





Abstract
Allogeneic haematopoietic stem cell transplantation (SCT) has emerged as a curative therapeutic option. However, the role of graft-versus-host disease in lung injury after SCT has yet to be determined.
In the present study, primary bronchial epithelial cells and the bronchial epithelial cell line BEAS-2B were used to investigate immune responses of allogeneic CD8+ T-cells directed against respiratory epithelial cells.
Following stimulation with irradiated bronchial epithelial cells, CD8+ T-cells produced significant amounts of interferon-γ, upregulated alloantigen activation markers and proliferated highly compared with T-cells stimulated with interleukin-2 alone. Furthermore, cytotoxicity assays demonstrated that bronchial epithelial cell-specific and granzyme B-mediated cytolytic activity was induced in CD8+ T-cells. Generation of natural killer (NK) T-cells, NK-like T-cells, cytokine-induced killer cells or lymphokine-activated killer cells could be excluded by phenotyping, culture conditions and neglectable lytic activity following stimulation with interleukin-2 alone. Inhibition experiments showed that lysis of bronchial epithelial cells was not major histocompatibility complex-I restricted, but depended on NK group 2 member D signalling; a stimulatory receptor initially shown to be expressed on NK cells.
The present data imply that the respiratory epithelium has an antigen presenting function and directly alloactivates cytotoxic CD8+ T-cells that show nonclassical effector function.
Diffuse lung injury is a major complication of allogeneic haematopoietic stem cell transplantation (SCT) and can account for up to 50% of transplant-related mortality 1. Bronchiolar inflammation with pronounced epithelial damage and lymphocytic bronchitis can be found along with idiopathic pneumonia syndrome (IPS), a widespread and mainly alveolar and interstitial lung injury within the first 100 days after SCT 2. The epithelial damage is thought to be an alloreactive immune response targeting bronchial epithelial cells 2.
The role of graft-versus-host disease (GvHD) and alloreactive T-cells in pathogenesis of acute and chronic lung injury following SCT still remains controversial 3. However, an association between lung injury and acute or chronic GvHD has been reported in clinical investigations as well as in animal models 4, 5. Alloactivation depends on T-cell receptor (TCR) stimulation and CD28 costimulation via CD80/CD86. Additional nonclassical pathways for alloactivation with the consequence of acute and chronic graft damage have also to be considered 6. Inhibition of CD70 prolongs allograft survival independently from CD28 7 and CD8+ T-cells have been shown to be directly activated by the tissue of the grafted organ, irrespective of antigen-presenting cells (APCs) 8.
Initially demonstrated to be a stimulatory receptor on natural killer (NK) cells, NK group 2 member D (NKG2D)-co-stimulation induces proliferation, survival, activation marker expression, cytokine production and cytotoxicity in CD8+ T-cells 9–12. Major histocompatibility complex (MHC) class I-related chains A and B (MIC A/B) are known ligands for NKG2D 13–15.
In the present study, bronchial epithelial cells were investigated as to whether they can function as nonclassical APCs, inducing epithelium directed allospecific cytotoxicity in CD8+ T-cells by triggering NKG2D in a bronchial epithelial cell line (BEAS-2B) as well as in primary bronchial epithelial cells (PBECs).
MATERIALS AND METHODS
Cell culture
The bronchial epithelial cell line BEAS-2B, immortalised by transfection of bronchial epithelial cells of a healthy human individual with adenovirus 12-SV40 hybrid virus, was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA; CRL-9609) 16. Cells were cultured in RPMI-1640 (Gibco, Paisley, UK) supplemented with 10% heat-inactivated foetal bovine serum (PAA; Cölbe, Germany), 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin (Gibco) at 37°C in a humidified atmosphere containing 5% CO2. Prior to use, BEAS-2B cells were harvested with Accutase (PAA).
K562 cells, a chronic myeloid leukaemia cell line without MHC class I expression (CCL-243; ATCC), were kept in suspension culture under the same conditions and used as NK-sensitive control targets.
Culture of PBECs
PBECs were isolated and cultured as previously described 17. In brief, human PBECs from healthy volunteers were obtained during bronchoscopy and cultured in serum-free bronchial epithelial cell growth medium (Promocell, Heidelberg, Germany) containing 100 U·mL−1 penicillin G, 100 μg·mL−1 streptomycin and 0.25 μg·mL−1 amphotericin-B (Invitrogen, Karlsruhe, Germany).
