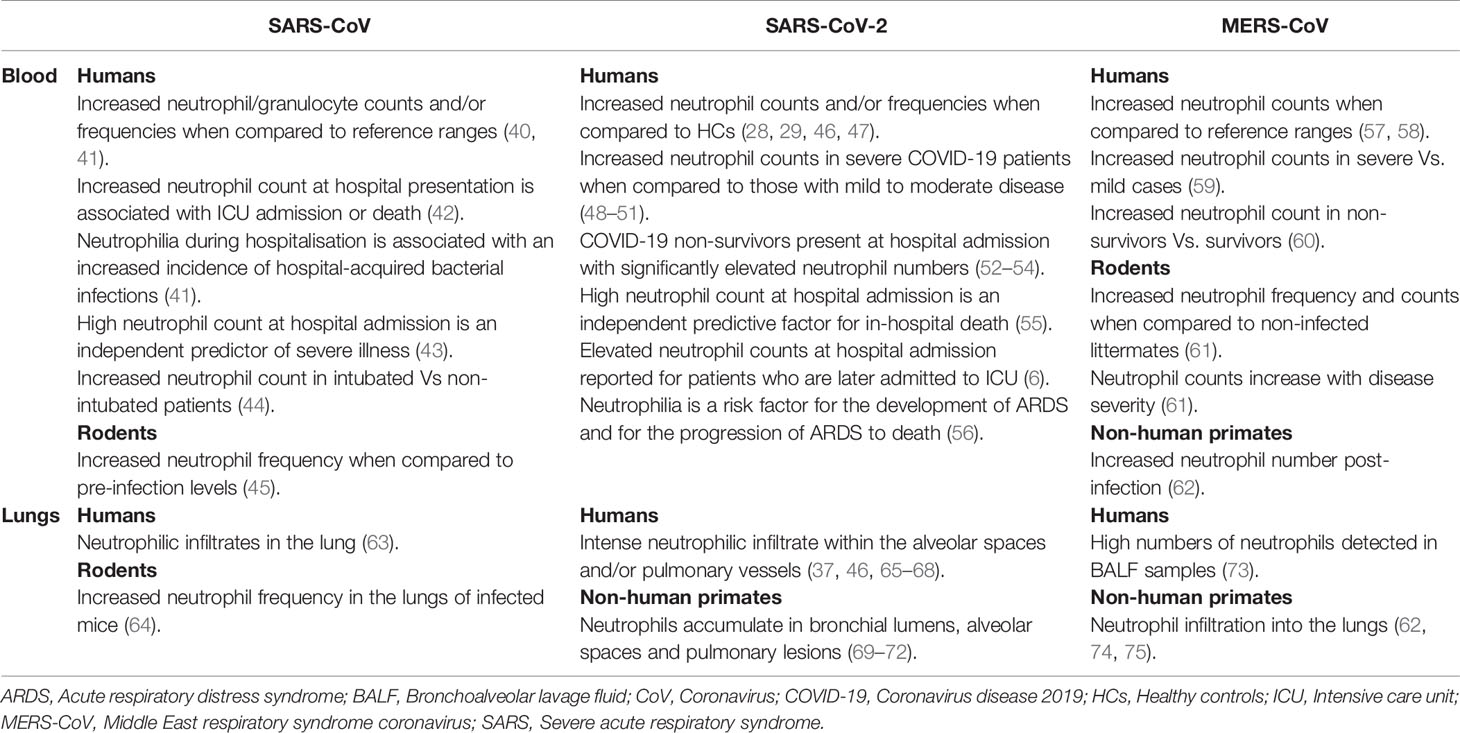
Table 1 Changes reported in the number/frequency of circulating or pulmonary neutrophils in patients, rodents or non-human primates infected with SARS-CoV, SARS-CoV-2 or MERS and their relationship with disease severity or clinical outcome.
Here, we provide a comprehensive overview of the changes that have been described in the composition, phenotype and function of the neutrophil pool in COVID-19 patients of differing disease severities, and discuss potential mediators of SARS-CoV-2-induced neutrophil dysfunction. With no specific anti-viral therapies currently approved for the treatment of SARS-CoV-2 infection, we conclude the review by discussing whether suppressing the production of or enhancing the clearance of NETs, or suppressing the release of immature neutrophils from the bone marrow represent viable therapeutic strategies for the treatment of severe COVID-19.
Neutrophils and COVID-19 Pathobiology – Animal Model Data
Animal models of infection provide valuable insights into the cellular and molecular mechanisms that underpin disease pathogenesis. Reminiscent of the pulmonary pathologies described for human COVID-19 patients with mild to severe disease, SARS-CoV-2 infection of non-human primates (e.g. African green monkeys, rhesus macaques, baboons and marmosets) results in lung consolidation, pulmonary edema, bronchointerstitial pneumonia, pleural adhesions, interstitial inflammation and diffuse alveolar damage (69–71, 76, 77). Across this spectrum of disease, analysis of post-mortem lung tissue has revealed that neutrophils accumulate in bronchial lumens, alveolar spaces and pulmonary lesions (69–72), with this infiltration coinciding with significantly elevated BALF concentrations of the chemokine IL-8 (70). Moreover, mirroring the changes reported in the composition of the pulmonary neutrophil pool in humans with severe COVID-19 (48, 78) immature “pre-neutrophils” have been detected in the lung sections of African green monkeys with high SARS-CoV-2 viral loads (79).
Angiotensin-converting enzyme 2 (ACE2), a type I integral membrane glycoprotein, is the receptor utilised by SARS-CoV-2 for cell entry (80). Due to the presence of ACE2 on the surface of cells that line their respiratory tract, ferrets have also emerged as a suitable host in which to study the pathogenesis of COVID-19 (81–83). Histological examination of lung tissue from SARS-CoV-2 infected ferrets has detected the presence of neutrophils in the alveolar septa, alveolar lumen and bronchiolar luminae (81, 83). Associated with bronchopneumonia, this neutrophilic infiltrate was observed primarily in ferrets infected with high doses of SARS-CoV-2 (81).
By replicating many of the pathological features observed in human disease, animal models of SARS-CoV-2 infection have been used to test potential therapies for the treatment of COVID-19 (71, 84, 85). Using a rhesus macaque infection model, Hoang et al showed that daily treatment with baricitinib, a selective JAK kinase inhibitor, significantly reduced the severity of SARS-CoV-2 infection (84). Associated with a milder inflammatory response, the beneficial effects of baricitinib treatment were attributed in part to reduced neutrophil-mediated immunopathology (84). For example, bulk RNA sequencing (RNA-seq) of BAL revealed a baricitinib-induced downregulation of genes associated with neutrophil degranulation, chemotaxis and extravasation (84). In line with these observations, flow cytometry analysis of BAL revealed significantly lower frequencies of neutrophils in baricitinib treated macaques when compared to untreated controls (84). Furthermore, baricitinib treatment was associated with reduced ex vivo NET formation and a virtual absence of NETs in post-mortem lung tissue (84). Pointing towards a prominent role for neutrophils in the pathology of severe COVID-19, these baricitinib-induced changes in neutrophil number and function coincided with a reduction in pulmonary lesions and lung pathology scores (84). Taken together, these findings offer potential mechanistic explanations for the improved clinical outcomes that have been reported for severe COVID-19 patients treated with baricitinib (86, 87).
The Neutrophil Response to SARS-CoV-2 Infection
Neutrophils in Blood
Neutrophil Number and the Neutrophil:Lymphocyte Ratio
The majority of studies that have compared full blood counts between COVID-19 patients and healthy controls (HCs) have reported a SARS-CoV-2-induced increase in the absolute number and/or frequency of circulating neutrophils (28, 29, 46–49). However, when accounting for disease severity, neutrophilia is not observed amongst all patient groups. For example, across several studies, no significant differences were reported in either the number or percentage of neutrophils between COVID-19 patients with mild or moderate disease and HCs (48–50). In contrast, severe COVID-19 patients exhibit neutrophil counts that are significantly greater than those recorded for both HCs and non-ICU patients with mild to moderate disease (48–51, 88–96). The exaggerated hyperinflammatory response that occurs in severe COVID-19 patients (5), particularly the systemic elevation in IL-6, has been suggested to drive this relationship between disease severity and elevated neutrophil counts (6, 15).
A handful of longitudinal studies that have performed serial sampling on hospitalised patients (51, 95, 97, 98) have shown the severity associated increase in neutrophil number persists over time. For example, across seven study time-points, ranging from days 1-15 post-hospitalisation, Ding et al observed significantly higher neutrophil counts in severe COVID-19 patients when compared to those with non-severe disease (51). Moreover, paired sample analyses have shown no significant difference in neutrophil counts between the hospital admission blood samples of severe COVID-19 patients, where neutrophilia is observed, and those obtained prior to discharge, suggesting a state of unresolved inflammation at the time of clinical recovery (95).
As patients with severe COVID-19 present at hospital admission with significantly higher neutrophil counts than those with mild to moderate disease (49, 51, 89, 91–93, 99–101), a number of studies have investigated whether neutrophilia at presentation can be used to assist in the diagnosis of severe COVID-19. Using a range of optimal cut off values (3.2-7.305 x 109/L), it has been demonstrated that neutrophil counts can discriminate non-severe from severe COVID-19 patients with a degree of accuracy ranging from poor to excellent (88, 95, 96, 102). In a meta-analysis of 26 studies, Soraya et al reported that a cut-off value of 3.65 x 109/L provided fair discrimination between severe and non-severe cases (AUROC, 0.79) (99). However, as observed in other meta-analyses (103), this model was outperformed by those built on lymphocyte counts or concentrations of acute phase proteins (99), suggesting that other clinical indices may be better placed to aid clinicians in the early identification of severe COVID-19 patients.
Relative to HCs, hospitalised COVID-19 patients, of all disease severities, exhibit significantly reduced numbers of circulating lymphocytes (28, 47, 49, 89). However, as with neutrophilia, the degree of lymphopenia is markedly influenced by disease severity, with severe COVID-19 patients presenting with significantly lower lymphocyte counts than those with mild to moderate disease (49–51, 89, 90, 92, 94–96, 100, 104). Suggested to be a consequence of systemic inflammation and/or SARS-CoV-2-induced cell death, this state of lymphopenia, when combined with maintained or elevated neutrophil counts, results in an elevated neutrophil:lymphocyte ratio (NLR) (89, 105). An increased NLR at hospital admission is emerging as a useful indicator of disease severity and a potential tool of diagnostic and prognostic utility (106, 107). For example, significantly higher baseline NLR values have been measured in COVID-19 patients who develop severe/critical disease when compared to mild or moderate cases (51, 90, 92, 95, 108–111), with this elevation persisting for up to 15 days post-hospital admission (51, 100, 112). In terms of its discriminatory power, NLR has been shown to distinguish severe COVID-19 patients from non-severe cases with a level of accuracy considered fair to good based on AUROC values (95, 100, 107, 109, 112, 113). Moreover, Liu et al reported that a significantly greater proportion of patients who presented with an NLR >3.13 progressed to critical illness when compared to those with an NLR <3.13, highlighting the potential of NLR as a marker by which to stratify patients based on their risk of disease progression (112). Thus, an early assessment of NLR may help identify patients that are likely to require more intensive monitoring and clinical care.
Neutrophil Phenotype
Until recently, neutrophils were considered a homogeneous population of terminally differentiated short-lived cells whose only role in host defence was the detection and elimination of pathogens. However, over the past decade it has been shown, under homeostatic and inflammatory conditions, that neutrophils are in fact a diverse cell population that exhibit phenotypic and functional heterogeneity (114, 115). Currently, it is unclear as to whether this heterogeneity is borne from the presence of distinct neutrophil “subsets”, or simply reflects differences in cell maturity and/or activation status (116). Indeed, whilst it is accepted that neutrophils possess pro-inflammatory, immunoregulatory and immune suppressive properties (114, 115), assigning these features to distinct subpopulations is difficult due to a lack of defined markers and the contrasting functions reported for these populations across different disease settings (116). In the context of systemic inflammation, two neutrophil populations are receiving significant attention. The first, termed low-density neutrophils (LDNs), are a heterogeneous group of immature and activated mature cells, whose functional capacities vary depending on the disease context. For example, in cancer, LDNs display T cell suppressive activity, and are often referred to as granulocytic myeloid-derived suppressor cells (114, 116, 117). Conversely, in the settings of autoimmunity (e.g. systemic lupus erythematosus) and infection, LDNs have been reported to exhibit a pro-inflammatory phenotype, as evidenced by increased type 1 IFN production, ROS generation, degranulation and an activated surface phenotype (118, 119). With a propensity for enhanced cytokine production and spontaneous NET generation, low density granulocytes (LDGs) are a second “distinct” class of neutrophils, whose functional hyperactivity has been extensively studied in the context of autoimmunity (114, 120, 121). Using a range of analytical approaches that have included single cell RNA-seq (scRNA-seq), high-dimensional flow cytometry and mass cytometry, a number of studies have reported marked differences in the composition of the circulating neutrophil pool between COVID-19 patients of differing disease severities (18, 29, 48, 49, 122–125). Supporting the idea that distinct innate immune responses underlie the different clinical trajectories of COVID-19, an accumulation of LDNs is emerging as a signature of severe disease (18, 21).
Defined by a broad range of surface phenotypes that have included CD16low/highCD10-, CD15+CD10-CD66b+, CD10-CD64+, CD10-, CD10-CD16-CD66b+ or CD10lowCD101+/-, significantly increased frequencies and absolute numbers of immature neutrophils have been detected in patients with COVID-19 when compared to HCs (48, 49, 122, 126–128). In depth characterisation of these cells has identified a CD16IntCD44LowCD11bInt subpopulation that possess characteristics reminiscent of LDGs. Referred to as low-density inflammatory band cells (LDIBs), these cells, whose circulating frequencies are significantly higher in patients with severe COVID-19 when compared to mild cases (29), exhibit spontaneous NET formation and LPS-induced pro-inflammatory cytokine production, with the generation of IL-6 and TNF-α significantly higher for LDIBs of severe COVID-19 cases when compared to mild patients (29). Further demonstrating severity-associated differences in LDNs, Combadiere et al detected an increased frequency of CD123+ and LOX1+ immature neutrophils in ICU patients when compared to non-ICU cases (125). Interestingly, suggesting that heterogeneity also exists within the neutrophil compartment of patients with severe disease, the authors reported an increased frequency of CD123+ and LOX1+ immature neutrophils in COVID-19 ICU patients that they classified as having more severe disease (defined as a sequential organ failure assessment (SOFA) score >8) when compared to ICU patients with a SOFA score <8 (125). In terms of longitudinal sampling, studies have reported a significant reduction in LDN frequencies over the course of COVID-19 (122), with convalescent patients presenting with absolute numbers of CD16low/highCD10- neutrophils that are significantly lower than those in patients with active disease and comparable to those recorded in HCs (126). Based on these observations, the presence of circulating LDNs is considered an early marker of SARS-CoV-2 infection (126).
ScRNA-seq of whole blood, peripheral blood mononuclear cells (PBMCs) and purified neutrophils isolated from COVID-19 patients of differing disease severities has revealed unique transcriptomic signatures for neutrophils of patients with severe disease (17, 18, 21, 124). Schulte-Schrepping et al identified two distinct subpopulations of LDNs in PBMC samples of patients with severe disease (18), with detailed analysis revealing eight distinct cell clusters consisting of pro-, pre- and mature neutrophils that expressed genes involved in NET formation (e.g. MPO, ELANE, PRTN3 and PADI4) and the suppression of adaptive immune responses (e.g. CD274 and ARG1) (18). In line with these observations, Aschenbrenner et al performed transcriptomic profiling on whole blood samples and purified neutrophils from mild and severe COVID-19 patients and showed signatures associated with neutrophil activation (e.g. CD177, PADI4, ELANE and MPO) and immune suppression (e.g. ARG1 and CD274) were enriched in patients with severe disease (17). Interestingly, the authors also showed that COVID-19 patients presented with combinations of gene clusters that were not detected in the whole blood transcriptome of patients with other infectious diseases or inflammatory-associated conditions (17). Moreover, mass cytometry of whole blood samples has shown that in contrast to the immune response of patients with SARS-CoV-2 negative flu-like illness, which was characterised by the presence of highly activated mature neutrophils, an accumulation of activated immature neutrophils with immune suppressive properties defined the granulocyte response of patients with severe COVID-19 (18). Taken together, these studies suggest that SARS-CoV-2 infection induces a distinct neutrophil response that is enriched with features of immaturity, activation and suppression.