Isolation, stimulation and activation of CD8+ lymphocytes
Peripheral blood mononuclear cells (PBMCs) were isolated from heparinised blood or apharesis products of healthy human donors according to a standard protocol using Ficoll-Hypaque density gradient centrifugation (Pharmacia, Freiburg, Germany). Subsequently, CD8+ T-cells were negatively selected from PBMCs using magnetically labelled beads following the manufacturer's protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). Interindividual differences between T-cell donors did not exceed interexperimental differences.
Irradiated (30 Gy) BEAS-2B or PBECs served as stimulator cells in the following experiments. For alloactivation, CD8+ T-cells from a given donor were mixed at a concentration of 0.5×106 cells·mL−1 with stimulator cells at a ratio of 1:1 and incubated in RPMI-1640 in the presence of low-dose interleukin (IL)-2 (100 U·mL−1). CD8+ T-cells were stimulated for 7 days or subsequently restimulated with fresh stimulator cells under the same conditions for an additional 3, 7 or 14 days. CD8+ T-cells cultured in the presence of IL-2 alone served as control. Supernatants of these cultures were collected for analysis by ELISA.
Cell surface expression analysis
Expression of cell surface molecules was assessed by flow cytometry with FACSCalibur and CellQuest analysis software (BD Biosciences, Heidelberg, Germany). Purity of isolated CD8+ T-cells was determined flow cytometrically before and after 7 days of stimulation with the Simultest IMK-Lymphocyte kit (BD Biosciences).
In order to determine expression of activation markers, freshly isolated and activated T-cells were stained with the following antibodies (BD Biosciences, unless stated otherwise): CD25 (M-A251), CD30 (BerH8), CD54 (HA58), CD69 (L78), CD70 (Ki-24), CD71 (YDJ1.2.2; Coulter Immunotech, Fullerton, CA, USA), CD80 (3771.11; R&D Systems, Minneapolis, MN, USA), CD86 (37301.111; R&D Systems), CD95 (ZB4; Immunotech), NKG2D (1D11; ebioscience, San Diego, CA, USA), human leukocyte antigen (HLA)-DR (B8.12.2; Immunotech), and the appropriate isotype controls.
Expression of MIC A/B on BEAS-2B and PBECs was determined by indirect immunofluorescence staining with anti-MIC A/B (clone 6D4; BD Biosciences). Cells were counterstained with propidium iodide (PI; Sigma-Aldrich, Deisenhofen, Germany) at a final concentration of 200 ng·mL−1, with only PI-negative cells being subjected to analysis.
ELISA
Colorimetric ELISA was performed according to the manufacturer's protocol for detection of interferon (IFN)-γ (Pierce Endogen, Rockford, IL, USA) in co-culture supernatants, after activation, and for detection of granzyme B (Hoelzel Diagnostics, Cologne, Germany) in the supernatants of cytotoxicity assays.
Succinimidyl ester of carboxyfluorescein proliferation assay
Freshly isolated CD8+ T-cells were labelled with carboxyfluorescein (CFSE; Invitrogen) at a final concentration of 2 µM, washed and incubated with IL-2 alone, with stimulator cells in the presence of IL-2 or with staphylococcal enterotoxin B (SEB; Sigma-Aldrich) at a final concentration of 100 ng·mL−1 as a positive control. After 7 days of activation, fluorescence intensity of CD8+ T-cells was analysed by fluorescence-activated cell sorting (FACS) and the percentage of proliferating cells was determined.
Functional cytotoxicity assay
T-cell-mediated cytotoxicity was measured with a standard 4-h chromium (51Cr) radioisotope assay. Target cells (BEAS-2B, PBECs or K562) were labelled with sodium chromate (51Cr; 100 µCi; Hartmann Analytics, Braunschweig, Germany) for 1.5 h, washed twice and then co-incubated with activated CD8+ T-cells as effectors, at descending effector:target cell (E:T) ratios. After 4 h of incubation at 37°C, cell free supernatants were harvested, radioactivity was determined by γ-counting and the percentage of specific lysis was calculated as follows:
((experimental release - spontaneous release)/(maximal release - spontaneous release))×100% (1)
The percentage of spontaneous release ((spontaneous release/maximal release)×100%) was <15% for each target cell in all experiments.
In order to determine granzyme B-release, supernatants were collected from a cytotoxicity assay performed in parallel to the described 51Cr-release assay in the absence of radioactivity.
The following blocking assays were performed: CD8+ T-cells were pre-incubated with functional-grade blocking antibodies directed against NKG2D (1D11; Biolegend, San Diego, CA USA) or immunoglobulin G1 (MG1-45; Biolegend) at a final concentration of 5 μg·mL−1, or labelled target cells were incubated with functional-grade blocking antibody directed against CD95 (ZB4; Immunotech), tumour necrosis factor (TNF; MAK195; Knoll AG, Ludwigshafen, Germany) or MHC class I antigens (W6/32). The cells were immediately used in standard 51Cr-release assays after a 30-min incubation period at room temperature. Control assays without blocking antibodies were always performed in parallel. For cold-target inhibition experiments, unlabelled (cold) targets as competitors were added to the labelled (hot) targets into a standard 51Cr-release assay, with increasing cold:hot ratios ranging 1–40 at a constant E:T ratio of 10:1.
Statistical analysis
If not stated otherwise, at least four independent experiments were performed and the mean±sem is shown. Means between two groups were tested for differences using a paired t-test for the 51Cr-release assay, unpaired t-test for all other comparisons or the nonparametric Mann–Whitney rank sum test, means of three groups were compared using ANOVA (following Dunn's or Tukey's post hoc test).
RESULTS
Characterisation of the lymphocyte population
The purified population of CD8+ T-cells obtained from PBMCs was controlled for its purity prior to use in all experiments. The population consisted of 89.4±5% CD3+CD8+ T-cells with only 1.8±1.7% CD3+CD4+ T-cells and <1% CD3-CD16/56+ NK cells (mean±sd of all experiments). No difference was found, in these phenotypic characteristics, between freshly isolated and the 7-day stimulated cells finally used in functional assays.
Allogeneic activation of purified CD8+ T-cells by bronchial epithelial cells
HLA-mismatched CD8+ T-cells were stimulated with irradiated BEAS-2B cells for 7 days in the presence of low dose IL-2 (100 U·mL−1). CD8+ T-cells from the same donor were cultured in IL-2 medium alone in order to determine levels of unspecific activation. Interferon (IFN)-γ release as a hallmark of CD8+ T-cell activation was measured using ELISA in the supernatant of both cell cultures. Irradiated BEAS-2B cells produced no IFN-γ by themselves (data not shown) but CD8+ T-cells cultured in IL-2 medium alone produced low levels of IFN-γ (fig. 1⇓). BEAS-2B cells stimulated CD8+ T-cells released significantly higher amounts of IFN-γ than control cells. Restimulation of activated CD8+ T-cells for an additional ≥3 days resulted in a further significant increase in IFN-γ release (fig. 1⇓).