The presence of immature neutrophils in the circulation of COVID-19 patients suggests SARS-CoV-2 infection triggers emergency granulopoiesis, the premature mobilisation of neutrophil precursors from the bone marrow. Emergency granulopoiesis has previously been reported in the setting of trauma and sepsis, where systemic inflammation is considered a key factor in driving the release of neutrophil progenitors from the bone marrow (129, 130). Suggesting that this may also be the case in COVID-19, significant positive associations have been reported between plasma concentrations of IL-6 and the frequency/absolute number of CD10-CD64+Lox1+ and CD16low/highCD10- neutrophils as well as CD16IntCD44LowCD11bInt LDIBs (29, 125, 126). Moreover, circulating concentrations of G-CSF, which is considered a key mediator of emergency granulopoiesis (129, 131), are significantly higher in COVID-19 patients when compared to HCs, with those with severe disease exhibiting higher concentrations than moderate cases (5).
Degranulation
Located in the membranes of primary, secondary and tertiary granules respectively, increased expression of CD63, CD11b or CD66b on the neutrophil cell surface is considered a surrogate marker of degranulation. Studies that have examined neutrophil degranulation via these changes in surface phenotype have reported conflicting results, with some groups describing a SARS-CoV-2-induced increase in the expression of CD63 and CD66b (26, 122, 128) and others reporting no difference in the expression of CD11b or CD66b between neutrophils isolated from COVID-19 patients of differing disease severities and HCs (122, 132). In contrast, studies that have measured the circulating concentrations of granule-derived proteins released by activated neutrophils [e.g. neutrophil elastase (NE), myeloperoxidase (MPO) and matrix metalloproteinase-9 (MMP-9)] have consistently reported robust neutrophil degranulation in response to SARS-CoV-2 infection.
NE is a serine protease stored within the primary granules of neutrophils. Compared to the levels recorded for HCs, both the concentration and activity of NE has been found to be significantly higher in blood samples from severe COVID-19 patients with ARDS and/or patients with disease severities ranging from mild to fatal (26, 122, 133, 134). When released into plasma, NE is rapidly inactivated by endogenous protease inhibitors. Interestingly, sera from COVID-19 patients has been shown to significantly inhibit the activity of exogenous NE in in vitro assays, suggesting that the increased activity of NE in the blood of COVID-19 patients reflects a degree of resistance of the protease to its endogenous inhibitors (26). On this note, NE is protected from plasma-mediated neutralisation when bound to DNA (135, 136). Thus, the significantly higher number of DNA-NE complexes that have been measured in SARS-CoV-2 infected patients offers a potential mechanistic explanation for the increased activity of NE in COVID-19 blood samples (26). In addition to causing bystander tissue damage that may contribute to the onset of acute lung injury (ALI) in COVID-19 patients, an elevation in the concentration and activity of NE may enhance SARS-CoV-2 infectivity (137, 138). SARS-CoV-2 enters cells directly from the cell surface when its surface expressed spike glycoprotein is cleaved by host proteases such as transmembrane serine protease-2 (TMPRSS-2), a modification that enables the virus to bind to its surface ACE2 (80). NE has been shown to cleave the spike glycoprotein of SARS-CoV viruses (139, 140). Thus, one can envisage a vicious cycle in patients with severe COVID-19 whereby SARS-CoV-2-induced activation of neutrophils triggers the release of NE, whose subsequent cleavage of the spike glycoprotein facilitates enhanced viral entry, which results in increased viral infectivity and disease pathogenesis (138).
When compared to HCs and patients with mild-to-moderate disease, significantly higher concentrations of MPO have been measured in blood samples acquired from severe COVID-19 patients (128, 141). Suggesting a relationship between increased neutrophil activation and disease severity, Ueland and colleagues reported a negative correlation between plasma MPO levels and Pa02/Fi02 (P/F) ratio, a marker of respiratory function for which low values indicate respiratory distress/failure (141). In a recent review article, in which it was suggested that MPO release is a consequence of the cytokine storm that is induced by SARS-CoV-2 infection, Goud et al addressed a potential role for elevated MPO levels in driving the clinical deterioration that precedes the onset of severe COVID-19 (142). Mechanistically, the authors suggested that by catalysing the generation of the oxidant hypochlorous acid and consuming nitric oxide, MPO may contribute to COVID-19 pathology by promoting oxidative stress, vasoconstriction, tissue injury and cell death (142).
Positively associated with neutrophil number, plasma concentrations of MMP-9 are significantly elevated in SARS-CoV-2 infected patients (105). A protease involved in the degradation of the extracellular matrix (ECM), activity of MMP-9 is increased when it is bound to the protein neutrophil gelatinase-associated lipocalin (NGAL), whilst its activity is negatively regulated by tissue inhibitor of metalloproteinase-1 (TIMP-1) (143, 144). Relative to HCs, significantly elevated concentrations of MMP-9:NGAL heterodimers and an increased MMP-9:TIMP-1 ratio have been detected in COVID-19 patients (105). Pointing towards enhanced plasma MMP-9 activity, which has previously been linked to the development of ARDS in critically-ill patients (145), these findings suggest that an increase in MMP-9-mediated proteolysis may, by promoting lung injury, contribute to the onset of severe COVID-19 (105). Supporting this idea, Ueland et al reported a significant negative correlation between plasma MMP-9 levels and P/F ratio in 39 hospitalised COVID-19 patients, with those who developed respiratory failure presenting at two study time-points (days 0-2 and 3-5 post-hospitalisation) with significantly higher circulating concentrations of MMP-9 than patients who did not experience respiratory failure (146). Interestingly, in a recent network-based systems biology study that set out to identify the molecular targets of drugs that have been proposed as potential candidates for the treatment of COVID-19, MMP-9 emerged as an interacting partner of both chloroquine and melatonin (147). Whilst current evidence suggests chloroquine treatment does not improve clinical outcomes in patients with COVID-19 (148), high dose melatonin has shown promise as an adjuvant therapy, with evidence of potential clinical benefit in hospitalised patients with COVID-19 pneumonia (149). Whilst the mechanism of action of melatonin is likely to be multi-factorial (150), its ability to significantly inhibit MMP-9 activity and expression (151) suggests that its clinical benefit could arise, at least in part, from a reduction in MMP-9 mediated inflammatory responses and lung damage.
NET Formation
NETs are an extracellular defence mechanism involved in the capture, neutralisation and elimination of invading pathogens (152). Renowned for their role in immune responses against bacteria and fungi (153, 154), data are now emerging demonstrating that NETs are also generated in response to viral challenge (155–158). Consisting of a DNA backbone decorated with histones and an array of granule-derived peptides and enzymes such as MPO, NE and gelatinase, NETs have significant cytotoxic and thrombotic potential (153). Thus, NETs have been proposed to represent “double-edged swords”, with their excessive or dysregulated production suggested to promote tissue damage, thrombotic complications and/or hyperinflammation, all of which offset their anti-microbial properties (159, 160). In the context of respiratory infections, aberrant NET formation has been associated with the development of ALI and ARDS (161–163), two conditions that, alongside multiple organ failure (MOF) are common secondary complications of severe COVID-19 (30, 164, 165). Thus, in an effort to establish whether NETs contribute to the pathogenesis of severe COVID-19, a number of studies have screened peripheral blood and tracheal aspirate samples from COVID-19 patients of differing disease severities for evidence of NET formation.
Concentrations of a range of NET-associated markers, including cell-free DNA (cfDNA), MPO/NE-DNA complexes and citrullinated histone H3 (CitH3), have all been shown to be significantly increased in the circulation and/or tracheal aspirates of patients with COVID-19 (26, 46, 65, 66, 105, 134, 166–170). As expected for a respiratory infection, NET levels are significantly higher in tracheal aspirates, bronchiolar lavage and BALF samples when compared to plasma (65, 66). In terms of disease severity, patients with severe disease present with significantly higher circulating levels of cfDNA, MPO-DNA complexes and CitH3 when compared to patients with mild to moderate COVID-19 (26, 105, 170), with one study showing a surge in plasma NET markers prior to ICU admission (170). Thus, in heterogeneous patient cohorts, plasma NET levels positively correlate with clinical severity scoring systems (65, 105, 170) and are negatively associated with the P/F ratio (65). Interestingly, concentrations of MPO-DNA complexes have been shown to be significantly higher in plasma samples from male patients when compared to their female counterparts (105), an observation worthy of further investigation given that male gender is a risk factor for ICU admission and death following SARS-CoV-2 infection (171). To date, few studies have measured NET generation longitudinally in COVID-19 patients, with those that have reporting conflicting observations (65, 66, 105). For example, Middleton et al measured comparable levels of MPO-DNA complexes in plasma samples obtained from convalescent patients and HCs (65), whereas Petito and co-workers detected similar concentrations of MPO-DNA complexes in plasma samples collected from patients during active infection and recovery (105). Thus, it remains to be determined whether patients who survive severe COVID-19 are at an increased long-term risk of NET-associated complications.
Treating neutrophils isolated from HCs with serum/plasma from COVID-19 patients triggers NET production in vitro, thereby demonstrating the presence of NET-inducing agents in the circulation of SARS-CoV-2 infected subjects (65, 91, 105, 167, 172, 173). Pro-inflammatory cytokines (e.g. IL-6, IL-8, G-CSF), the complement factor C5a, antigen-antibody complexes and platelet derived factors (e.g. platelet factor 4 (PF4) and RANTES), whose circulating levels are all increased in patients with severe COVID-19 (5, 65, 91, 167, 174, 175), have been proposed as potential mediators of NET formation (65, 91, 167, 173). Interestingly, adding to a growing list of viruses, which includes chikungunya virus and human immunodeficiency virus (156, 157), SARS-CoV-2 directly promotes NET release (46, 172). Dependent upon the activity of peptidylarginine deiminase 4 (PAD4), TMPRSS-2 and the expression of ACE2 (46), SARS-CoV-2 triggers a level of NET production comparable to that observed for neutrophils treated with the potent protein kinase C agonist phorbol 12-myristate 13-acetate (172). Via its involvement in the detection of single-stranded RNA viruses, the intracellular pathogen recognition receptor toll-like receptor (TLR)-7 has been implicated in SARS-CoV-2-induced NET formation (46), although this remains to be proven.
In the absence of exogenous stimulation, neutrophils isolated from COVID-19 patients exhibit enhanced NET generation in vitro (46, 65, 168, 175). Mechanistically, this increase in spontaneous NET formation may reflect SARS-CoV-2-induced changes in the composition of the circulating neutrophil pool, specifically the presence of hyperactive neutrophils and/or immature LDNs. Indeed, preliminary data from Parackova and colleagues has shown evidence of increased activation of p38 and c-Jun N-terminal kinase, two kinases important for NET production (176, 177), in resting neutrophils isolated from COVID-19 patients (122). Moreover, LDNs, whose circulating frequencies are increased in patients with severe disease (128), exhibit enhanced spontaneous NET formation when compared to mature neutrophils (121, 178).
Metabolism
Results of metabolomic and lipidomic profiling on plasma and serum samples from COVID-19 patients of varying disease severities suggest that metabolic dysregulation contributes to disease pathogenesis, with perturbations reported in such pathways as tryptophan metabolism, the tricarboxylic acid (TCA) cycle, mobilisation of polyunsaturated fatty acids and eicosanoid biosynthesis (32, 179–184). Alongside these changes in the systemic metabolome, data are accumulating that suggest SARS-CoV-2 infection promotes metabolomic reprogramming in neutrophils (128, 185) and PBMCs (181).
In a study that compared metabolic processes between neutrophils isolated from HCs and severe COVID-19 patients, McElvaney et al demonstrated a disease associated increase in the cytosolic levels of pyruvate kinase M2, an enzyme that catalyses the final step of glycolysis, and the nuclear expression of hypoxia-inducible factor-1α, a transcription factor that promotes the expression of glycolytic enzymes (185). A profile that would be predicted to enhance glycolytic flux, the authors found cytosolic lactate levels and the lactate:pyruvate ratio were significantly higher in neutrophils of COVID-19 patients, suggesting a SARS-CoV-2-induced shift of neutrophil metabolism towards glycolysis (185). Critical for neutrophil anti-microbial responses, glycolysis and lactate generation are essential steps in the formation of NETs (186, 187). Thus, this elevated glycolytic activity of COVID-19 neutrophils could be a mechanistic explanation for the aforementioned increase in spontaneous ex vivo NET generation by neutrophils isolated from patients with severe disease (46, 65, 168, 175). However, in an independent study, Reyes et al provided evidence of reduced glycolytic flux and a switch to oxidative metabolism in neutrophils isolated from COVID-19 patients with ARDS (128). Reflecting the infancy of this field of research, these conflicting observations are likely to be a consequence of small cohort sizes (n=3-8) and differences in methodological approach. Thus, further studies enrolling a larger number of patients with varying disease severities are required to fully understand how SARS-CoV-2 infection impacts upon neutrophil metabolism.
Pulmonary Neutrophil Response
As a lower respiratory tract infection, the greatest insight into the immune response to SARS-CoV-2 has been provided by studies that have directly examined lung tissue, via histological staining of post-mortem COVID-19 lung samples, or analysed the pulmonary microenvironment via the acquisition of BALF. Although hampered by small sample sizes and the decision to often use samples collected from HCs as a reference cohort rather than patients with non-COVID-19 respiratory infections, current data suggest that robust chemokine production and dysregulated myeloid responses, which include the infiltration and activation of neutrophils, characterise the pulmonary immune responses of patients who develop severe COVID-19.
Analysis of BALF
Our current understanding of how SARS-CoV-2 infection influences the neutrophil pool within the lung is based almost entirely upon the findings of studies that have performed scRNA-seq on BALF samples obtained from COVID-19 patients of different disease severities (48, 78, 97, 188–193). Pathway analysis of the differentially expressed genes that have been identified between the BALF transcriptomes of COVID-19 patients and HCs has revealed chemokine signalling to be one of the most up-regulated pathways in SARS-CoV-2 infected subjects (189). In terms of specific transcripts, the expression levels of genes relating to neutrophil chemotaxis such as CXCL1, CXCL2, CXCL6, CXCL8 and CCL2 have consistently been found to be up-regulated in COVID-19 patients (189–192). Moreover, suggesting that SARS-CoV-2 triggers a robust neutrophilic response, the up-regulation of neutrophil chemokine associated genes in COVID-19 patients is significantly greater than that reported in BALF from patients with viral or non-viral community-acquired pneumonia (189). A combination of ex vivo data (188, 193) and results from in vitro cell culture systems (191, 194, 195) suggest that this chemokine signature of SARS-CoV-2 infection is expressed predominantly within lung macrophages and both alveolar and bronchial epithelial cells. Interestingly, suggesting a breakdown in regulatory negative feedback loops, infection of primary human lung epithelial cells in vitro with SARS-CoV-2 was found to result in down-regulation of the gene encoding for CXCL14 (195). A negative regulator of immune cell recruitment, it has been suggested that, if replicated in vivo, reduced expression of CXCL14 would result in enhanced recruitment of neutrophils to the lung (195). In line with these transcriptomic data, increased concentrations of IL-8 have been measured in the BALF of patients with severe COVID-19 when compared to those with moderate disease (188).
As expected for a microenvironment enriched in neutrophil chemoattractants, neutrophil associated transcripts are more abundant in the BALF samples of severe COVID-19 patients when compared to those of HCs (48, 189, 190), subjects with mild disease (48, 190, 196) or patients with non SARS-CoV-2-induced pneumonia (78, 189). This enrichment in neutrophil signatures has been shown to be independent of lung injury, age, gender and underlying co-morbidities (78), suggesting that rather than simply representing a marker of severe lung inflammation, neutrophils contribute to the immunopathology that underpins the onset of COVID-19 pneumonia (78). Akin to the findings reported in whole blood (17, 18, 21), heterogeneity has also been reported in the pulmonary neutrophil pool (48, 78). Wauters et al clustered BALF residing neutrophils into five distinct phenotypes and found the frequency of both “progenitor” and “inflammatory mature” neutrophils were significantly higher in samples from mild/critical COVID-19 patients when compared to subjects with non-COVID-19 pneumonia (78). Interestingly, phenotypic profiling of immature CD16IntCD44LowCD11bInt LDIBs, whose circulating frequencies are increased in severe COVID-19 patients, has shown that this subpopulation exhibits decreased surface expression of CD44 (29). As low CD44 expression is associated with increased neutrophil migration to the lungs (197), it may be that pulmonary infiltration of LDIBs contributes to the “progenitor” phenotype of neutrophils detected in the BALF samples of critical COVID-19 patients (78).
In terms of functional responses, gene sets related to neutrophil degranulation and NETs are up-regulated in lung/BALF samples of COVID-19 patients (97). Interaction network analysis has shown these NET associated genes interact with negative regulators of T and natural killer cell function as well as genes linked to ROS formation (97). Thus, it has been suggested that in addition to promoting direct tissue injury, NET formation within the lungs of COVID-19 patients may hasten viral replication and disease pathogenesis by impairing host anti-viral immune responses (97).
Analysis of Lung Tissue
Histopathological staining of post-mortem lung tissue has demonstrated intense neutrophilic infiltrate within the alveolar spaces and/or pulmonary vessels of patients with severe COVID-19 (37, 46, 65–68). Furthermore, and in agreement with data obtained from total RNA-seq (198), NETs have been identified throughout the lungs of fatal COVID-19 cases. Defined as extracellular areas of DNA staining positive for MPO and CitH3, NETs have been detected in the airway, interstitial, alveolar and vascular compartments of COVID-19 lungs (26, 46, 65, 66, 199). Suggesting that NET formation occurs only in the lungs, Radermecker et al failed to detect evidence of NETs in tissue sections obtained from the liver, pancreas, kidney or heart of SARS-CoV-2 infected patients (199). However, in contrast, Leppkes and colleagues showed the presence of aggregated NETs in both kidney and liver tissue samples, leading them to propose that widespread NET generation may contribute to multi-organ damage in cases of severe COVID-19 (26). To address these conflicting observations, and establish whether NET formation in peripheral organs is indeed a feature of severe disease, additional studies with larger sample sizes are required.
Although SARS-CoV-2 can directly induce NET formation (46, 172), only a small number of viral transcripts have been detected in post-mortem lung samples (198), suggesting that other agonists may be triggering pulmonary NET formation in severe COVID-19. Potential candidates that have been proposed thus far include PF4, whose gene expression is increased ~5.5 fold in lung tissue of fatal COVID-19 cases (198), and an array of NET-inducing cytokines and chemokines such as IL-6, IL-8, TNF-α, IL-1β and CCL20 whose expression levels are up-regulated in COVID-19 BALF samples and/or human lung epithelial cells infected with SARS-CoV-2 (188, 189, 194, 195). The low viral burden present in post-mortem lung tissue has led to the suggestion that rather than an ongoing active viral infection, tissue injury and inflammation arising from dysregulated and overexuberant host immune responses is responsible for patient death (198). Due to their cytotoxic and pro-coagulant nature (159, 160), aberrant NET formation may be one factor contributing to the diffuse alveolar damage and interstitial lesions that have been detected in the lung samples of fatal COVID-19 cases (37, 46, 199, 200).
In an elegant study that investigated the early innate immune response to SARS-CoV-2 infection, Guo et al found a greater number of genes related to “cellular responses to lipopolysaccharide (LPS)” and “neutrophil chemotaxis” were up-regulated in lung tissue samples obtained from rhesus macaques and mice at days 3 and 5 post-infection when compared to genes involved in anti-viral immunity (201). Not observed in mice infected with other RNA or DNA viruses, SARS-CoV-2 triggered the infiltration of immature CD45+11b+Ly6Gvariable aberrant neutrophils into the lung, which was associated with fatal outcome (201). Mechanistically, this anti-bacterial response to viral challenge was attributed to robust induction of S100A8, an alarmin that upon binding to S100A9 forms calprotectin, a damage associated molecular pattern (DAMP) that promotes neutrophil chemotaxis and activates TLR4, the pathogen recognition receptor for LPS (201–203). Indeed, associated with increased survival rates, significantly fewer immature neutrophils and reduced viral loads were recovered from lung samples of SARS-CoV-2 infected mice treated with paquinimod, an inhibitor of S100A8/S100A9 signalling (201). In addition to the lungs, these aberrant immature neutrophils, which were suggested to be granulocytic myeloid-derived suppressor cells, were also detected in the blood and bone marrow of SARS-CoV-2 infected mice, demonstrating systemic dysregulation of the neutrophil pool (201). Interestingly, relative to HCs, increased expression of S100A8 has also been detected in lung tissue samples from patients with severe COVID-19 (198, 201), who, when compared to moderate cases, also present with significantly higher circulating concentrations of calprotectin (48, 204, 205). Thus, based on the observations of Guo et al (201), and data that has shown calprotectin stimulates granulopoiesis (206), neutrophil chemotaxis (202) and NET formation (207), the systemic and pulmonary elevation that occurs in the levels of this DAMP could promote the premature mobilisation and infiltration of immature neutrophils into the lungs, where they contribute to tissue injury and pulmonary failure.
Neutrophil Tolerance in Severe COVID-19
With an incidence rate of 13.5-56%, hospital-acquired infections are emerging as a common secondary complication amongst COVID-19 patients (175, 208–210). More frequently observed in those with severe disease (209), secondary bacterial and fungal superinfections have been shown to be significantly associated with an increased hospital and ICU length of stay, an increased need for mechanical ventilation and a lower 28-day ventilator-free survival rate (208). This susceptibility to fungal and bacterial infections suggests that neutrophils of COVID-19 patients exhibit impaired anti-microbial functions upon secondary challenge. Supporting this idea of functional tolerance/exhaustion, significantly reduced ROS production, degranulation and NET formation have been reported for neutrophils isolated from COVID-19 patients following in vitro stimulation with bacteria (e.g. Staphylococcus aureus, Streptococcus pneumonia or Escherichia coli) or bacterial products (LPS), all of which culminate in reduced bactericidal activity (18, 26, 65, 175).
Suggesting the presence of an inhibitory/tolerising factor in the circulation of COVID-19 patients, Shambat et al demonstrated that pre-treating neutrophils isolated from HCs with COVID-19 patient plasma resulted in significantly impaired functional responses upon bacterial challenge (175). Mechanistically, the authors suggested IL-4 and IL-10, two anti-inflammatory cytokines that inhibit neutrophil function (211, 212), and whose circulating concentrations are significantly increased in SARS-CoV-2 infected patients (5, 175), may be responsible for the plasma-driven reduction they observed in neutrophil activity (175). However, it is unlikely that these cytokines are wholly responsible. For instance, pre-treatment of HC neutrophils with the complement factor C5a, whose circulating concentrations are increased in severe COVID-19 patients (91, 174), has been shown to reduce both phagocytic and bactericidal activity (213, 214). Moreover, due to the inflammation-induced tissue damage that would occur in patients with severe COVID-19, we suggest that the release of mitochondrial-derived damage associated molecular patterns (mtDAMPs) may also contribute to the SARS-CoV-2-induced induction of neutrophil tolerance.
Passively released from damaged/necrotic tissue and secreted by activated immune cells, damage associated molecular patterns (DAMPs) are a heterogeneous collection of nuclear, cytosol and mitochondrial-derived proteins, lipids and DNA (215). Owing to their prokaryotic origins, mtDAMPs are potent immune activators, with their stimulation of neutrophils linked to the induction of systemic inflammatory responses and organ injury (216–218). However, in addition to immune activation, we and others have shown that mtDAMPs induce functional tolerance, with neutrophils pre-treated with mtDAMPs or mtDAMP mimetics exhibiting significantly impaired calcium mobilisation, chemotaxis and NET generation upon secondary stimulation with inflammatory or pharmacological agonists (216, 219–221). To date, whilst several studies have reported evidence of cellular injury and inflammatory cell death in patients with severe COVID-19 (23, 26, 222), few have directly screened blood samples for the presence of DAMPs. However, in those that have, significantly elevated levels of the nuclear-derived DAMPs high mobility group box-1 (HMGB-1) and extracellularly-secreted nicotinamide phosphoribosyl-transferase (eNAMPT), as well as mitochondrial-derived DNA (mtDNA) were detected in the plasma of severe COVID-19 patients when compared to HCs and/or mild to moderate COVID-19 cases (205, 223, 224). Given that N-formylated peptides, which we have shown are responsible for mediating mtDAMP-induced neutrophil tolerance (219), are released alongside mtDNA, then these preliminary data suggest that tolerising mtDAMPs are present in the circulation of patients with severe COVID-19. However, studies that quantify N-formylated peptides (e.g. NADH-ubiquinone oxidoreductase chain 6 (ND6)) in COVID-19 patient plasma and assess their relationship with ex vivo neutrophil function are needed to support our hypothesis that mtDAMPs contribute to SARS-CoV-2-induced neutrophil tolerance.
In the setting of non-COVID related ARDS, where elevated BALF concentrations of mtDNA and N-formylated peptides have been implicated in disease pathogenesis (225, 226), pharmacological manipulation of mtDAMP signalling has been suggested as a potential treatment for neutrophil-mediated lung injury (225). Similarly, with the aim of suppressing pulmonary inflammation and preventing the onset of severe disease, blocking DAMP signalling was recently proposed as a treatment for COVID-19 (223, 227). We believe that this approach, particularly when focussed upon mtDAMPs, may have the additional benefit of reducing the susceptibility of COVID-19 patients to superinfections by preventing mtDAMP-induced neutrophil tolerance. This viewpoint is shared by researchers in other fields of critical care who have proposed that mtDAMP exposure underlies the susceptibility of traumatically-injured patients to pulmonary infections and that blocking the formyl peptide receptor would, by restoring neutrophil function, enhance immune responses to bacterial challenge (221). Finally, whilst this discussion has focussed on neutrophil responses, it should be noted that monocytes of COVID-19 patients also exhibit impaired anti-microbial responses ex vivo and that pre-treating HC monocytes with COVID-19 patient plasma significantly reduced their bactericidal activity (175). Given that exposure to mtDAMPs also induces functional tolerance in monocytes (228), then it is reasonable to speculate that targeting mtDAMP signalling may restore the effector functions of two key innate immune cells, thereby reducing the susceptibility of COVID-19 patients to hospital-acquired superinfections.
Are Neutrophils a Potential Therapeutic Target for the Treatment of COVID-19?
If a granulocytic signature characterised by immature, hyperactive and pro-inflammatory neutrophils is a feature of the dysregulated peripheral and pulmonary immune responses that contribute to the transition from mild to severe COVID-19 (17–21), then does the neutrophil represent a potential therapeutic target for the treatment of hospitalised COVID-19 patients? This question has been raised in a number of previous perspective and review articles, where an approach that targets specific signalling pathways and/or neutrophil products (e.g. NETs, ROS or proteases) appears to be favoured over strategies that would result in the complete suppression of neutrophil function (36, 37, 137, 138, 191, 229–231). Indeed, given the abovementioned susceptibility of hospitalised COVID-19 patients to bacterial and fungal superinfections (175, 208–210), a therapeutic strategy that suppresses neutrophil hyperactivity whilst preserving anti-microbial function would be best placed to reduce neutrophil-mediated bystander tissue damage without increasing the risk of secondary infections (231). In this section, we consider whether targeting NETs or suppressing the release of immature neutrophils from the bone marrow represent potential therapeutic strategies for the treatment of COVID-19.
NETs
To date, discussions regarding the potential use of anti-neutrophil therapies for the treatment of COVID-19 have centred on targeting NETs, whose enhanced generation is likely to contribute to both the pulmonary and extrapulmonary manifestations of severe COVID-19 (231–233). Implicating NETs in the clotting associated complications that have been suggested to account for more than 70% of COVID-19 deaths (234), significantly higher plasma levels of cfDNA, MPO-DNA complexes and CitH3 have been detected in patients who developed thrombotic events when compared to those who did not (105, 170). In addition, excessive NET formation in the lungs has been shown to result in vascular occlusion and disturbed microcirculation (26). Through their ability to induce macrophages to secrete IL-1β (235), which is itself a stimulus for NET formation (236, 237), the presence of NETs within the lungs could foster a self-perpetuating and deleterious positive feedback loop that, by promoting a state of hyperinflammation, would accelerate the formation of microthrombi and the onset of respiratory failure (37). Moreover, with recent evidence demonstrating that NET bound NE inhibits efferocytosis by cleaving the integrins avβ3 and avβ5 from the surface of macrophages (238), increased NET formation may also delay the resolution phase of pulmonary immune responses in patients with severe COVID-19. Thus, via their potential involvement in facilitating immunothrombosis, pulmonary and systemic hyperinflammation and respiratory failure, NETs represent an ideal therapeutic target for treating a number of the complications experienced by severe COVID-19 patients. Potentially achievable via the repurposing of existing therapies, two strategies currently under consideration for alleviating NET-associated pathologies in SARS-CoV-2 infected patients are to (i) suppress NET formation or (ii) enhance NET breakdown.
Suppression of NET Formation
Promoting chromatin decondensation via the citrullination of histone H3, PAD4 is a major regulator of NET formation (239). Having shown promise in murine models of infection and inflammation, where inhibition of PAD4 resulted in reduced NET-associated lung injury (162, 240), systemic or pulmonary administration of PAD4 inhibitors has been proposed as a potential treatment for severe COVID-19 (36). Current data that would support such an approach are the results of studies that have shown in vitro treatment with Cl-amidine, an irreversible pan-PAD inhibitor, significantly reduced SARS-CoV-2-induced NET production by HC neutrophils (46) as well as the enhanced spontaneous NET generation of neutrophils isolated from COVID-19 patients (172).
Treating HC neutrophils in vitro with COVID-19 patient plasma/serum induces NET formation (65, 91, 105, 167, 172, 173). To date, C5a and antigen-antibody complexes have been proposed to be some of the inflammatory agonists that trigger this response (91, 173). If correct, then targeting these mediators directly or blocking their signalling pathways may represent another therapeutic approach by which to suppress NET production. In this area of research, Strich et al recently demonstrated that via inhibition of spleen tyrosine kinase (SYK), which signals downstream of FCγRIIA receptors, pre-treating neutrophils with R406, the metabolically active component of the food and drug administration (FDA)-approved drug fostamatinib, significantly reduced NET formation by HC neutrophils stimulated with COVID-19 patient plasma (173). Hypothesising that a reduction in NET generation may be one mechanism by which inhibition of SYK could improve the outcome of patients with COVID-19, a phase II, randomised, double-blind, placebo controlled trial of fostamatinib for the treatment of hospitalised COVID-19 patients is currently underway (ClinicalTrials.gov identifier: NCT04579393). Another drug under investigation in ongoing COVID-19 clinical trials is eculizumab (ClinicalTrials.gov identifiers: NCT04346797, NCT04355494, NCT04288713). A humanized IgG monoclonal antibody that inhibits the cleavage of the complement protein C5, eculizumab treatment has been shown to significantly reduce neutrophil activation in patients with a history of thrombosis (241, 242), which in one study was associated with a decline in circulating markers of NETs (242). Interestingly, clinical recovery following eculizumab treatment was recently reported in a case series of four COVID-19 ICU patients with severe pneumonia or ARDS, with reduced neutrophil activation and infiltration into the lungs proposed as potential mechanistic explanations (243). Follow-up studies that measure markers of neutrophil activation and NET generation in plasma and/or BALF samples of COVID-19 patients treated with eculizumab would help address this hypothesis. Alongside these potential therapies, inhibition of the abovementioned IL-1β-NET positive feedback loop could be achieved by treatment with anakinra. A human IL-1 receptor antagonist, anakinra is approved for use in patients with rheumatoid arthritis and has been shown to significantly reduce NET formation in vitro (236). Furthermore, in a murine model of airway inflammation, anakinra treatment resulted in a significant decrease in the BALF levels of the DAMP HMGB-1 (244). An inducer of NET formation (245, 246), significantly higher concentrations of HMGB-1 have been reported in severe COVID-19 patients when compared to both HCs and mild/moderate cases (205). Thus, via a number of mechanisms, antagonism of the IL-1 receptor may reduce the NET burden of COVID-19 patients, a hypothesis that can be tested by studying the results of an ongoing clinical trial that is measuring DNA-MPO complexes in serial blood samples acquired from severe COVID-19 patients receiving anakinra treatment (ClinicalTrials.gov identifiers: NCT04594356).