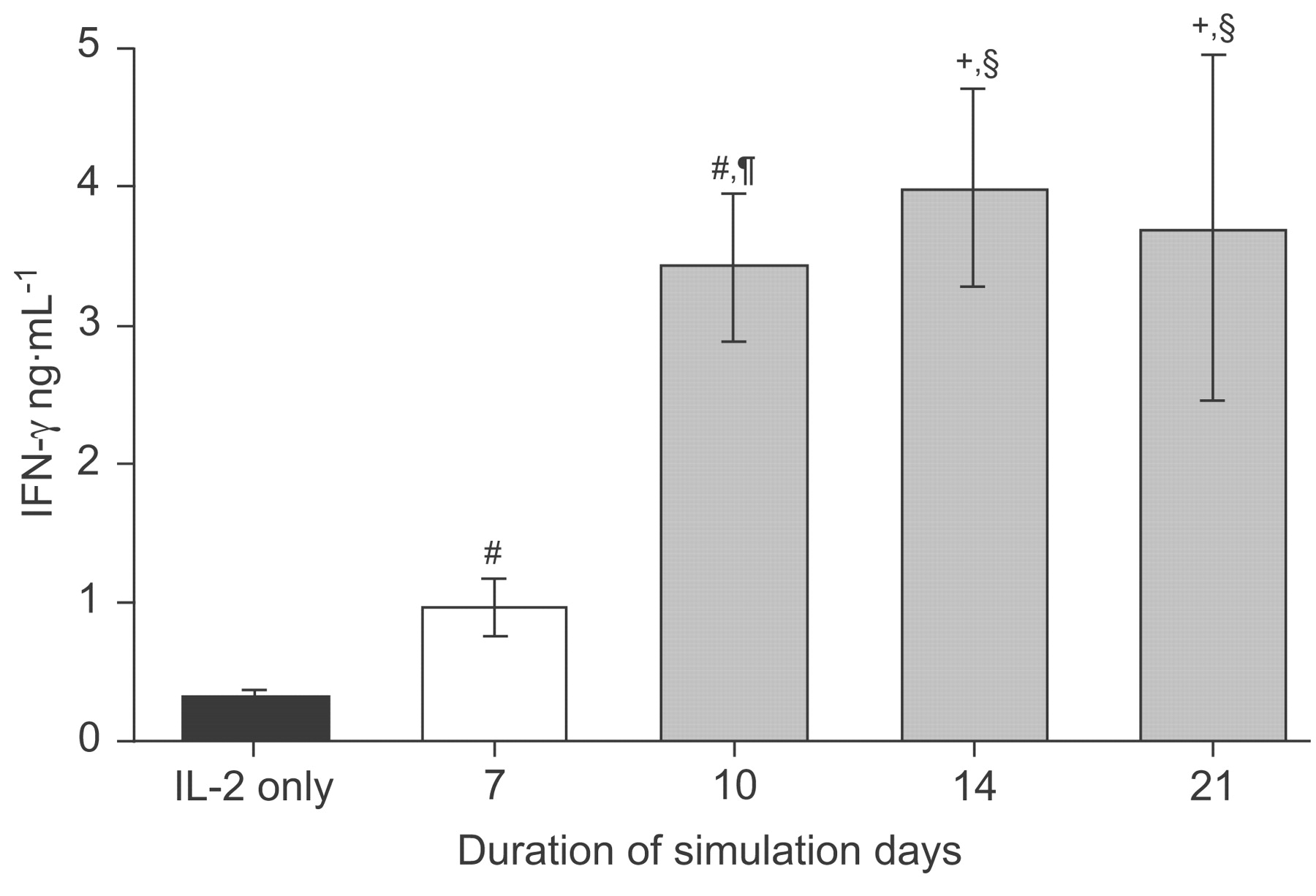
Interferon (IFN)-γ production by CD8+ T-lymphocytes during stimulation. Stimulation with irradiated BEAS-2B cells in the presence of low dose interleukin (IL)-2 (100 U·mL−1; ▪) activates CD8+ T-cells to secrete IFN-γ in significantly higher amounts than control CD8+ T-cells incubated with IL-2 alone. Additional restimulation of BEAS-2B-activated CD8+ T-cells (▓) significantly enhances IFN-γ secretion compared with a stimulation of 7 days (□). n = 11, 18, 16, 4 and 5, for IL-2 only and 7, 10, 14 and 21 days, respectively. #: p<0.001 versus IL-2 only; ¶: p<0.001 versus 7-day stimulation with irradiated BEAS-2B; +: p<0.01 versus IL-2 only; §: p<0.01 versus 7-day stimulation with irradiated BEAS-2B.
In addition, the surface expression of the activation markers CD25, CD30, CD54, CD69, CD70, CD71, CD80, CD86, CD95 and HLA-DR was analysed on CD8+ T-cells before and after 7 days of stimulation with irradiated BEAS-2B. Freshly isolated CD8+ T-cells showed either no, or only neglectable expression of the analysed markers (data not shown). Following IL-2 stimulation alone, the percentage of positive cells and the mean fluorescence intensity (reflecting the antigen density on the cells) of positive cells were slightly but significantly upregulated for all activation markers except for CD86 (fig. 2⇓). In contrast, stimulation with irradiated BEAS-2B induced the expression of not only the alloactivation markers CD30 and CD69, but also CD25, CD70, CD71 and CD80, significantly, with respect to both the percentage of positive cells as well as the antigen density on those cells (fig. 2⇓). This demonstrates that BEAS-2B cells activated CD8+ T-cells stronger than IL-2 alone.

Upregulation of activation markers on CD8+ T-cells during stimulation. Expression of the activation markers on CD8+ T-cells was measured using fluorescence-activated cell sorting analysis immediately after isolation (naïve; - - -) and after 7 days of stimulation with irradiated BEAS-2B cells in presence of interleukin (IL)-2 (▪) or with IL-2 alone (□). Mean±sem of a) induction of mean fluorescence intensity (MFI) of positive cells and b) percentages of positively (+ve) stained cells out of six independent experiments given as n-fold of naïve. #: p≤0.05 versus IL-2 alone; ¶: p≤0.005 versus IL-2 alone.