Compared to subjects who received standard anti-viral and steroid therapy, an open label study of 31 severe COVID-19 patients reported improved clinical indices and remission rates for patients co-treated with the FDA-approved drug dipyridamole (247). An anti-thrombotic medication, dipyridamole was recently shown to suppress NET formation in vitro via activation of the adenosine A2A receptor (248). Based on these observations, it was suggested that a randomised trial that would assess the impact of dipyridamole treatment on NET formation and its relationship with clinical outcome in COVID-19 patients would be of interest (167). Since this proposal, Petito et al have shown that pre-treatment of HC neutrophils with dipyridamole failed to inhibit NET formation triggered by COVID-19 patient plasma (105). Thus, whilst dipyridamole may not suppress NET generation in SARS-CoV-2 infected subjects, the idea of activating signalling pathways that negatively regulate NET formation is worth pursuing as a therapy for COVID-19. Under consideration as a strategy for reducing NET generation in other inflammatory conditions is treatment with prostaglandin E2 (PGE2), which potently suppresses NET formation by increasing intracellular levels of cyclic adenosine monophosphate (249, 250). However, due to the dysregulated lipid profiles of COVID-19 patients (32) and the potent immunomodulatory properties of PGE2, which extend beyond the inhibition of NET formation, more studies are required to address the practicalities of such an approach in SARS-CoV-2-infected patients.
Whilst the idea of blocking NET formation is gaining traction as a potential therapy for the treatment of severe COVID-19 (36, 37, 173), we feel it is important to highlight a potential caveat of such an approach: reduced anti-microbial immunity. Although deleterious when generated in excess, NETs play an important role in the entrapment, neutralisation and eradication of bacterial and fungal pathogens (153, 154). Thus, the use of PAD4 inhibitors for example, may increase the susceptibility of severe COVID-19 patients to secondary infections. Indeed, whilst it has been shown that the ability of neutrophils isolated from PAD4-/- mice to phagocytose and generate ROS is comparable to that of neutrophils from wild type littermates (251), PAD4-/- mice showed impaired bacterial clearance in a model of pneumonia-induced ALI (162). Thus, the reported benefit of reduced NET-mediated lung injury in PAD4-/- mice was offset by an increased bacterial burden (162). Such factors as dosage, timing and duration of therapy may therefore prove to be critical in determining the outcome of any strategy that attempts to eradicate NET formation for the treatment of COVID-19.
Degradation of NETs
Responsible for the breakdown of 90% of circulating cfDNA, deoxyribonuclease-1 (DNase-1) is the predominant endonuclease in bodily fluids (252). Relative to HCs, patients with severe, but not mild, COVID-19 exhibit significantly reduced circulating levels of this endonuclease (166), whose plasma concentrations have also been shown to be significantly lower in COVID-19 non-survivors when compared to survivors (169). In terms of function, significantly impaired total DNase activity has been measured in serum samples acquired from COVID-19 patients with mild and severe disease (233). Thus, the associations that have been reported between elevated NET markers, clinical deterioration and mortality could be attributable in part to a severity-associated impairment in DNase-1 mediated clearance of cfDNA (65, 105, 167, 169).
Used in the treatment of cystic fibrosis to aid in mucociliary clearance, dornase alfa is an FDA-approved recombinant human DNase-1 that digests extracellular DNA. Designed in part on the idea that exaggerated NET formation contributes to pulmonary hyperinflammation, a prospective randomised multi-centre clinical trial entitled COVIDornase is underway to investigate the effect of twice daily aerosolized intra-tracheal administration of dornase alfa on the severity and progression of ARDS in critically ill ICU COVID-19 patients (253). Potential benefits that have been proposed may arise from DNase-1 therapy in COVID-19 patients are enhanced clearance of mucous secretions, improved ventilation and a reduced risk of secondary infections (37, 253). However, whilst a promising therapy, it is important to highlight some potential barriers that we feel need to be considered and overcome if DNase-1 treatment is to be of clinical benefit for patients with severe COVID-19. These are:
1. Timing of treatment – In a murine model of sepsis, Mai et al demonstrated that despite reducing plasma levels of cfDNA, early, but not late, administration of DNase resulted in increased plasma concentrations of IL-6, enhanced inflammatory cell infiltration into the lungs and multi-organ damage (254). In the context of COVID-19, particularly patients with secondary infections, inappropriate timing of DNase therapy could therefore potentially exacerbate the pre-existing states of systemic and pulmonary hyperinflammation.
2. Substrate – Relative to HCs, both the circulating concentration and pulmonary expression of PF4 have been shown to be increased in COVID-19 patients (65, 198). Under flow conditions in vitro, PF4 binds directly to NETs resulting in their compaction, a morphological change that confers resistance to degradation by DNase-1 (255). Thus, the efficiency of DNase-1-mediated breakdown of NETs could be reduced in COVID-19 patients.
3. Microenvironment – Released into the circulation as a consequence of cell damage or death, actin is a negative regulator of DNase-1 activity (256, 257). Responsible for the clearance of circulating actin is the extracellular actin scavenger system (EASS), which comprises of two plasma proteins: vitamin D binding protein (VDBP) and gelsolin (GSN) (258). In the setting of thermal injury, we have recently demonstrated dysregulation of the EASS, where reduced circulating concentrations of GSN and VDBP were associated with impaired serum DNase activity and an accumulation of cfDNA (259). A similar scenario is emerging in COVID-19, where a significant reduction in total serum DNase activity (233) is accompanied by increased and decreased circulating concentrations of actin and GSN respectively (179, 260, 261), with GSN levels negatively associated with disease severity (260). Thus, we hypothesise that exogenous DNase-1 would be administered into an environment rich in circulating actin, which would result in the formation of DNase1-actin complexes and an impairment in endonuclease activity. A potential approach that could circumvent this issue is the use of AIR DNase™, an actin-resistant endonuclease whose safety, tolerability and efficacy has been tested in phase 2 trials of cystic fibrosis patients (ClinicalTrials.gov identifier: NCT02722122). Alternatively, a reduction in NET levels could be achieved by replenishing the EASS via supplementation with recombinant human plasma GSN (Rhu-pGSN), an approach that could restore the activity of endogenous DNase by promoting its dissociation from actin (262). A phase 2, randomised, double-blind, placebo-controlled trial of Rhu-pGSN in severe COVID-19 patients with pneumonia that will measure inflammatory biomarkers is currently underway (ClinicalTrials.gov identifier: NCT04358406).
4. Generation of toxic degradation products – As an endonuclease, DNase-1 is unable to degrade NET-associated proteins. Indeed, in a murine model of bloodstream infection, histones and NE remained attached to the walls of liver sinusoids following intravenous DNase treatment (263). Combined with the results of an in vitro study that demonstrated NET degradation by DNase-1 resulted in enhanced NE activity (264), then it is conceivable that DNase-1 treatment in COVID-19 patients could potentiate organ damage by exposing endothelial cells to cytotoxic histones (265) and releasing proteolytically active NE from NETs. A combined therapeutic approach of DNase-1 with either histone blocking antibodies (266), small molecular inhibitors of NE (e.g. sivelestat or alvelestat) or heparin could help eradicate any potential bystander tissue damage that would arise from DNase-1-mediated digestion of NETs. Of these strategies, co-therapy with heparin may be particularly beneficial given that this anti-coagulant has been shown to (i) reduce histone cytotoxicity towards endothelial cells (267), (ii) accelerate DNase-1-mediated breakdown of NETs (26) and (iii) reduce NET formation triggered by IL-8 and HMGB-1 (268), two pro-inflammatory agonists whose circulating concentrations are increased in patients with severe COVID-19 (5, 205).
Immature Neutrophils
Calculated by subtracting the fraction of mature neutrophils from the sum of MPO-reactive cells, the delta neutrophil index (DNI) reflects the circulating fraction of immature neutrophils. In a study of 388 ICU patients, Birben et al found that the DNI, calculated at the time of hospital/ICU presentation, was significantly higher in COVID-19 patients who died within the first 30 days of ICU admission when compared to those who survived (269). Mechanistically, this relationship between increased neutrophil immaturity and poor outcome may be attributable to the pro-inflammatory nature of immature neutrophils, whose propensity for enhanced NET formation and cytokine production could contribute to systemic inflammation and tissue damage.
Compared to HCs, COVID-19 ICU patients exhibit significantly increased plasma concentrations of GM-CSF (5). Systemic elevation of this growth factor triggers the premature release of immature myeloid cells from the bone marrow. Thus, could inhibiting the activity of GM-CSF improve the outcome of COVID-19 patients by suppressing the early mobilisation of pro-inflammatory immature neutrophils into the circulation? Currently, a series of randomised placebo-controlled clinical trials are assessing the efficacy and safety of GM-CSF based therapies in patients with severe COVID-19 (270). Using a range of GM-CSF targeting monoclonal antibodies that include lenzilumab, otilimab, namilumab and gimsilumab, these trials are investigating the impact of GM-CSF neutralisation on such clinical outcomes as mortality, hospital/ICU LOS and ventilator-free days (270). Besides regulating haematopoiesis, GM-CSF is a potent modulator of innate immunity, promoting the proliferation, differentiation, activation and/or survival of macrophages and neutrophils (271, 272). Thus, it has been suggested that any potential benefits of GM-CSF based therapies in COVID-19 patients are likely to arise from the suppression of hyperactive innate immune responses (270, 273). In line with this idea, we suggest that, by suppressing emergency granulopoiesis, neutralisation of GM-CSF would dampen the systemic inflammatory response in COVID-19 patients by reducing the mobilisation of pro-inflammatory immature neutrophils into the circulation. Moreover, as immature neutrophils exhibit impaired anti-microbial responses (274, 275), an additional benefit of a more mature circulating neutrophil pool could be a reduced susceptibility to nosocomial infections.
Conclusions
Several studies of circulating and pulmonary immune cells in patients with COVID-19 have suggested that a hyperactive and dysregulated neutrophil response underpins the development of severe disease. The neutrophil response, including excessive NET generation with compromised removal of cfDNA, and higher frequencies of immature neutrophils with an immunosuppressive phenotype, represent novel therapeutic targets. With few specific treatments approved for COVID-19 other than dexamethasone, we conclude that trials targeting the generation of immature neutrophils, for example by inhibiting the actions of GM-CSF, or improving the removal of cfDNA using gelsolin or AIR DNase™ offer novel and rational approaches to reducing the severity of COVID-19 symptoms. Indeed the advances in our understanding of neutrophil biology and neutrophil mediated pathology as a result of COVID-19 research will likely provide benefit to other clinical conditions considered to be mediated by neutrophils, such as chronic obstructive pulmonary disease.
Author Contributions
JH wrote the manuscript and JL critically appraised and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
JH is supported by the National Institute for Health Research (NIHR) Surgical Reconstruction and Microbiology Research Centre (SRMRC). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cucinotta D, Vanelli M. WHO Declares COVID-19 a Pandemic. Acta Biomed (2020) 91:157–60. doi: 10.23750/abm.v91i1.9397
2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A Novel Coronavirus From Patients With Pneumonia in China, 2019. N Engl J Med (2020) 382:727–33. doi: 10.1056/nejmoa2001017
3. Who Coronavirus Disease (Covid-19) Dashboard | WHO Coronavirus Disease (Covid-19) Dashboard. Available at: https://covid19.who.int/ (Accessed May 10, 2021).
4. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (Covid-19) Outbreak in China. JAMA (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
5. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
6. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
7. Mo P, Xing Y, Xiao Y, Deng L, Zhao Q, Wang H, et al. Clinical Characteristics of Refractory COVID-19 Pneumonia in Wuhan, China. Clin Infect Dis (2020) ciaa270. doi: 10.1093/cid/ciaa270
8. Ramanathan K, Antognini D, Combes A, Paden M, Zakhary B, Ogino M, et al. Planning and Provision of ECMO Services for Severe ARDS During the COVID-19 Pandemic and Other Outbreaks of Emerging Infectious Diseases. Lancet Respir Med (2020) 8:518–26. doi: 10.1016/S2213-2600(20)30121-1
9. Bonanad C, García-Blas S, Tarazona-Santabalbina F, Sanchis J, Bertomeu-González V, Fácila L, et al. The Effect of Age on Mortality in Patients With Covid-19: A Meta-Analysis With 611,583 Subjects. J Am Med Dir Assoc (2020) 21:915–8. doi: 10.1016/j.jamda.2020.05.045
10. Li J, Huang DQ, Zou B, Yang H, Hui WZ, Rui F, et al. Epidemiology of COVID-19: A Systematic Review and Meta-Analysis of Clinical Characteristics, Risk Factors, and Outcomes. J Med Virol (2020) 93:1449–58. doi: 10.1002/jmv.26424
11. Wang X, Fang X, Cai Z, Wu X, Gao X, Min J, et al. Comorbid Chronic Diseases and Acute Organ Injuries are Strongly Correlated With Disease Severity and Mortality Among COVID-19 Patients: A Systemic Review and Meta-Analysis. Research (2020) 2020:2402961. doi: 10.34133/2020/2402961
12. Liu H, Chen S, Liu M, Nie H, Lu H. Comorbid Chronic Diseases are Strongly Correlated With Disease Severity Among COVID-19 Patients: A Systematic Review and Meta-Analysis. Aging Dis (2020) 11:668–78. doi: 10.14336/AD.2020.0502
13. Severe Outcomes Among Patients With Coronavirus Disease 2019 (Covid-19) — United States, February 12–March 16, 2020 | Mmwr. Available at: https://www.cdc.gov/mmwr/volumes/69/wr/mm6912e2.htm (Accessed March 2, 2021).
14. Cunningham JW, Vaduganathan M, Claggett BL, Jering KS, Bhatt AS, Rosenthal N, et al. Clinical Outcomes in Young Us Adults Hospitalized With COVID-19. JAMA Intern Med (2020) 181:379–81. doi: 10.1001/jamainternmed.2020.5313
15. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine Storm and Leukocyte Changes in Mild Versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J Leukoc Biol (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
16. Gustine JN, Jones D. Immunopathology of Hyperinflammation in COVID-19. Am J Pathol (2021) 191:4–17. doi: 10.1016/j.ajpath.2020.08.009
17. Aschenbrenner AC, Mouktaroudi M, Krämer B, Antonakos N, Oestreich M, Gkizeli K, et al. Disease Severity-Specific Neutrophil Signatures in Blood Transcriptomes Stratify COVID-19 Patients. Genome Med (2021) 13(1):7. doi: 10.1186/s13073-020-00823-5
18. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is Marked by a Dysregulated Myeloid Cell Compartment. Cell (2020) 182:1419–40. doi: 10.1016/j.cell.2020.08.001
19. Meizlish ML, Pine AB, Bishai JD, Goshua G, Nadelmann ER, Simonov M, et al. A Neutrophil Activation Signature Predicts Critical Illness and Mortality in COVID-19. Blood Adv (2021) 5:1164–77. doi: 10.1182/bloodadvances.2020003568
20. Vitte J, Diallo AB, Boumaza A, Lopez A, Michel M, Allardet-Servent J, et al. A Granulocytic Signature Identifies COVID-19 and its Severity. J Infect Dis (2020) 222:1985–96. doi: 10.1093/infdis/jiaa591
21. Wilk AJ, Lee MJ, Wei B, Parks B, Pi R, Martínez-Colón GJ, et al. Multi-Omic Profiling Reveals Widespread Dysregulation of Innate Immunity and Hematopoiesis in COVID-19. bioRxiv (2020). doi: 10.1101/2020.12.18.423363
22. Stetson DB, Medzhitov R. Type I Interferons in Host Defense. Immunity (2006) 25:373–81. doi: 10.1016/j.immuni.2006.08.007
23. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science (2020) 369:718–24. doi: 10.1126/science.abc6027
24. Schönrich G, Raftery MJ, Samstag Y. Devilishly Radical NETwork in COVID-19: Oxidative Stress, Neutrophil Extracellular Traps (Nets), and T Cell Suppression. Adv Biol Regul (2020) 77:100741. doi: 10.1016/j.jbior.2020.100741
25. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An Inflammatory Cytokine Signature Predicts COVID-19 Severity and Survival. Nat Med (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
26. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
27. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (Covid-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
28. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired Immune Cell Cytotoxicity in Severe COVID-19 is IL-6 Dependent. J Clin Invest (2020) 130:4694–703. doi: 10.1172/JCI138554
29. Morrissey SM, Geller AE, Hu X, Tieri D, Cooke EA, Ding C, et al. Emergence of Low-Density Inflammatory Neutrophils Correlates With Hypercoagulable State and Disease Severity in COVID-19 Patients. medRxiv (2020) 2020:5. doi: 10.1101/2020.05.22.20106724
30. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
31. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical Predictors of Mortality Due to COVID-19 Based on an Analysis of Data of 150 Patients From Wuhan, China. Intensive Care Med (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
32. Schwarz B, Sharma L, Roberts L, Peng X, Bermejo S, Leighton I, et al. Cutting Edge: Severe SARS-CoV-2 Infection in Humans is Defined by a Shift in the Serum Lipidome, Resulting in Dysregulation of Eicosanoid Immune Mediators. J Immunol (2021) 206:329–34. doi: 10.4049/jimmunol.2001025
33. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of COVID-19: Current State of the Science. Immunity (2020) 52:910–41. doi: 10.1016/j.immuni.2020.05.002
34. Azkur AK, Akdis M, Azkur D, Sokolowska M, van de Veen W, Brüggen MC, et al. Immune Response to SARS-CoV-2 and Mechanisms of Immunopathological Changes in COVID-19. Allergy Eur J Allergy Clin Immunol (2020) 75:1564–81. doi: 10.1111/all.14364
35. Cavalcante-Silva LHA, Carvalho DCM, Lima É de A, Galvão JGFM, da Silva JSF, Sales-Neto JM, et al. Neutrophils and COVID-19: The Road So Far. Int Immunopharmacol (2021) 90:107223. doi: 10.1016/j.intimp.2020.107233
36. Narasaraju T, Tang BM, Herrmann M, Muller S, Chow VTK, Radic M. Neutrophilia and NETopathy as Key Pathologic Drivers of Progressive Lung Impairment in Patients With Covid-19. Front Pharmacol (2020) 11:870. doi: 10.3389/fphar.2020.00870
37. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J Exp Med (2020) 217:e20200652. doi: 10.1084/jem.20200652
38. Galani IE, Andreakos E. Neutrophils in Viral Infections: Current Concepts and Caveats. J Leukoc Biol (2015) 98:557–64. doi: 10.1189/jlb.4vmr1114-555r
39. Drescher B, Bai F. Neutrophil in Viral Infections, Friend or Foe? Virus Res (2013) 171:1–7. doi: 10.1016/j.virusres.2012.11.002
40. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple Organ Infection and the Pathogenesis of SARS. J Exp Med (2005) 202:415–24. doi: 10.1084/jem.20050828
41. Wong RSM, Wu A, To KF, Lee N, Lam CWK, Wong CK, et al. Haematological Manifestations in Patients With Severe Acute Respiratory Syndrome: Retrospective Analysis. Br Med J (2003) 326:1358–62. doi: 10.1136/bmj.326.7403.1358
42. Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, et al. A Major Outbreak of Severe Acute Respiratory Syndrome in Hong Kong. N Engl J Med (2003) 348:1986–94. doi: 10.1056/nejmoa030685
43. Leung C, Kwan Y, Ko P, Chiu SS, Loung P, Fong N, et al. Severe Acute Respiratory Syndrome Among Children. Pediatrics (2004) 113(6):e535–543. doi: 10.1542/peds.113.6.e535
44. Wang YH, Lin AS, Chao TY, Lu SN, Liu JW, Chen SS, et al. A Cluster of Patients With Severe Acute Respiratory Syndrome in a Chest Ward in Southern Taiwan. Intensive Care Med (2004) 30:1228–31. doi: 10.1007/s00134-004-2311-8
45. Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L, et al. A Mouse-Adapted SARS-coronavirus Causes Disease and Mortality in BALB/c Mice. PloS Pathog (2007) 3:0023–37. doi: 10.1371/journal.ppat.0030005
46. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. Sars-CoV-2-triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J Exp Med (2020) 217:e20201129. doi: 10.1084/jem.20201129
47. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe COVID-19. Nature (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
48. Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe From Mild Covid-19. Cell (2020) 182:1401–18. doi: 10.1016/j.cell.2020.08.002
49. Chevrier S, Zurbuchen Y, Cervia C, Adamo S, Raeber ME, de Souza N, et al. A Distinct Innate Immune Signature Marks Progression From Mild to Severe COVID-19. Cell Rep Med (2021) 2:100166. doi: 10.1016/j.xcrm.2020.100166
50. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive Mapping of Immune Perturbations Associated With Severe COVID-19. Sci Immunol (2020) 5:eabd7114. doi: 10.1126/sciimmunol.abd7114
51. Ding X, Yu Y, Lu B, Huo J, Chen M, Kang Y, et al. Dynamic Profile and Clinical Implications of Hematological Parameters in Hospitalized Patients With Coronavirus Disease 2019. Clin Chem Lab Med (2020) 58(8):1365–71. doi: 10.1515/cclm-2020-0411
52. Cui N, Yan R, Qin C, Zhao J. Clinical Characteristics and Immune Responses of 137 Deceased Patients With Covid-19: A Retrospective Study. Front Cell Infect Microbiol (2020) 10:595333. doi: 10.3389/fcimb.2020.595333
53. Chen L, Yu J, He W, Chen L, Yuan G, Dong F, et al. Risk Factors for Death in 1859 Subjects With COVID-19. Leukemia (2020) 34(8):2173–83. doi: 10.1038/s41375-020-0911-0
54. Zhao Y, Nie HX, Hu K, Wu XJ, Zhang YT, Wang MM, et al. Abnormal Immunity of non-Survivors With COVID-19: Predictors for Mortality. Infect Dis Poverty (2020) 9(1):108. doi: 10.1186/s40249-020-00723-1
55. Wang JH, Chen RD, Yang HK, Zeng LC, Chen H, Hou YY, et al. Inflammation-Associated Factors for Predicting in-Hospital Mortality in Patients With COVID-19. J Med Virol (2021) 93:2908–17. doi: 10.1002/jmv.26771
56. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
57. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a Novel Coronavirus From a Man With Pneumonia in Saudi Arabia. N Engl J Med (2012) 367:1814–20. doi: 10.1056/nejmoa1211721
58. Min CK, Cheon S, Ha NY, Sohn KM, Kim Y, Aigerim A, et al. Comparative and Kinetic Analysis of Viral Shedding and Immunological Responses in MERS Patients Representing a Broad Spectrum of Disease Severity. Sci Rep (2016) 6:25359. doi: 10.1038/srep25359
59. Kim ES, Choe PG, Park WB, Oh HS, Kim EJ, Nam EY, et al. Clinical Progression and Cytokine Profiles of Middle East Respiratory Syndrome Coronavirus Infection. J Korean Med Sci (2016) 31:1717–25. doi: 10.3346/jkms.2016.31.11.1717
60. Alfaraj SH, Al-Tawfiq JA, Assiri AY, Alzahrani NA, Alanazi AA, Memish ZA. Clinical Predictors of Mortality of Middle East Respiratory Syndrome Coronavirus (Mers-CoV) Infection: A Cohort Study. Travel Med Infect Dis (2019) 29:48–50. doi: 10.1016/j.tmaid.2019.03.004
61. Leist SR, Jensen KL, Baric RS, Sheahan TP. Increasing the Translation of Mouse Models of MERS Coronavirus Pathogenesis Through Kinetic Hematological Analysis. PloS One (2019) 14:e0220126. doi: 10.1371/journal.pone.0220126
62. De Wit E, Rasmussen AL, Falzarano D, Bushmaker T, Feldmann F, Brining DL, et al. Middle East Respiratory Syndrome Coronavirus (MerscoV) Causes Transient Lower Respiratory Tract Infection in Rhesus Macaques. Proc Natl Acad Sci U S A (2013) 110:16598–603. doi: 10.1073/pnas.1310744110
63. van den Brand JMA, Haagmans BL, van Riel D, Osterhaus ADME, Kuiken T. The Pathology and Pathogenesis of Experimental Severe Acute Respiratory Syndrome and Influenza in Animal Models. J Comp Pathol (2014) 151:83–112. doi: 10.1016/j.jcpa.2014.01.004
64. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. MBio (2018) 9(5):e01753–18. doi: 10.1128/mBio.01753-18
65. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood (2020) 136(10):1169–79. doi: 10.1182/blood.2020007008
66. Ouwendijk WJD, Raadsen MP, van Kampen JJA, Verdijk RM, von der Thusen JH, Guo L, et al. Neutrophil Extracellular Traps Persist at High Levels in the Lower Respiratory Tract of Critically Ill COVID-19 Patients. J Infect Dis (2021) 223(9):1512–21. doi: 10.1093/infdis/jiab053
67. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
68. Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, et al. Pulmonary Post-Mortem Findings in a Series of COVID-19 Cases From Northern Italy: A Two-Centre Descriptive Study. Lancet Infect Dis (2020) 20(10):1135–40. doi: 10.1016/S1473-3099(20)30434-5
69. Chandrashekar A, Liu J, Martino AJ, McMahan K, Mercad NB, Peter L, et al. Sars-CoV-2 Infection Protects Against Rechallenge in Rhesus Macaques. Science (2020) 369:812–7. doi: 10.1126/science.abc4776
70. Singh DK, Singh B, Ganatra SR, Gazi M, Cole J, Thippeshappa R, et al. Responses to Acute Infection With SARS-CoV-2 in the Lungs of Rhesus Macaques, Baboons and Marmosets. Nat Microbiol (2021) 6:73–86. doi: 10.1038/s41564-020-00841-4
71. Rauch S, Gooch K, Hall Y, Salguero FJ, Dennis MJ, Gleeson FV, et al. mRNA Vaccine CVnCoV Protects non-Human Primates From SARS-CoV-2 Challenge Infection. bioRxiv (2020) 2020.12.23.424138. doi: 10.1101/2020.12.23.424138
72. Rockx B, Kuiken T, Herfst S, Bestebroer T, Lamers MM, Munnink BBO, et al. Comparative Pathogenesis of COVID-19, MERS, and SARS in a Nonhuman Primate Model. Science (2020) 368:1012–5. doi: 10.1126/science.abb7314
73. Guery B, Poissy J, El Mansouf L, Séjourné C, Ettahar N, Lemaire X, et al. Clinical Features and Viral Diagnosis of Two Cases of Infection With Middle East Respiratory Syndrome Coronavirus: A Report of Nosocomial Transmission. Lancet (2013) 381:2265–72. doi: 10.1016/S0140-6736(13)60982-4
74. Baseler LJ, Falzarano D, Scott DP, Rosenke R, Thomas T, Munster VJ, et al. An Acute Immune Response to Middle East Respiratory Syndrome Coronavirus Replication Contributes to Viral Pathogenicity. Am J Pathol (2016) 186:630–8. doi: 10.1016/j.ajpath.2015.10.025
75. Yu P, Xu Y, Deng W, Bao L, Huang L, Xu Y, et al. Comparative Pathology of Rhesus Macaque and Common Marmoset Animal Models With Middle East Respiratory Syndrome Coronavirus. PloS One (2017) 12(2):e0172093. doi: 10.1371/journal.pone.0172093
76. Blair RV, Vaccari M, Doyle-Meyers LA, Roy CJ, Russell-Lodrigue K, Fahlberg M, et al. Acute Respiratory Distress in Aged, SARS-Cov-2–Infected African Green Monkeys But Not Rhesus Macaques. Am J Pathol (2021) 191:274–82. doi: 10.1016/j.ajpath.2020.10.016
77. Munster VJ, Feldmann F, Williamson BN, van Doremalen N, Pérez-Pérez L, Schulz J, et al. Respiratory Disease in Rhesus Macaques Inoculated With SARS-Cov-2. Nature (2020) 585:268–72. doi: 10.1038/s41586-020-2324-7
78. Wauters E, Van Mol P, Garg AD, Jansen S, Van Herck Y, Vanderbeke L, et al. Discriminating Mild From Critical COVID-19 by Innate and Adaptive Immune Single-Cell Profiling of Bronchoalveolar Lavages. Cell Res (2021) 31(3):272–90. doi: 10.1038/s41422-020-00455-9
79. Fahlberg MD, Blair RV, Doyle-Meyers LA, Midkiff CC, Zenere G, Russell-Lodrigue KE, et al. Cellular Events of Acute, Resolving or Progressive COVID-19 in SARS-CoV-2 Infected non-Human Primates. Nat Commun (2020) 11(1):6078. doi: 10.1038/s41467-020-19967-4
80. Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of Angiotensin-Converting Enzyme 2 (ACE2) in COVID-19. Crit Care (2020) 24(1):422. doi: 10.1186/s13054-020-03120-0
81. Ryan KA, Bewley KR, Fotheringham SA, Slack GS, Brown P, Hall Y, et al. Dose-Dependent Response to Infection With SARS-CoV-2 in the Ferret Model and Evidence of Protective Immunity. Nat Commun (2021) 12(1):81. doi: 10.1038/s41467-020-20439-y
82. Kim Y, Kim SG, Kim SM, Kim EH, Park SJ, Yu KM, et al. Infection and Rapid Transmission of SARS-CoV-2 in Ferrets. Cell Host Microbe (2020) 27:704–9.e2. doi: 10.1016/j.chom.2020.03.023
83. Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B, et al. Susceptibility of Ferrets, Cats, Dogs, and Other Domesticated Animals to SARS-coronavirus 2. Science (2020) 368:1016–20. doi: 10.1126/science.abb7015
84. Hoang TN, Pino M, Boddapati AK, Viox EG, Starke CE, Upadhyay AA, et al. Baricitinib Treatment Resolves Lower-Airway Macrophage Inflammation and Neutrophil Recruitment in SARS-CoV-2-infected Rhesus Macaques. Cell (2021) 184:460–75.e21. doi: 10.1016/j.cell.2020.11.007