Proliferation of CD8+ T-cells after alloactivation with bronchial epithelial cells
Following stimulation with BEAS-2B cells, 49.5±12.4% of the CD8+ T-cell population showed proliferation (n = 5; fig. 3⇓; p<0.05 versus IL-2 alone). Similar results were obtained in the SEB control (50.3±8.6%), whereas IL-2 alone did not initiate relevant proliferation of cells (5.4±2.6%).

Proliferation of CD8+ T-cells. Proliferation was measured using succinimidyl ester of carboxyflurescein (CFSE)incorporation and fluorescence-activated cell sorting analysis in CD8+ T-cells stimulated with a) interleukin (IL)-2 alone as a control, b) with staphylococcal enterotoxin B as a positive control or c) with irradiated BEAS-2B cells in the presence of IL-2 for 7 days. Histogram plots of one representative of five independent experiments are shown. M1: proliferating cells.
Cytolytic activity of alloactivated CD8+ T-cells against bronchial epithelial cells
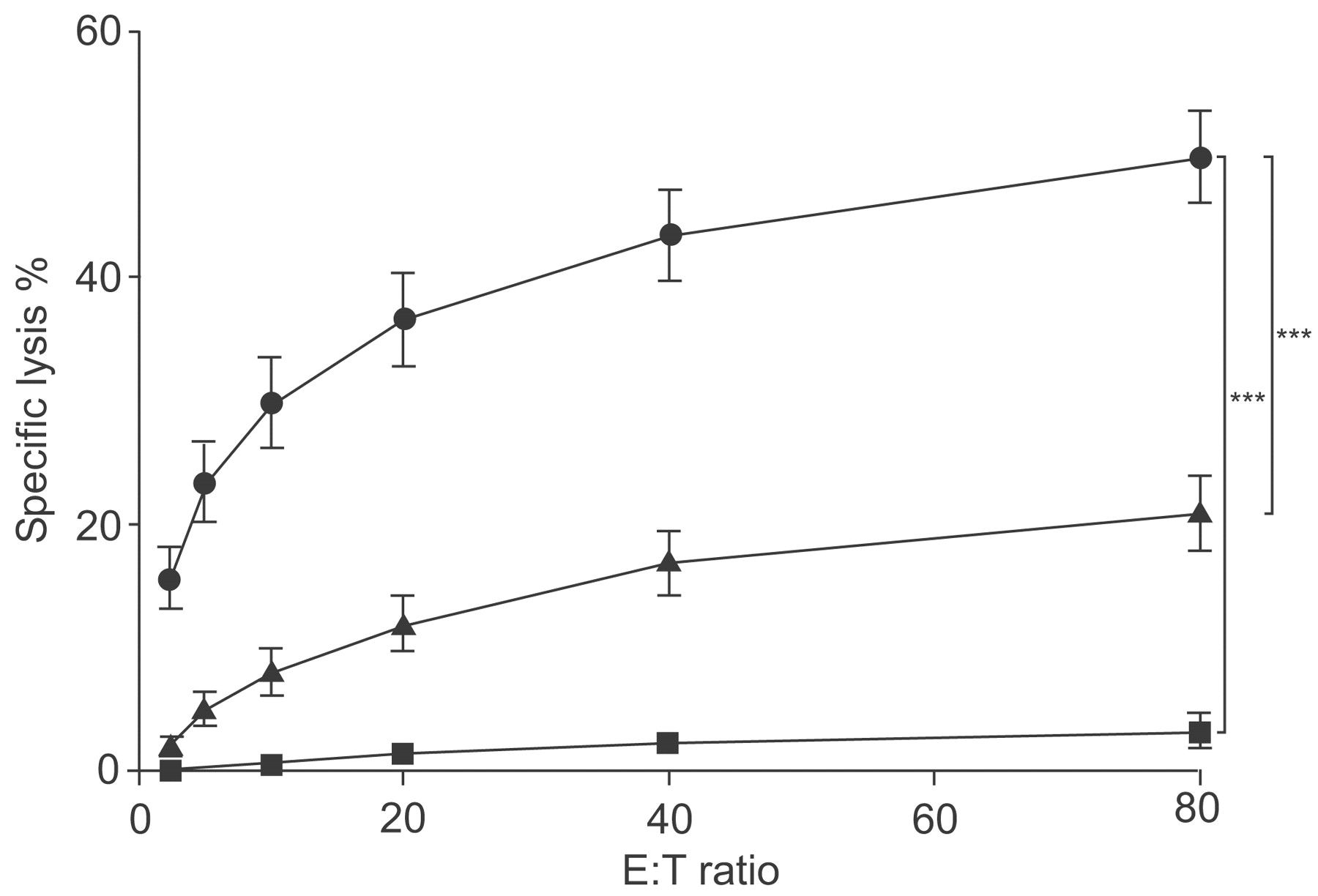
Alloantigen-activated CD8+ T-cells showed high, specific lysis of BEAS-2B target cells depending on the E:T ratio (fig. 4⇓). At the highest E:T ratio of 80:1, specific lysis of target cells amounted to a mean value of 50% within 4 h. IL-2-stimulated CD8+ T-cells showed only minor and significantly lower lytic activity against BEAS-2B, compared with BEAS-2B cell-activated CD8+ T-cells (p≤0.001; data not shown). Freshly isolated, naïve CD8+ T-cells were not able to kill BEAS-2B cells (fig. 4⇓). As BEAS-2B cell-activated CD8+ T-cells showed low lytic activity against the MHC class I-negative NK-cell target K562, cold-target inhibition assays were performed to exclude residual NK cell- or lymphokine-activated killer (LAK) activity and to test whether BEAS-2B and K562 cells were damaged by the same or different effector cells. Both BEAS-2B and K562 cells were simultaneously co-cultured with the effectors, one target was 51Cr labelled (hot target), the other added unlabelled (cold) for competition. Lysis of BEAS-2B cells was not inhibited by the presence of K562 competitors at any cold:hot target ratio tested, whereas addition of cold BEAS-2B cells resulted in a concentration-dependent inhibition. However, addition of BEAS-2B competitors to hot K562 target cells inhibited K562 lysis, of a similar effectiveness as the cold K562 itself (fig. 5⇓).