85. Yu J, Tostanosk LH, Peter L, Mercad NB, McMahan K, Mahrokhia SH, et al. DNA Vaccine Protection Against SARS-CoV-2 in Rhesus Macaques. Science (2020) 369:806–11. doi: 10.1126/science.abc6284
86. Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D. Baricitinib Therapy in COVID-19: A Pilot Study on Safety and Clinical Impact. J Infect (2020) 81:318–56. doi: 10.1016/j.jinf.2020.04.017
87. Bronte V, Ugel S, Tinazzi E, Vella A, de Sanctis F, Canè S, et al. Baricitinib Restrains the Immune Dysregulation in Patients With Severe COVID-19. J Clin Invest (2020) 130:6409–16. doi: 10.1172/JCI141772
88. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of SARS-CoV-2 Infected Patients. EBioMedicine (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
89. Song CY, Xu J, He JQ, Lu YQ. Immune Dysfunction Following COVID-19, Especially in Severe Patients. Sci Rep (2020) 10:15838. doi: 10.1038/s41598-020-72718-9
90. Yu G, Zhang Q, Wang R, Jiang S. Predictive Value of Neutrophil-to-Lymphocyte Ratio and Other Inflammatory Indicators in Estimating Clinical Severity of Coronavirus Disease. World J Emerg Med (2021) 12:79–80. doi: 10.5847/wjem.j.1920-8642.2021.01.014
91. Busch MH, Timmermans SAMEG, Nagy M, Visser M, Huckriede J, Aendekerk JP, et al. Neutrophils and Contact Activation of Coagulation as Potential Drivers of COVID-19. Circulation (2020) 142:1787–90. doi: 10.1161/CIRCULATIONAHA.120.050656
92. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis (2020) 71:762–8. doi: 10.1093/cid/ciaa248
93. Ouyang Y, Yin J, Wang W, Shi H, Shi Y, Xu B, et al. Downregulated Gene Expression Spectrum and Immune Responses Changed During the Disease Progression in Patients With COVID-19. Clin Infect Dis (2020) 71:2052–60. doi: 10.1093/cid/ciaa462
94. Kong M, Zhang H, Cao X, Mao X, Lu Z. Higher Level of Neutrophil-to-Lymphocyte is Associated With Severe COVID-19. Epidemiol Infect (2020) 148:e139. doi: 10.1017/S0950268820001557
95. Shi S, Nie B, Chen X, Cai Q, Lin C, Zhao G, et al. Clinical and Laboratory Characteristics of Severe and non-Severe Patients With COVID-19: A Retrospective Cohort Study in China. J Clin Lab Anal (2021) 35:e23692. doi: 10.1002/jcla.23692
96. Tahtasakal CA, Oncul A, Sevgi DY, Celik E, Ocal M, Turkkan HM, et al. Could We Predict the Prognosis of the COVID-19 Disease? J Med Virol (2020) 93:2420–30. doi: 10.1002/jmv.26751
97. Wang J, Li Q, Yin Y, Zhang Y, Cao Y, Lin X, et al. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front Immunol (2020) 11:2063. doi: 10.3389/fimmu.2020.02063
98. Payen D, Cravat M, Maadadi H, Didelot C, Prosic L, Dupuis C, et al. A Longitudinal Study of Immune Cells in Severe Covid-19 Patients. Front Immunol (2020) 11:580250. doi: 10.3389/fimmu.2020.580250
99. Soraya GV, Ulhaq ZS. Crucial Laboratory Parameters in COVID-19 Diagnosis and Prognosis: An Updated Meta-Analysis. Med Clin (Barc) (2020) 155:143–51. doi: 10.1016/j.medcli.2020.05.017
100. Fu J, Kong J, Wang W, Wu M, Yao L, Wang Z, et al. The Clinical Implication of Dynamic Neutrophil to Lymphocyte Ratio and D-dimer in COVID-19: A Retrospective Study in Suzhou China. Thromb Res (2020) 192:3–8. doi: 10.1016/j.thromres.2020.05.006
101. Li Q, Xie Y, Cui Z, Tang S, Yuan B, Huang H, et al. Analysis of Peripheral Blood IL-6 and Leukocyte Characteristics in 364 COVID-19 Patients of Wuhan. Front Immunol (2020) 11:559716. doi: 10.3389/fimmu.2020.559716
102. Gong J, Dong H, Xia Q, Huang Z, Wang D, Zhao Y, et al. Correlation Analysis Between Disease Severity and Inflammation-related Parameters in Patients With COVID-19: A Retrospective Study. BMC Infect Dis (2020) 20(1):963. doi: 10.1186/s12879-020-05681-5
103. Elshazli RM, Toraih EA, Elgaml A, El-Mowafy M, El-Mesery M, Amin MN, et al. Diagnostic and Prognostic Value of Hematological and Immunological Markers in COVID-19 Infection: A Meta-Analysis of 6320 Patients. PloS One (2020) 15:e0238160. doi: 10.1371/journal.pone.0238160
104. Zeng F, Li L, Zeng J, Deng Y, Huang H, Chen B, et al. Can We Predict the Severity of Coronavirus Disease 2019 With a Routine Blood Test? Polish Arch Intern Med (2020) 130:400–6. doi: 10.20452/pamw.15331
105. Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al. Association of Neutrophil Activation, More Than Platelet Activation, With Thrombotic Complications in Coronavirus Disease 2019. J Infect Dis (2020) 223(6):933–44. doi: 10.1093/infdis/jiaa756
106. Simadibrata DM, Calvin J, Wijaya AD, Ibrahim NAA. Neutrophil-to-Lymphocyte Ratio on Admission to Predict the Severity and Mortality of COVID-19 Patients: A Meta-Analysis. Am J Emerg Med (2021) 42:60–9. doi: 10.1016/j.ajem.2021.01.006
107. Li X, Liu C, Mao Z, Xiao M, Wang L, Qi S, et al. Predictive Values of Neutrophil-to-Lymphocyte Ratio on Disease Severity and Mortality in COVID-19 Patients: A Systematic Review and Meta-Analysis. Crit Care (2020) 24:674. doi: 10.1186/s13054-020-03374-8
108. Taj S, Kashif A, Arzinda Fatima S, Imran S, Lone A, Ahmed Q. Role of Hematological Parameters in the Stratification of COVID-19 Disease Severity. Ann Med Surg (2021) 62:68–72. doi: 10.1016/j.amsu.2020.12.035
109. Yang AP, Liu JP, Tao WQ, Li HM. The Diagnostic and Predictive Role of NLR, d-NLR and PLR in COVID-19 Patients. Int Immunopharmacol (2020) 84:106504. doi: 10.1016/j.intimp.2020.106504
110. Lagunas-Rangel FA. Neutrophil-to-Lymphocyte Ratio and lymphocyte-to-C-reactive Protein Ratio in Patients With Severe Coronavirus Disease 2019 (COVID-19): A Meta-Analysis. J Med Virol (2020) 92:1733–4. doi: 10.1002/jmv.25819
111. Piwowarczyk P, Szczukocka M, Kutnik P, Borys M, Mikłaszewska A, Kiciak S, et al. Risk Factors and Outcomes for Acute Respiratory Failure in Coronavirus Disease 2019: An Observational Cohort Study. Adv Clin Exp Med (2020) 30:165–71. doi: 10.17219/acem/130603
112. Liu J, Liu Y, Xiang P, Pu L, Xiong H, Li C, et al. Neutrophil-to-Lymphocyte Ratio Predicts Critical Illness Patients With 2019 Coronavirus Disease in the Early Stage. J Transl Med (2020) 18:206. doi: 10.1186/s12967-020-02374-0
113. Song CY, Xu J, He JQ, Lu YQ. Covid-19 Early Warning Score: A Multi-Parameter Screening Tool to Identify Highly Suspected Patients. medRxiv (2020) 2020.03.05.20031906. doi: 10.1101/2020.03.05.20031906
114. Silvestre-Roig C, Fridlender ZG, Glogauer M, Scapini P. Neutrophil Diversity in Health and Disease. Trends Immunol (2019) 40:565–83. doi: 10.1016/j.it.2019.04.012
115. Rosales C. Neutrophil: A Cell With Many Roles in Inflammation or Several Cell Types? Front Physiol (2018) 9:113. doi: 10.3389/fphys.2018.00113
116. Deniset JF, Kubes P. Neutrophil Heterogeneity: Bona Fide Subsets or Polarization States? J Leukoc Biol (2018) 103:829–38. doi: 10.1002/JLB.3RI0917-361R
117. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
118. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A Distinct Subset of Proinflammatory Neutrophils Isolated From Patients With Systemic Lupus Erythematosus Induces Vascular Damage and Synthesizes Type I Ifns. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
119. Davis RE, Sharma S, Conceição J, Carneiro P, Novais F, Scott P, et al. Phenotypic and Functional Characteristics of HLA-DR + Neutrophils in Brazilians With Cutaneous Leishmaniasis. J Leukoc Biol (2017) 101:739–49. doi: 10.1189/jlb.4a0915-442rr
120. Carmona-Rivera C, Kaplan MJ. Low-Density Granulocytes: A Distinct Class of Neutrophils in Systemic Autoimmunity. Semin Immunopathol (2013) 35:455–63. doi: 10.1007/s00281-013-0375-7
121. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187(1):538–52. doi: 10.4049/jimmunol.1100450
122. Parackova Z, Zentsova I, Bloomfield M, Vrabcova P, Smetanova J, Klocperk A, et al. Disharmonic Inflammatory Signatures in COVID-19: Augmented Neutrophils’ But Impaired Monocytes’ and Dendritic Cells’ Responsiveness. Cells (2020) 9:2206. doi: 10.3390/cells9102206
123. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al. A Single-Cell Atlas of the Peripheral Immune Response in Patients With Severe COVID-19. Nat Med (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
124. Combes AJ, Courau T, Kuhn NF, Hu KH, Ray A, Chen WS, et al. Global Absence and Targeting of Protective Immune States in Severe Covid-19. Nature (2021) 591(7848):124–30. doi: 10.1038/s41586-021-03234-7
125. Combadiere B, Adam L, Quentric P, Rosenbaum P, Dorgham K, Bonduelle O, et al. LOX-1 + Immature Neutrophils Predict Severe COVID-19 Patients at Risk of Thrombotic Complications. bioRxiv (2020) 2020.09.15.293100. doi: 10.1101/2020.09.15.293100
126. Carissimo G, Xu W, Kwok I, Abdad MY, Chan YH, Fong SW, et al. Whole Blood Immunophenotyping Uncovers Immature Neutrophil-to-VD2 T-cell Ratio as an Early Marker for Severe COVID-19. Nat Commun (2020) 11:5243. doi: 10.1038/s41467-020-19080-6
127. Parackova Z, Bloomfield M, Klocperk A, Sediva A. Neutrophils Mediate Th17 Promotion in COVID-19 Patients. J Leukoc Biol (2021) 109:73–6. doi: 10.1002/JLB.4COVCRA0820-481RRR
128. Reyes L, Sanchez-Garcia MA, Morrison T, Howden AJM, Watts ER, Arienti S, et al. Proteomics Identifies a Type I IFN, Prothrombotic Hyperinflammatory Circulating COVID-19 Neutrophil Signature Distinct From non-COVID-19 Ards. medRxiv (2020) 2020.09.15.20195305. doi: 10.1101/2020.09.15.20195305
129. Manz MG, Boettcher S. Emergency Granulopoiesis. Nat Rev Immunol (2014) 14:302–14. doi: 10.1038/nri3660
130. Fuchs A, Monlish DA, Ghosh S, Chang S-W, Bochicchio GV, Schuettpelz LG, et al. Trauma Induces Emergency Hematopoiesis Through IL-1/MyD88–Dependent Production of G-CSF. J Immunol (2019) 202:3020–32. doi: 10.4049/jimmunol.1801456
131. Pedersen CC, Borup R, Fischer-Nielsen A, Mora-Jensen H, Fossum A, Cowland JB, et al. Changes in Gene Expression During G-CSF–Induced Emergency Granulopoiesis in Humans. J Immunol (2016) 197:1989–99. doi: 10.4049/jimmunol.1502690
132. Peruzzi B, Bencini S, Capone M, Mazzoni A, Maggi L, Salvati L, et al. Quantitative and Qualitative Alterations of Circulating Myeloid Cells and Plasmacytoid DC in SARS-CoV-2 Infection. Immunology (2020) 161:345–53. doi: 10.1111/imm.13254
133. Seren S, Derian L, Keleş I, Guillon A, Lesner A, Gonzalez L, et al. Proteinase Release From Activated Neutrophils in Mechanically Ventilated Patients With non-COVID-19 and COVID-19 Pneumonia. Eur Respir J (2021) 57(4):2003755. doi: 10.1183/13993003.03755-2020
134. Guéant J, Guéant-Rodriguez R, Fromonot J, Oussalah A, Louis H, Chery C, et al. Elastase and Exacerbation of Neutrophil Innate Immunity are Involved in Multi-Visceral Manifestations of COVID-19. Allergy (2021) 10.1111/all.14746. doi: 10.1111/all.14746
135. Barbosa da Cruz D, Helms J, Aquino LR, Stiel L, Cougourdan L, Broussard C, et al. DNA-Bound Elastase of Neutrophil Extracellular Traps Degrades Plasminogen, Reduces Plasmin Formation, and Decreases Fibrinolysis: Proof of Concept in Septic Shock Plasma. FASEB J (2019) 33:14270–80. doi: 10.1096/fj.201901363RRR
136. Belorgey D, Bieth JG. Effect of Polynucleotides on the Inhibition of Neutrophil Elastase by Mucus Proteinase Inhibitor and α1-Proteinase Inhibitor. Biochemistry (1998) 37:16416–22. doi: 10.1021/bi981536o
137. Thierry AR, Roch B. Neutrophil Extracellular Traps and By-Products Play a Key Role in COVID-19: Pathogenesis, Risk Factors, and Therapy. J Clin Med (2020) 37:16416–22. doi: 10.3390/jcm9092942
138. Thierry AR. Anti-Protease Treatments Targeting Plasmin(Ogen) and Neutrophil Elastase may be Beneficial in Fighting Covid-19. Physiol Rev (2020) 100:1597–8. doi: 10.1152/physrev.00019.2020
139. Watanabe R, Matsuyama S, Shirato K, Maejima M, Fukushi S, Morikawa S, et al. Entry From the Cell Surface of Severe Acute Respiratory Syndrome Coronavirus With Cleaved S Protein as Revealed by Pseudotype Virus Bearing Cleaved S Protein. J Virol (2008) 82:11985–91. doi: 10.1128/jvi.01412-08
140. Belouzard S, Madu I, Whittaker GR. Elastase-Mediated Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein at Discrete Sites Within the S2 Domain. J Biol Chem (2010) 285:22758–63. doi: 10.1074/jbc.M110.103275
141. Ueland T, Heggelund L, Lind A, Holten AR, Tonby K, Michelsen AE, et al. Elevated Plasma sTIM-3 Levels in Patients With Severe COVID-19. J Allergy Clin Immunol (2021) 147:92–8. doi: 10.1016/j.jaci.2020.09.007
142. Goud PT, Bai D, Abu-Soud HM. A Multiple-Hit Hypothesis Involving Reactive Oxygen Species and Myeloperoxidase Explains Clinical Deterioration and Fatality in Covid-19. Int J Biol Sci (2020) 17:62–72. doi: 10.7150/ijbs.51811
143. Yan L, Borregaard N, Kjeldsen L, Moses MA. The High Molecular Weight Urinary Matrix Metalloproteinase (MMP) Activity is a Complex of Gelatinase B/MMP-9 and Neutrophil Gelatinase-Associated Lipocalin (NGAL): Modulation of MMP-9 Activity by NGAL. J Biol Chem (2001) 276:37258–65. doi: 10.1074/jbc.M106089200
144. Brew K, Nagase H. The Tissue Inhibitors of Metalloproteinases (Timps): An Ancient Family With Structural and Functional Diversity. Biochim Biophys Acta - Mol Cell Res (2010) 1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003
145. Hsu AT, Barrett CD, DeBusk GM, Ellson CD, Gautam S, Talmor DS, et al. Kinetics and Role of Plasma Matrix Metalloproteinase-9 Expression in Acute Lung Injury and the Acute Respiratory Distress Syndrome. Shock (2015) 44:128–36. doi: 10.1097/SHK.0000000000000386
146. Ueland T, Holter JC, Holten AR, Müller KE, Lind A, Bekken GK, et al. Distinct and Early Increase in Circulating MMP-9 in COVID-19 Patients With Respiratory Failure: MMP-9 and Respiratory Failure in COVID-19. J Infect (2020) 81:e41–3. doi: 10.1016/j.jinf.2020.06.061
147. Hazra S, Chaudhuri AG, Tiwary BK, Chakrabarti N. Matrix Metallopeptidase 9 as a Host Protein Target of Chloroquine and Melatonin for Immunoregulation in COVID-19: A Network-Based Meta-Analysis. Life Sci (2020) 257:118096. doi: 10.1016/j.lfs.2020.118096
148. Elavarasi A, Prasad M, Seth T, Sahoo RK, Madan K, Nischal N, et al. Chloroquine and Hydroxychloroquine for the Treatment of COVID-19: A Systematic Review and Meta-Analysis. J Gen Intern Med (2020) 35:3308–14. doi: 10.1007/s11606-020-06146-w
149. View of Melatonin as Adjuvant Treatment for Coronavirus Disease 2019 Pneumonia Patients Requiring Hospitalization (MAC-19 PRO): A Case Series. Available at: https://www.melatonin-research.net/index.php/MR/article/view/88/599 (Accessed March 2, 2021).