Cytolytic activity of CD8+ T-cells. CD8+ T-cells were stimulated with irradiated BEAS-2B cells in the presence of interleukin (IL)-2 and subsequently subjected as effectors (E) to a 51Cr-release assay with BEAS-2B (n = 46; •) or K562 (n = 46; ▴) as target cells (T) in increasing E:T ratios, or CD8+ T-cells were left naïve and subjected to a 51Cr-release assay with BEAS-2B cells shortly after separation (n = 4; ▪). Mean±sem of the percentage of specific lysis at varying E:T ratios of independent experiments are shown. ***: p<0.001.

Cytolytic activity of CD8+ T-cells. Standard 51Cr-release assay with activated CD8+ T-cells and 51Cr-labelled BEAS-2B or K562 cells as targets was performed at an effector:target ratio of 10 (control). Additional unlabelled (cold) BEAS-2B or K562 were added, as competitors, to each labelled (hot) target in increasing cold:hot target ratios. Percentage of specific lysis of the labelled targets is given as n-fold of control lysis at varying cold:hot ratios. •: hot BEAS-2B and cold K562; ○: hot BEAS-2B and cold BEAS-2B; ▪: hot K562 and cold BEAS-2B; □: hot K562 and cold K562, n = 4.
Mechanism of the cytolytic activity of alloactivated CD8+ T-cells
In order to assess the mechanism of the lysis, the classical mediators of cytotoxicity (perforin/granzyme B, CD95 and TNF) were investigated. Granzyme B was detectable in significant amounts in the supernatants of the co-cultures of cytotoxicity assays, increasing with E:T ratios. Alloactivated CD8+ T-cells did not produce granzyme B in the absence of BEAS-2B targets (fig. 6a⇓). In blocking experiments, pre-incubation of targets with a neutralising antibody against CD95 or TNF did not show any effect on lysis (fig. 6b⇓ and c). Interestingly, inhibition of MHC class I also did not inhibit the lysis of BEAS-2B cells (fig. 6d⇓).

Mechanism of cytolytic activity of CD8+ T-cells against the respiratory target cell line BEAS-2B. a) Activated CD8+ T-cells release granzyme B during lysis of BEAS-2B depending on the effector:target (E:T) ratio in a cytotoxicity assay performed in parallel to a 51Cr-release assay. Blocking experiments were performed by incubating BEAS-2B target cells with b) anti-CD95 (n = 5), c) anti-tumour necrosis factor (TNF; n = 3) or d) anti-major histocompatibility complex (MHC) class I (n = 23), prior to use in a standard 51Cr-release assay with activated CD8+ T-cells. Mean±sem of independent experiments are shown. •: BEAS-2B cells alone; ○: BEAS-2B pre-incubated with neutralising antibodies against b) CD95, c) TNF or d) MHC class I. #: p≤0.01 versus CD8+ only; ¶: p≤0.001 versus CD8+ only; +: p≤0.005 versus E:T ratio of 20.
FACS analysis demonstrated MIC A/B expression on BEAS-2B cells as well as NKG2D expression on CD8+ T-cells (fig. 7a⇓ and b), with the latter being increased during activation (fig. 7c⇓). Consequently, pre-incubation of CD8+ T-cells with anti-NKG2D antibody resulted in a significant inhibition of ≤25% of BEAS-2B cell lysis in a standard 51Cr-release assay; an isotype-matched control antibody had no effect on lysis. The aforementioned low lysis levels of K562 without MHC class I expression were inhibited to ∼40% of controls with anti-NKG2D (fig. 7d⇓).

Expression of natural killer group 2 member D (NKG2D) on CD8+ T-cells and two of its ligands MIC A/B on BEAS-2B. a) BEAS-2B cells were stained with anti-MIC A/B and b) CD8+ T-cells were stained with anti-NKG2D-specific antibody and analysed by flow cytometry (FL2-H). Histogram plots show one representative (▓) out of six independent experiments with isotype control (□). c) Mean±sem of five independent experiments showing induction of NKG2D on positive CD8+ T-cells during stimulation. #: p≤0.01 versus control. Inhibition of the cytolytic activity of CD8+ T cells with anti-NKG2D. d) Activated CD8+ T-cells were incubated with anti-NKG2D antibody (1D11) or a nonspecific isotype control antibody, or BEAS-2B were incubated with anti-major histocompatibility complex (MHC) class I (W6/32) for 30 min prior to use in a standard 51Cr-release assay. Mean±sem of the percentage of specific lysis of independent experiments are shown for varying effector:target (E:T) ratios and given as n-fold of control at a constant E:T = 20. M1:proliferating cells; Ig: immunoglobulin. ¶: p≤0.001 versus nonspecific isotope control antibody.
In order to exclude cell-type nonspecific, artificial killing of BEAS-2B due to the viral immortalisation, PBECs were used as activator and target cells in cytotoxicity assays. PBECs, which expressed MHC class I (data not shown) and MIC A/B (fig. 8a⇓) were specifically lysed by PBEC-alloactivated CD8+ T-cells. In accordance with the results seen in BEAS-2B cells, lysis was significantly inhibited by the anti-NKG2D antibody; however, blocking of MHC class I did not reduce lysis of PBECs (fig. 8b⇓ and c).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of MICA/B on primary bronchial epithelial cells (PBEC). a) PBEC were stained with anti-MIC A/B-specific antibody and analysed by flow cytometry (FL2-H). Histogram plot of one representative out of five independent experiments with isotype control (□) is shown. Cytolytic activity of CD8+ T-cells against the PBEC. CD8+ T-cells were stimulated with irradiated PBEC in the presence of interleukin (IL)-2 and subsequently subjected to a 51Cr-release assay as effectors (E) in increasing E:target (T) ratios. b) Activated CD8+ T-cells were left untreated or incubated with anti-NKG2D antibody (1D11) prior to use in a standard 51Cr-release assay with PBEC (•) as targets. c) PBEC were left untreated or incubated with anti-MHC I (W6/32; ○) prior to use as target cells in a standard 51Cr-release assay.
DISCUSSION
In the present study, it has been demonstrated that human bronchial epithelial cells are potent direct activators of cytotoxic CD8+ T-cells as well as targets for alloactivated CD8+ T-cells. This is of importance because T-cells of donor origin have direct contact with lung epithelium, e.g. after cytokine-induced lymphocyte extravasation or due to TNF-induced endothelial apoptosis during the course of IPS 18. Furthermore, the presence of donor T-cells in bronchoalveolar fluids of patients after SCT underlines the relevance of the results of the present study 19. The findings support the hypothesis that alloreactive CD8+ T-cells can account for directly mediated lung injuries seen after SCT.