150. Zhang R, Wang X, Ni L, Di X, Ma B, Niu S, et al. Covid-19: Melatonin as a Potential Adjuvant Treatment. Life Sci (2020) 250:117583. doi: 10.1016/j.lfs.2020.117583
151. Rudra DS, Pal U, Maiti NC, Reiter RJ, Swarnakar S. Melatonin Inhibits Matrix Metalloproteinase-9 Activity by Binding to its Active Site. J Pineal Res (2013) 54:398–405. doi: 10.1111/jpi.12034
152. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18:134–47. doi: 10.1038/nri.2017.105
153. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
154. Urban CF, Nett JE. Neutrophil Extracellular Traps in Fungal Infection. Semin Cell Dev Biol (2019) 89:47–57. doi: 10.1016/j.semcdb.2018.03.020
155. Cortjens B, De Boer OJ, De Jong R, Antonis AFG, Sabogal Piñeros YS, Lutter R, et al. Neutrophil Extracellular Traps Cause Airway Obstruction During Respiratory Syncytial Virus Disease. J Pathol (2016) 238:401–11. doi: 10.1002/path.4660
156. Hiroki CH, Toller-Kawahisa JE, Fumagalli MJ, Colon DF, Figueiredo LTM, Fonseca BALD. Et al. Neutrophil Extracellular Traps Effectively Control Acute Chikungunya Virus Infection. Front Immunol (2020) 10:3108. doi: 10.3389/fimmu.2019.03108
157. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil Extracellular Traps Mediate a Host Defense Response to Human Immunodeficiency Virus-1. Cell Host Microbe (2012) 12:109–16. doi: 10.1016/j.chom.2012.05.015
158. Muraro SP, De Souza GF, Gallo SW, Da Silva BK, De Oliveira SD, Vinolo MAR, et al. Respiratory Syncytial Virus Induces the Classical ROS-dependent Netosis Through PAD-4 and Necroptosis Pathways Activation. Sci Rep (2018) 8:14166. doi: 10.1038/s41598-018-32576-y
159. Kaplan MJ, Radic M. Neutrophil Extracellular Traps: Double-Edged Swords of Innate Immunity. J Immunol (2012) 189:2689–95. doi: 10.4049/jimmunol.1201719
160. Sørensen OE, Borregaard N. Neutrophil Extracellular Traps - The Dark Side of Neutrophils. J Clin Invest (2016) 126:1612–20. doi: 10.1172/JCI84538
161. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
162. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive Role of Neutrophil Extracellular Traps in Pathogen-Induced Lung Injury. JCI Insight (2018) 3:e98178. doi: 10.1172/jci.insight.98178
163. Zhang N, Zhu L, Zhang Y, Zhou C, Song R, Yang X, et al. Circulating Rather Than Alveolar Extracellular Deoxyribonucleic Acid Levels Predict Outcomes in Influenza. J Infect Dis (2020) 222:1145–54. doi: 10.1093/infdis/jiaa241
164. Mokhtari T, Hassani F, Ghaffari N, Ebrahimi B, Yarahmadi A, Hassanzadeh G. Covid-19 and Multiorgan Failure: A Narrative Review on Potential Mechanisms. J Mol Histol (2020) 51:613–28. doi: 10.1007/s10735-020-09915-3
165. Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382:1708–20. doi: 10.1056/nejmoa2002032
166. Park HH, Park W, Lee YY, Kim H, Seo HS, Choi DW, et al. Bioinspired DNase-I-Coated Melanin-Like Nanospheres for Modulation of Infection-Associated Netosis Dysregulation. Adv Sci (2020) 7:2001940. doi: 10.1002/advs.202001940
167. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5:e138999. doi: 10.1172/jci.insight.138999
168. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and Tissue Factor–Enriched Neutrophil Extracellular Traps are Key Drivers in COVID-19 Immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1172/JCI141374
169. Lee YY, Park HH, Park W, Kim H, Jang JG, Hong KS, et al. Long-Acting Nanoparticulate DNase-1 for Effective Suppression of SARS-CoV-2-mediated Neutrophil Activities and Cytokine Storm. Biomaterials (2021) 267:120389. doi: 10.1016/j.biomaterials.2020.120389
170. Fernández-Pérez MP, Águila S, Reguilón-Gallego L, los Reyes-García AM, Miñano A, Bravo-Pérez C, et al. Neutrophil Extracellular Traps and Von Willebrand Factor are Allies That Negatively Influence COVID-19 Outcomes. Clin Transl Med (2021) 11:e268. doi: 10.1002/ctm2.268
171. Peckham H, de Gruijter NM, Raine C, Radziszewska A, Ciurtin C, Wedderburn LR, et al. Male Sex Identified by Global COVID-19 Meta-Analysis as a Risk Factor for Death and ITU Admission. Nat Commun (2020) 11:6317. doi: 10.1038/s41467-020-19741-6
172. Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, dos Reis MC, de Castro GMM, et al. The Emerging Role of Neutrophil Extracellular Traps in Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Sci Rep (2020) 10:19630. doi: 10.1038/s41598-020-76781-0
173. Strich JR, Ramos-Benitez MJ, Randazzo D, Stein SR, Babyak A, Davey RT, et al. Fostamatinib Inhibits Neutrophils Extracellular Traps Induced by COVID-19 Patient Plasma: A Potential Therapeutic. J Infect Dis (2020) 223(6):981–4. doi: 10.1093/infdis/jiaa789
174. Carvelli J, Demaria O, Vély F, Batista L, Chouaki Benmansour N, Fares J, et al. Association of COVID-19 Inflammation With Activation of the C5a–C5aR1 Axis. Nature (2020) 588:146–50. doi: 10.1038/s41586-020-2600-6
175. Shambat SM, Gómez-Mejia A, Schweizer TA, Huemer M, Chang CC, Acevedo C, et al. Neutrophil and Monocyte Dysfunctional Effector Response Towards Bacterial Challenge in Critically-Ill COVID-19 Patients. bioRxiv (2020) 2020.12.01.406306. doi: 10.1101/2020.12.01.406306
176. Gan T, Yang Y, Hu F, Chen X, Zhou J, Li Y, et al. Tlr3 Regulated Poly I:C-Induced Neutrophil Extracellular Traps and Acute Lung Injury Partly Through P38 MAP Kinase. Front Microbiol (2018) 9:3174. doi: 10.3389/fmicb.2018.03174
177. Khan MA, Farahvash A, Douda DN, Licht JC, Grasemann H, Sweezey N, et al. Jnk Activation Turns on LPS-And Gram-Negative Bacteria-Induced Nadph Oxidase-Dependent Suicidal Netosis. Sci Rep (2017) 7:3409. doi: 10.1038/s41598-017-03257-z
178. Mauracher L-M, Krall M, Roiß J, Hell L, Koder S, Hofbauer TM, et al. Neutrophil Subpopulations and Their Activation Potential in Patients With Antiphospholipid Syndrome and Healthy Individuals. Rheumatology (2020) 60(4):1687–99. doi: 10.1093/rheumatology/keaa532
179. Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell (2020) 182:59–72. doi: 10.1016/j.cell.2020.05.032
180. Song JW, Lam SM, Fan X, Cao WJ, Wang SY, Tian H, et al. Omics-Driven Systems Interrogation of Metabolic Dysregulation in COVID-19 Pathogenesis. Cell Metab (2020) 32:188–202. doi: 10.1016/j.cmet.2020.06.016
181. Gardinassi LG, Souza COS, Sales-Campos H, Fonseca SG. Immune and Metabolic Signatures of COVID-19 Revealed by Transcriptomics Data Reuse. Front Immunol (2020) 11:1636. doi: 10.3389/fimmu.2020.01636
182. Barberis E, Timo S, Amede E, Vanella VV, Puricelli C, Cappellano G, et al. Large-Scale Plasma Analysis Revealed New Mechanisms and Molecules Associated With the Host Response to Sars-Cov-2. Int J Mol Sci (2020) 21:8623. doi: 10.3390/ijms21228623
183. Blasco H, Bessy C, Plantier L, Lefevre A, Piver E, Bernard L, et al. The Specific Metabolome Profiling of Patients Infected by SARS-COV-2 Supports the Key Role of Tryptophan-Nicotinamide Pathway and Cytosine Metabolism. Sci Rep (2020) 10:16824. doi: 10.1038/s41598-020-73966-5
184. Wu D, Shu T, Yang X, Song J-X, Zhang M, Yao C, et al. Plasma Metabolomic and Lipidomic Alterations Associated With COVID-19. Natl Sci Rev (2020) 7:1157–68. doi: 10.1093/nsr/nwaa086
185. McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med (2020) 202:812–21. doi: 10.1164/rccm.202005-1583OC
186. Awasthi D, Nagarkoti S, Sadaf S, Chandra T, Kumar S, Dikshit M. Glycolysis Dependent Lactate Formation in Neutrophils: A Metabolic Link Between NOX-dependent and Independent Netosis. Biochim Biophys Acta - Mol Basis Dis (2019) 1865:165542. doi: 10.1016/j.bbadis.2019.165542
187. Rodríguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MMB, López-Villegas EO, Sánchez-García FJ. Metabolic Requirements for Neutrophil Extracellular Traps Formation. Immunology (2015) 145:213–24. doi: 10.1111/imm.12437
188. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
189. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe (2020) 27:883–90. doi: 10.1016/j.chom.2020.04.017
190. Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe Covid-19. Cells (2020) 9:2374. doi: 10.3390/cells9112374
191. Didangelos A. Covid-19 Hyperinflammation: What About Neutrophils? mSphere (2020) 5:e00367. doi: 10.1128/msphere.00367-20
192. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in COVID-19 Patients. Emerg Microbes Infect (2020) 9:761–70. doi: 10.1080/22221751.2020.1747363
193. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. Covid-19 Severity Correlates With Airway Epithelium–Immune Cell Interactions Identified by Single-Cell Analysis. Nat Biotechnol (2020) 38:970–9. doi: 10.1038/s41587-020-0602-4
194. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181:1036–45. doi: 10.1016/j.cell.2020.04.026
195. Maxwell AJ, Ding J, You Y, Dong Z, Chehade H, Alvero A, et al. Identification of Key Signaling Pathways Induced by SARS-CoV2 That Underlie Thrombosis and Vascular Injury in COVID-19 Patients. J Leukoc Biol (2021) 109:35–47. doi: 10.1002/JLB.4COVR0920-552RR
196. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-Viral Infection Maps Reveal Signatures of Severe Covid-19 Patients. Cell (2020) 181:1475–88. doi: 10.1016/j.cell.2020.05.006
197. Wang Q, Teder P, Judd NP, Noble PW, Doerschuk CM. CD44 Deficiency Leads to Enhanced Neutrophil Migration and Lung Injury in Escherichia Coli Pneumonia in Mice. Am J Pathol (2002) 161:2219–28. doi: 10.1016/S0002-9440(10)64498-7
198. Wu M, Chen Y, Xia H, Wang C, Tan CY, Cai X, et al. Transcriptional and Proteomic Insights Into the Host Response in Fatal COVID-19 Cases. Proc Natl Acad Sci USA (2020) 117:28336–43. doi: 10.1073/pnas.2018030117
199. Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d’Emal C, et al. Neutrophil Extracellular Traps Infiltrate the Lung Airway, Interstitial, and Vascular Compartments in Severe COVID-19. J Exp Med (2020) 217:e20201012. doi: 10.1084/jem.20201012
200. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological Study of the 2019 Novel Coronavirus Disease (COVID-19) Through Postmortem Core Biopsies. Mod Pathol (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
201. Guo Q, Zhao Y, Li J, Liu J, Yang X, Guo X, et al. Induction of Alarmin S100A8/A9 Mediates Activation of Aberrant Neutrophils in the Pathogenesis of COVID-19. Cell Host Microbe (2021) 29:222–35. doi: 10.1016/j.chom.2020.12.016
202. Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory Activities of S100: Proteins S100A8, S100A9, and S100A8/A9 Induce Neutrophil Chemotaxis and Adhesion. J Immunol (2003) 170:3233–42. doi: 10.4049/jimmunol.170.6.3233
203. Ehrchen JM, Sunderkötter C, Foell D, Vogl T, Roth J. The Endogenous Toll-like Receptor 4 Agonist S100A8/S100A9 (Calprotectin) as Innate Amplifier of Infection, Autoimmunity, and Cancer. J Leukoc Biol (2009) 86:557–66. doi: 10.1189/jlb.1008647
204. Shi H, Zuo Y, Yalavarthi S, Gockman K, Zuo M, Madison JA, et al. Neutrophil Calprotectin Identifies Severe Pulmonary Disease in COVID-19. J Leukoc Biol (2021) 109:67–72. doi: 10.1002/JLB.3COVCRA0720-359R
205. Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y, et al. Elevated Serum Levels of S100A8/A9 and HMGB1 at Hospital Admission are Correlated With Inferior Clinical Outcomes in COVID-19 Patients. Cell Mol Immunol (2020) 17:992–4. doi: 10.1038/s41423-020-0492-x
206. Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation (2020) 141:1080–94. doi: 10.1161/CIRCULATIONAHA.119.043833
207. Wang Y, Gao H, Kessinger CW, Schmaier A, Jaffer FA, Simon DI. Myeloid-Related protein-14 Regulates Deep Vein Thrombosis. JCI Insight (2017) 2:e91356. doi: 10.1172/jci.insight.91356
208. Buehler PK, Zinkernagel AS, Hofmaenner DA, García PDW, Acevedo CT, Gómez-Mejia A, et al. Bacterial Pulmonary Superinfections are Associated With Unfavourable Outcomes in Critically Ill COVID-19 Patients. Cell Rep Med (2021) 2(4):100229. doi: 10.1016/j.xcrm.2021.100229
209. Feng Y, Ling Y, Bai T, Xie Y, Huang J, Li J, et al. Covid-19 With Different Severities: A Multicenter Study of Clinical Features. Am J Respir Crit Care Med (2020) 201:1380–8. doi: 10.1164/rccm.202002-0445OC
210. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical Course and Outcomes of Critically Ill Patients With SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir Med (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
211. Woytschak J, Keller N, Krieg C, Impellizzieri D, Thompson RW, Wynn TA, et al. Type 2 Interleukin-4 Receptor Signaling in Neutrophils Antagonizes Their Expansion and Migration During Infection and Inflammation. Immunity (2016) 45:172–84. doi: 10.1016/j.immuni.2016.06.025
212. Cassatella MA. The Neutrophil: One of the Cellular Targets of Interleukin-10. Int J Clin Lab Res (1998) 28:148–61. doi: 10.1007/s005990050036
213. Conway Morris A, Kefala K, Wilkinson TS, Dhaliwal K, Farrell L, Walsh T, et al. C5a Mediates Peripheral Blood Neutrophil Dysfunction in Critically Ill Patients. Am J Respir Crit Care Med (2009) 180:19–28. doi: 10.1164/rccm.200812-1928OC
214. Morris AC, Brittan M, Wilkinson TS, McAuley DF, Antonelli J, McCulloch C, et al. C5a-Mediated Neutrophil Dysfunction is RhoA-dependent and Predicts Infection in Critically Ill Patients. Blood (2011) 117:5178–88. doi: 10.1182/blood-2010-08-304667
215. Roh JS, Sohn DH. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw (2018) 18:e27. doi: 10.4110/in.2018.18.e27
216. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature (2010) 464:104–7. doi: 10.1038/nature08780
217. Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM. N-Formyl Peptides Drive Mitochondrial Damage Associated Molecular Pattern Induced Neutrophil Activation Through ERK1/2 and P38 MAP Kinase Signalling Pathways. Injury (2015) 46:975–84. doi: 10.1016/j.injury.2015.03.028
218. Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial Damage Associated Molecular Patterns From Femoral Reamings Activate Neutrophils Through Formyl Peptide Receptors and P44/42 MAP Kinase. J Orthop Trauma (2010) 24:534–8. doi: 10.1097/BOT.0b013e3181ec4991
219. Hazeldine J, Dinsdale RJ, Harrison P, Lord JM. Traumatic Injury and Exposure to Mitochondrial-Derived Damage Associated Molecular Patterns Suppresses Neutrophil Extracellular Trap Formation. Front Immunol (2019) 10:685. doi: 10.3389/fimmu.2019.00685
220. Gabl M, Sundqvist M, Holdfeldt A, Lind S, Mårtensson J, Christenson K, et al. Mitocryptides From Human Mitochondrial DNA–Encoded Proteins Activate Neutrophil Formyl Peptide Receptors: Receptor Preference and Signaling Properties. J Immunol (2018) 200:3269–82. doi: 10.4049/jimmunol.1701719
221. Kaczmarek E, Hauser CJ, Kwon WY, Riça I, Chen L, Sandler N, et al. A Subset of Five Human Mitochondrial Formyl Peptides Mimics Bacterial Peptides and Functionally Deactivates Human Neutrophils. J Trauma Acute Care Surg (2018) 95:936–43. doi: 10.1097/TA.0000000000001971
222. Szarpak L, Ruetzler K, Safiejko K, Hampel M, Pruc M, Kanczuga - Koda L, et al. Lactate Dehydrogenase Level as a COVID-19 Severity Marker. Am J Emerg Med (2020) S0735-6757(20):31034-2. doi: 10.1016/j.ajem.2020.11.025
223. Bime C, Casanova NG, Nikolich-Zugich J, Knox KS, Camp SM, Garcia JGN. Strategies to DAMPen Covid-19-mediated Lung and Systemic Inflammation and Vascular Injury. Transl Res (2021) 232:37–48. doi: 10.1016/j.trsl.2020.12.008
224. Scozzi D, Cano M, Ma L, Zhou D, Zhu JH, O’Halloran JA, et al. Circulating Mitochondrial DNA is an Early Indicator of Severe Illness and Mortality From COVID-19. JCI Insight (2021) 6:143299. doi: 10.1172/jci.insight.143299
225. Dorward DA, Lucas CD, Doherty MK, Chapman GB, Scholefield EJ, Conway Morris A, et al. Novel Role for Endogenous Mitochondrial Formylated Peptide-Driven Formyl Peptide Receptor 1 Signalling in Acute Respiratory Distress Syndrome. Thorax (2017) 72:928–36. doi: 10.1136/thoraxjnl-2017-210030
226. Grazioli S, Dunn-Siegrist I, Pauchard LA, Blot M, Charles PE, Pugin J. Mitochondrial Alarmins are Tissue Mediators of Ventilator-Induced Lung Injury and ARDS. PloS One (2019) 14:e0225468. doi: 10.1371/journal.pone.0225468
227. Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil Extracellular Traps (Nets) and Damage-Associated Molecular Patterns (Damps): Two Potential Targets for COVID-19 Treatment. Mediators Inflamm (2020) 2020:7527953. doi: 10.1155/2020/7527953
228. Fernández-Ruiz I, Arnalich F, Cubillos-Zapata C, Hernández-Jiménez E, Moreno-González R, Toledano V, et al. Mitochondrial DAMPs Induce Endotoxin Tolerance in Human Monocytes: An Observation in Patients With Myocardial Infarction. PloS One (2014) 9:e95073. doi: 10.1371/journal.pone.0095073
229. Janiuk K, Jabłońska E, Garley M. Significance of NETs Formation in COVID-19. Cells (2021) 10:151. doi: 10.3390/cells10010151
230. Koenig LM, Boehmer DFR, Metzger P, Schnurr M, Endres S, Rothenfusser S. Blocking Inflammation on the Way: Rationale for CXCR2 Antagonists for the Treatment of COVID-19. J Exp Med (2020) 217:e20201342. doi: 10.1084/jem.20201342
231. Chiang CC, Korinek M, Cheng WJ, Hwang TL. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front Pharmacol (2020) 11:572009. doi: 10.3389/fphar.2020.572009
232. Bohn MK, Hall A, Sepiashvili L, Jung B, Steele S, Adeli K. Pathophysiology of COVID-19: Mechanisms Underlying Disease Severity and Progression. Physiology (2020) 35:288–301. doi: 10.1152/physiol.00019.2020
233. Guéant JL, Fromonot J, Guéant-Rodriguez RM, Lacolley P, Guieu R, Regnault V. Blood myeloperoxidase-DNA, a Biomarker of Early Response to SARS-CoV-2 Infection? Allergy Eur J Allergy Clin Immunol (2020) 76:892–6. doi: 10.1111/all.14533
234. Pujhari S, Paul S, Ahluwalia J, Rasgon JL. Clotting Disorder in Severe Acute Respiratory Syndrome Coronavirus 2. Rev Med Virol (2021) 31(3):e2177. doi: 10.1002/rmv.2177
235. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil Extracellular Trap–Associated Protein Activation of the NLRP3 Inflammasome Is Enhanced in Lupus Macrophages. J Immunol (2013) 190:1217–26. doi: 10.4049/jimmunol.1202388
236. Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, et al. Novel Role of IL (Interleukin)-1β in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler Thromb Vasc Biol (2018) 38:843–53. doi: 10.1161/ATVBAHA.117.309897
237. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PloS One (2012) 7:e48111. doi: 10.1371/journal.pone.0048111
238. Chen K, Murao A, Arif A, Takizawa S, Jin H, Jiang J, et al. Inhibition of Efferocytosis by Extracellular Cirp–Induced Neutrophil Extracellular Traps. J Immunol (2020) 206:797–806. doi: 10.4049/jimmunol.2000091
239. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone Hypercitrullination Mediates Chromatin Decondensation and Neutrophil Extracellular Trap Formation. J Cell Biol (2009) 184:205–13. doi: 10.1083/jcb.200806072
240. Du M, Yang L, Gu J, Wu J, Ma Y, Wang T. Inhibition of Peptidyl Arginine Deiminase-4 Prevents Renal Ischemia-Reperfusion-Induced Remote Lung Injury. Mediators Inflammation (2020) 2020):1724206. doi: 10.1155/2020/1724206
241. van Bijnen STA, Wouters D, van Mierlo GJ, Muus P, Zeerleder S. Neutrophil Activation and Nucleosomes as Markers of Systemic Inflammation in Paroxysmal Nocturnal Hemoglobinuria: Effects of Eculizumab. J Thromb Haemost (2015) 13:2004–11. doi: 10.1111/jth.13125
242. Zeerleder S, van Bijnen S, Wouters D, van Mierlo GJ, Muus P. Neutrophil Extracellular Trap Formation in PNH Patients With and Without a History of Thrombosis - Effects of Eculizumab. Blood (2013) 122:1235–5. doi: 10.1182/blood.v122.21.1235.1235
243. Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab Treatment in Patients With COVID-19: Preliminary Results From Real Life ASL Napoli 2 Nord Experience. Eur Rev Med Pharmacol Sci (2020) 24:4040–7. doi: 10.26355/EURREV_202004_20875
244. Ullah MA, Loh Z, Gan WJ, Zhang V, Yang H, Li JH, et al. Receptor for Advanced Glycation End Products and its Ligand High-Mobility Group Box-1 Mediate Allergic Airway Sensitization and Airway Inflammation. J Allergy Clin Immunol (2014) 134:440–50. doi: 10.1016/j.jaci.2013.12.1035
245. Tadie J-M, Bae H-B, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 Promotes Neutrophil Extracellular Trap Formation Through Interactions With Toll-like Receptor 4. Am J Physiol Lung Cell Mol Physiol (2013) 304:L342–9. doi: 10.1152/ajplung.00151.2012
246. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J Thromb Haemost (2014) 12:2074–88. doi: 10.1111/jth.12710
247. Liu X, Li Z, Liu S, Sun J, Chen Z, Jiang M, et al. Potential Therapeutic Effects of Dipyridamole in the Severely Ill Patients With COVID-19. Acta Pharm Sin B (2020) 10:1205–15. doi: 10.1016/j.apsb.2020.04.008
248. Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, et al. Adenosine Receptor Agonism Protects Against NETosis and Thrombosis in Antiphospholipid Syndrome. Nat Commun (2019) 10:1916. doi: 10.1038/s41467-019-09801-x
249. Mutua V, Gershwin LJ. A Review of Neutrophil Extracellular Traps (Nets) in Disease: Potential Anti-NETs Therapeutics. Clin Rev Allergy Immunol (2020) 1–18. doi: 10.1007/s12016-020-08804-7
250. Shishikura K, Horiuchi T, Sakata N, Trinh DA, Shirakawa R, Kimura T, et al. Prostaglandin E2 Inhibits Neutrophil Extracellular Trap Formation Through Production of Cyclic AMP. Br J Pharmacol (2016) 173:319–31. doi: 10.1111/bph.13373
251. Biron BM, Chung C-S, Chen Y, Wilson Z, Fallon EA, Reichner JS, et al. Pad4 Deficiency Leads to Decreased Organ Dysfunction and Improved Survival in a Dual Insult Model of Hemorrhagic Shock and Sepsis. J Immunol (2018) 200:1817–28. doi: 10.4049/jimmunol.1700639
252. Nadano D, Yasuda T, Kishi K. Measurement of Deoxyribonuclease I Activity in Human Tissues and Body Fluids by a Single Radial Enzyme-Diffusion Method. Clin Chem (1993) 39:448–52. doi: 10.1093/clinchem/39.3.448
253. Desilles JP, Gregoire C, Le Cossec C, Lambert J, Mophawe O, Losser MR, et al. Efficacy and Safety of Aerosolized Intra-Tracheal Dornase Alfa Administration in Patients With SARS-CoV-2-induced Acute Respiratory Distress Syndrome (ARDS): A Structured Summary of a Study Protocol for a Randomised Controlled Trial. Trials (2020) 21:548. doi: 10.1186/s13063-020-04488-8
254. Mai SHC, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ, et al. Delayed But Not Early Treatment With Dnase Reduces Organ Damage and Improves Outcome in a Murine Model of Sepsis. Shock (2015) 44:166–72. doi: 10.1097/SHK.0000000000000396
255. Gollomp K, Kim M, Johnston I, Hayes V, Welsh J, Arepally GM, et al. Neutrophil Accumulation and NET Release Contribute to Thrombosis in HIT. JCI Insight (2018) 3:e99445. doi: 10.1172/jci.insight.99445
256. Eulitz D, Mannherz HG. Inhibition of Deoxyribonuclease I by Actin is to Protect Cells From Premature Cell Death. Apoptosis (2007) 12:1511–21. doi: 10.1007/s10495-007-0078-4
257. Lazarides E, Lindberg U. Actin is the Naturally Occurring Inhibitor of Deoxyribonuclease I. Proc Natl Acad Sci U S A (1974) 71:4742–6. doi: 10.1073/pnas.71.12.4742
258. Lee WM, Galbraith RM. The Extracellular Actin-Scavenger System and Actin Toxicity. N Engl J Med (1992) 326:1335–41. doi: 10.1056/nejm199205143262006
259. Dinsdale RJ, Hazeldine J, Al Tarrah K, Hampson P, Devi A, Ermogenous C, et al. Dysregulation of the Actin Scavenging System and Inhibition of DNase Activity Following Severe Thermal Injury. Br J Surg (2020) 107:391–401. doi: 10.1002/bjs.11310
260. Overmyer KA, Shishkova E, Miller IJ, Balnis J, Bernstein MN, Peters-Clarke TM, et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst (2021) 12:23–40. doi: 10.1016/j.cels.2020.10.003
261. Messner CB, Demichev V, Wendisch D, Michalick L, White M, Freiwald A, et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst (2020) 11:11–24. doi: 10.1016/j.cels.2020.05.012
262. Chhabra D, Nosworthy NJ, Dos Remedios CG. The N-terminal Fragment of Gelsolin Inhibits the Interaction of Dnase I With Isolated Actin, But Not With the Cofilin-Actin Complex. Proteomics (2005) 5:3131–6. doi: 10.1002/pmic.200401127
263. Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular Mechanisms of NET Formation and Degradation Revealed by Intravital Imaging in the Liver Vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
264. Podolska MJ, Mahajan A, Hahn J, Knopf J, Maueröder C, Petru L, et al. Treatment With DNases Rescues Hidden Neutrophil Elastase From Aggregated Nets. J Leukoc Biol (2019) 106:1359–66. doi: 10.1002/JLB.3AB0918-370R
265. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PloS One (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
266. Chen R, Kang R, Fan XG, Tang D. Release and Activity of Histone in Diseases. Cell Death Dis (2014) 5:e1370. doi: 10.1038/cddis.2014.337
267. Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Kadota K, Sato K. Heparins Attenuated Histone-Mediated Cytotoxicity In Vitro and Improved the Survival in a Rat Model of Histone-Induced Organ Dysfunction. Intensive Care Med Exp (2015) 3:36. doi: 10.1186/s40635-015-0072-z
268. Manfredi AA, Rovere-Querini P, D’Angelo A, Maugeri N. Low Molecular Weight Heparins Prevent the Induction of Autophagy of Activated Neutrophils and the Formation of Neutrophil Extracellular Traps. Pharmacol Res (2017) 123:146–56. doi: 10.1016/j.phrs.2016.08.008
269. Birben B, Birben OD, Akın T, Akkurt G, Surel AA, Yakısık E, et al. Efficacy of the Delta Neutrophil Index in Predicting 30-Day Mortality in COVID-19 Patients Requiring Intensive Care. Int J Clin Pract (2021) 75(5):e13970. doi: 10.1111/ijcp.13970
270. Lang FM, Lee KMC, Teijaro JR, Becher B, Hamilton JA. Gm-CSF-based Treatments in COVID-19: Reconciling Opposing Therapeutic Approaches. Nat Rev Immunol (2020) 20:507–14. doi: 10.1038/s41577-020-0357-7
271. Bhattacharya P, Thiruppathi M, Elshabrawy HA, Alharshawi K, Kumar P, Prabhakar BS. Gm-Csf: An Immune Modulatory Cytokine That can Suppress Autoimmunity. Cytokine (2015) 75:261–71. doi: 10.1016/j.cyto.2015.05.030
272. Shiomi A, Usui T. Pivotal Roles of GM-CSF in Autoimmunity and Inflammation. Mediators Inflamm (2015) 2015:568543. doi: 10.1155/2015/568543
273. Mehta P, Porter JC, Manson JJ, Isaacs JD, Openshaw PJM, McInnes IB, et al. Therapeutic Blockade of Granulocyte Macrophage Colony-Stimulating Factor in COVID-19-associated Hyperinflammation: Challenges and Opportunities. Lancet Respir Med (2020) 8:822–30. doi: 10.1016/S2213-2600(20)30267-8
274. Drifte G, Dunn-Siegrist I, Tissières P, Pugin J. Innate Immune Functions of Immature Neutrophils in Patients With Sepsis and Severe Systemic Inflammatory Response Syndrome. Crit Care Med (2013) 41:820–32. doi: 10.1097/CCM.0b013e318274647d
275. Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JRB, Hazeldine J, et al. Neutrophil Dysfunction, Immature Granulocytes, and Cell-free DNA are Early Biomarkers of Sepsis in Burn-injured Patients: A Prospective Observational Cohort Study. Ann Surg (2016) 265:1241–9. doi: 10.1097/SLA.0000000000001807
Keywords: COVID-19, immune dysregulation, neutrophils, SARS-CoV-2, neutrophil extracellular traps
Citation: Hazeldine J and Lord JM (2021) Neutrophils and COVID-19: Active Participants and Rational Therapeutic Targets. Front. Immunol. 12:680134. doi: 10.3389/fimmu.2021.680134
Received: 13 March 2021; Accepted: 17 May 2021;
Published: 02 June 2021.
Edited by:
Kristina De Paris (Abel), University of North Carolina at Chapel Hill, United StatesReviewed by:
Veronica Schmitz, Oswaldo Cruz Foundation (Fiocruz), BrazilShetty Ravi Dyavar, University of Nebraska Medical Center, United States
Copyright © 2021 Hazeldine and Lord. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jon Hazeldine, J.Hazeldine@bham.ac.uk