BEAS-2B-activated CD8+ T-cells showed significant production of IFN-γ, which was increased when the CD8+ T-cells were re-stimulated. Upregulation of the activation markers CD25, CD30, CD54, CD69, CD70, CD71, CD80, CD86, CD95 and HLA-DR on the BEAS-2B-activated CD8+ T-cells was significantly higher than after IL-2 stimulation alone, which points towards an alloantigen-specific stimulation 20–22. In particular, CD30 was reported to be present on alloreactive T-cells, CD69 and HLA-DR were shown to be increased after alloantigen stimulation and CD69 expression correlates with graft rejection 21, 23–25.
The observed alloactivation is paralleled by the proliferative response of CD8+ T-cells during stimulation with irradiated BEAS-2B cells, which was as high as the SEB-induced positive control. In contrast, no proliferation was found after stimulation with IL-2 only. Irradiated stimulator cells were also found to be rendered metabolically inactive and thus no longer able to secrete any T-cell-activating proteins. All these results suggest a need for physical contact (e.g. MHC-TCR allospecific interaction) between effector and stimulator cells for efficient induction of proliferation (and hence alloactivation) to occur.
It is unlikely that the presence of NK cells or NK T-cells during BEAS-2B-stimulation accounts for the observed low-level lysis of the classical NK-cell target K562. The phenotype of the population, i.e. CD3+ CD4- CD8+ cells, lacking the expression of the NK cell and NK T-cell markers CD16/CD56, did not change during stimulation. The low dose IL-2 used for stimulation also strongly argues against the generation of LAK or cytokine-induced killer cells capable of lysing any cell type in an equal efficient way 26–28. The cold-target inhibition assays demonstrated that BEAS-2B cells inhibited the lysis of K562, but not vice versa, showing that BEAS-2B-stimulated effector cells have a higher affinity for BEAS-2B than for K562 cells, which is in contradiction to a NK cell-, NK T-cell- or LAK cell-mediated killing. Interestingly, cold-target inhibition also demonstrated that K562 cells are lysed by the same effector population killing the BEAS-2B cells, albeit to a lesser extent, and the residual lysis of K562 cells is also inhibited when NKG2D is blocked.
Killing of BEAS-2B cells or PBECs by the CD8+ T-cell effector population was not impaired in the presence of the MHC class I blocking antibody W6/32. It was demonstrated by FACS that W6/32 binds to BEAS-2B cells and PBECs and the blocking function of the used lots of W6/32 was confirmed in parallel cytotoxicity assays with human vascular endothelial cells as targets (data not shown) 29.
The impact of NKG2D in an effector function is emphasised by significantly decreased cytotoxicity when blocking the NKG2D-receptor on CD8+ T-cells and the finding that BEAS-2B cells and PBECs express the NKG2D ligands MIC A/B. This is paralleled by upregulation of NKG2D on CD8+ T-cells during the course of alloantigen-specific activation. Importantly, experiments performed with PBECs gave the same results as those performed with BEAS-2B cells, which excludes cell-type nonspecific-induced effector function due to an immune response against the virally immortalised cell line BEAS-2B.
Inhibition of NKG2D did not completely abolish lysis; therefore, additional mechanisms have to be assumed for co-stimulation, target recognition and lysis. As CD70 expression is increased on alloactivated CD8+ T-cells, the CD70–CD27 interaction might contribute to the co-stimulation, as has already been shown 7. In the present study, cell lysis was very likely caused by the granzyme B/perforin pathway. Interestingly, neither neutralising CD95 nor TNF, inhibited CD8+ T-cell cytotoxicity, although both are well known for their cytotoxic activity.
In summary, the results of the present study affirm that respiratory epithelial cells can function as nonclassical antigen-presenting cells and represent a target of cytotoxic T-cell reactivity. This supports the hypothesis of a lung directed graft-versus-host disease following allogeneic stem-cell transplantation. Interestingly, respiratory epithelial-cell specific cytotoxic T-cells show nonclassical partially natural killer group 2 member D-mediated T-cell effector function but not major histocompatibility complex I restriction, although they express classical activation markers upon allogeneic activation. This might be of clinical relevance for understanding the course of graft-versus-host disease and lung injury seen in patients following stem cell transplantation.
Support statement
This study was supported by a grant from the Wilhelm Sander Foundation (Munich, Germany).
Statement of interest
None declared.
- Received July 29, 2007.
- Accepted April 7, 2008.
- © ERS Journals Ltd
References










